Regulation of Cancer Stem Cell Metabolism by Secreted Frizzled-Related Protein 4 (sFRP4)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
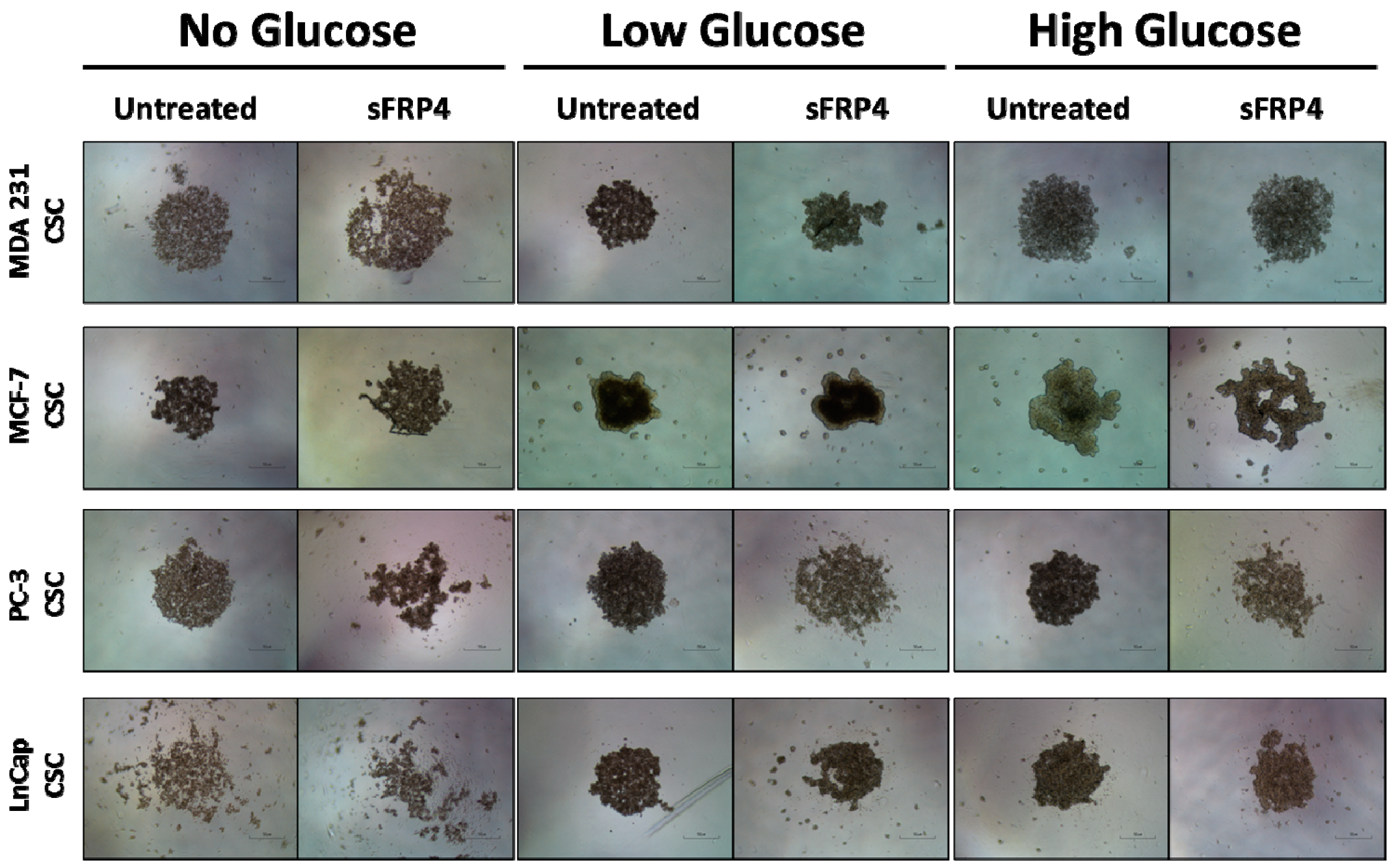
2.1. The Sphere Forming Capacity of CSCs Is Reduced by sFRP4 Irrespective of Glucose Content
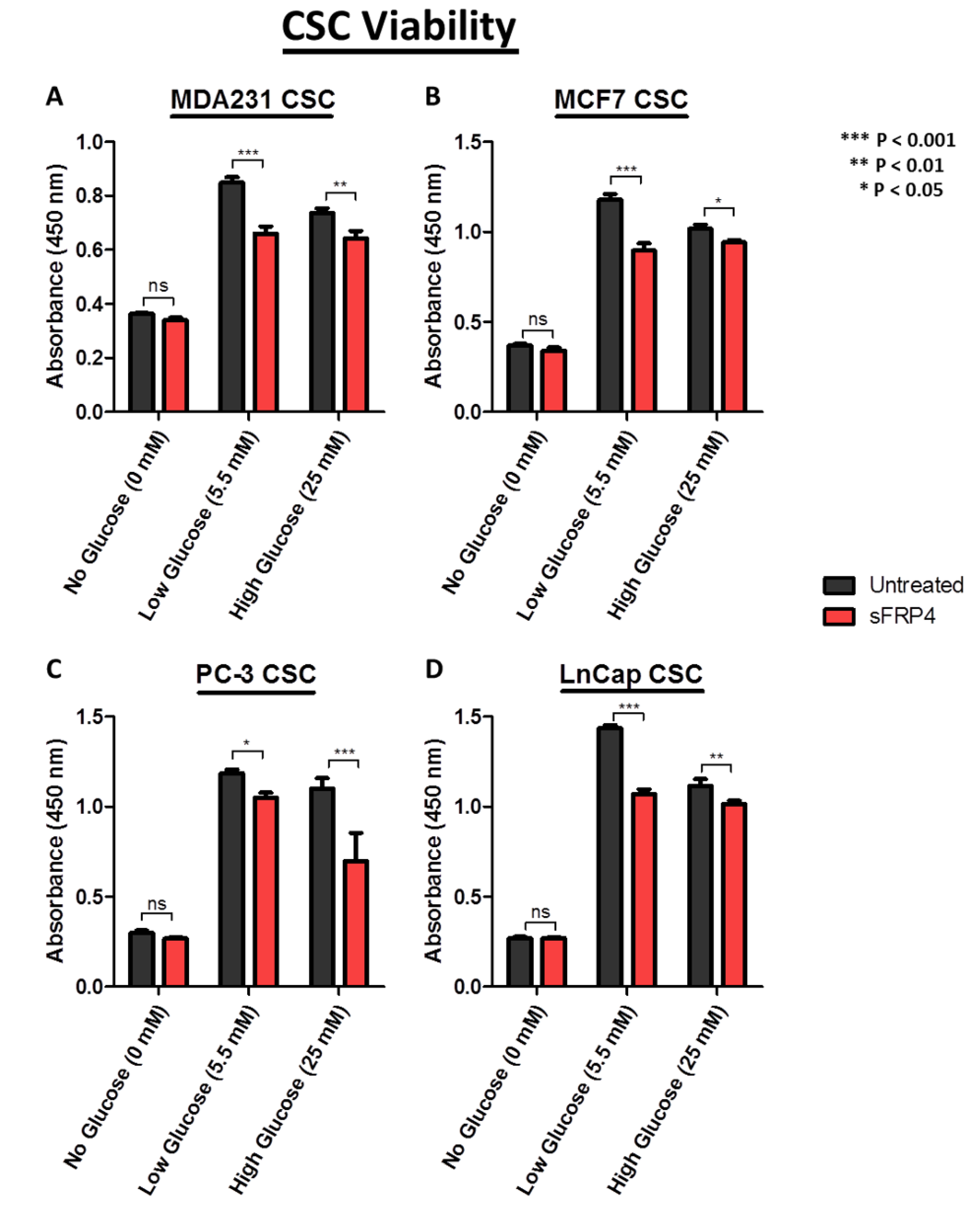
2.2. CSC Viability Is Reduced by sFRP4 in Low and High Glucose Conditions
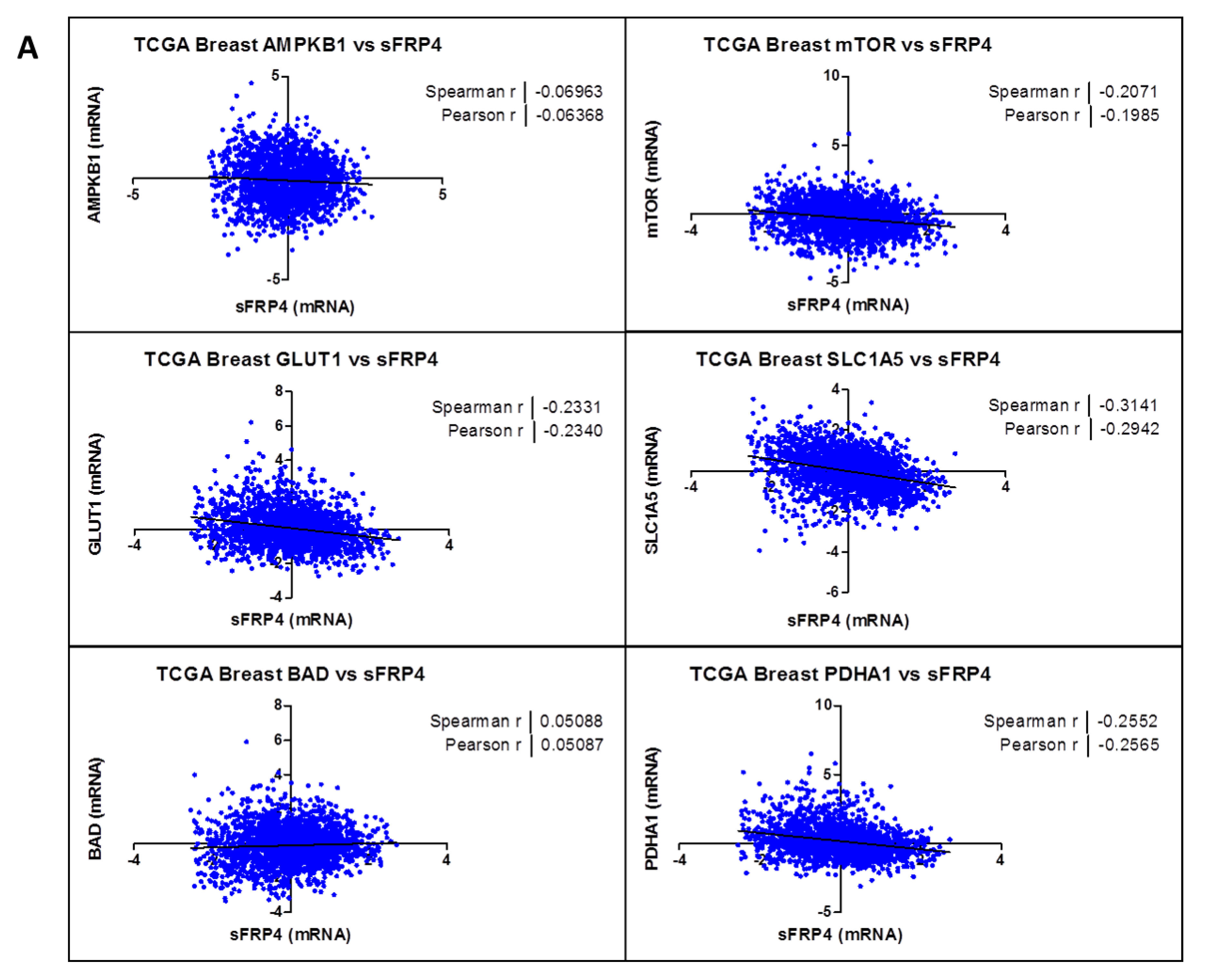
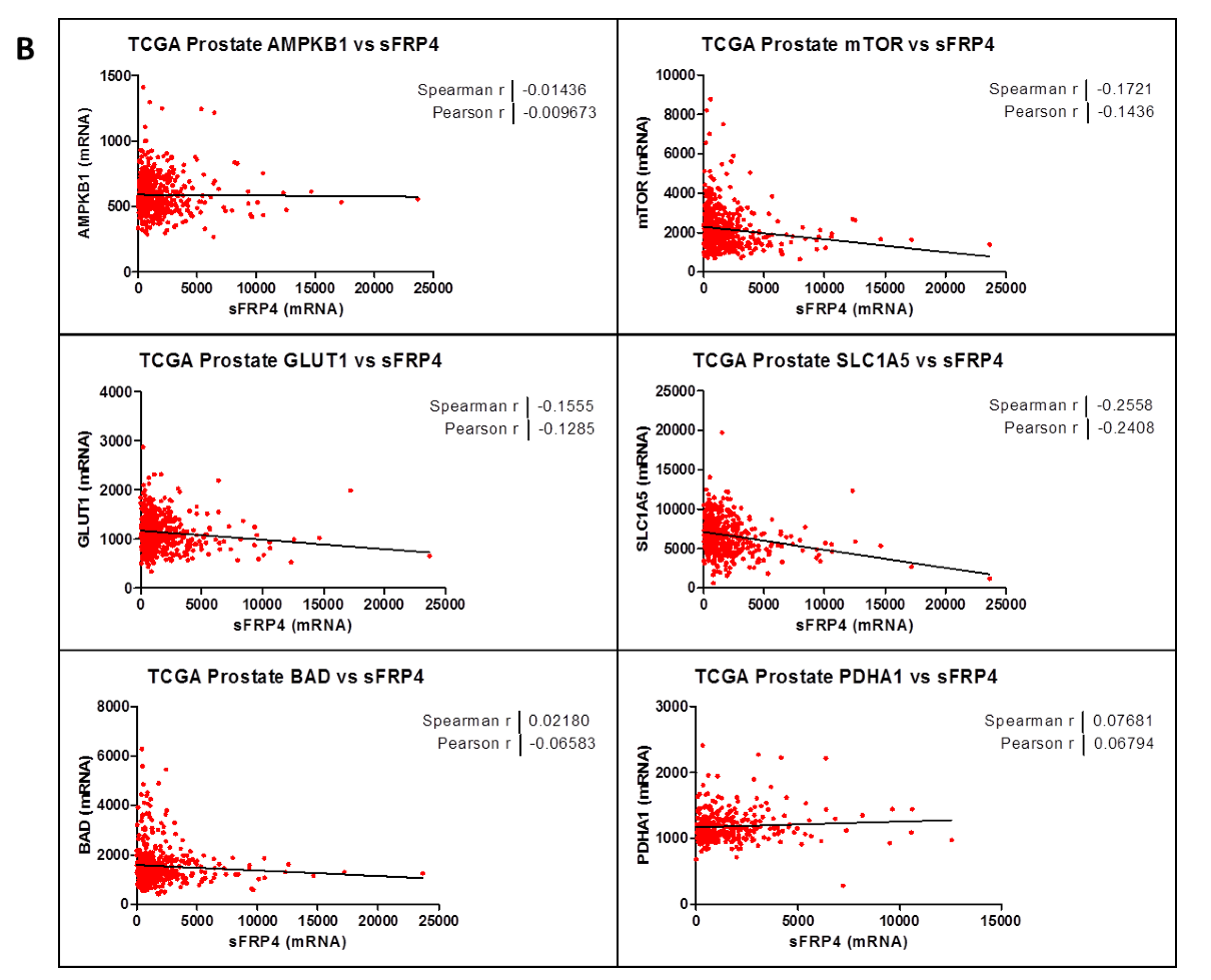
2.3. Correlation Coefficient of sFRP4 with Metabolic Gene-Set
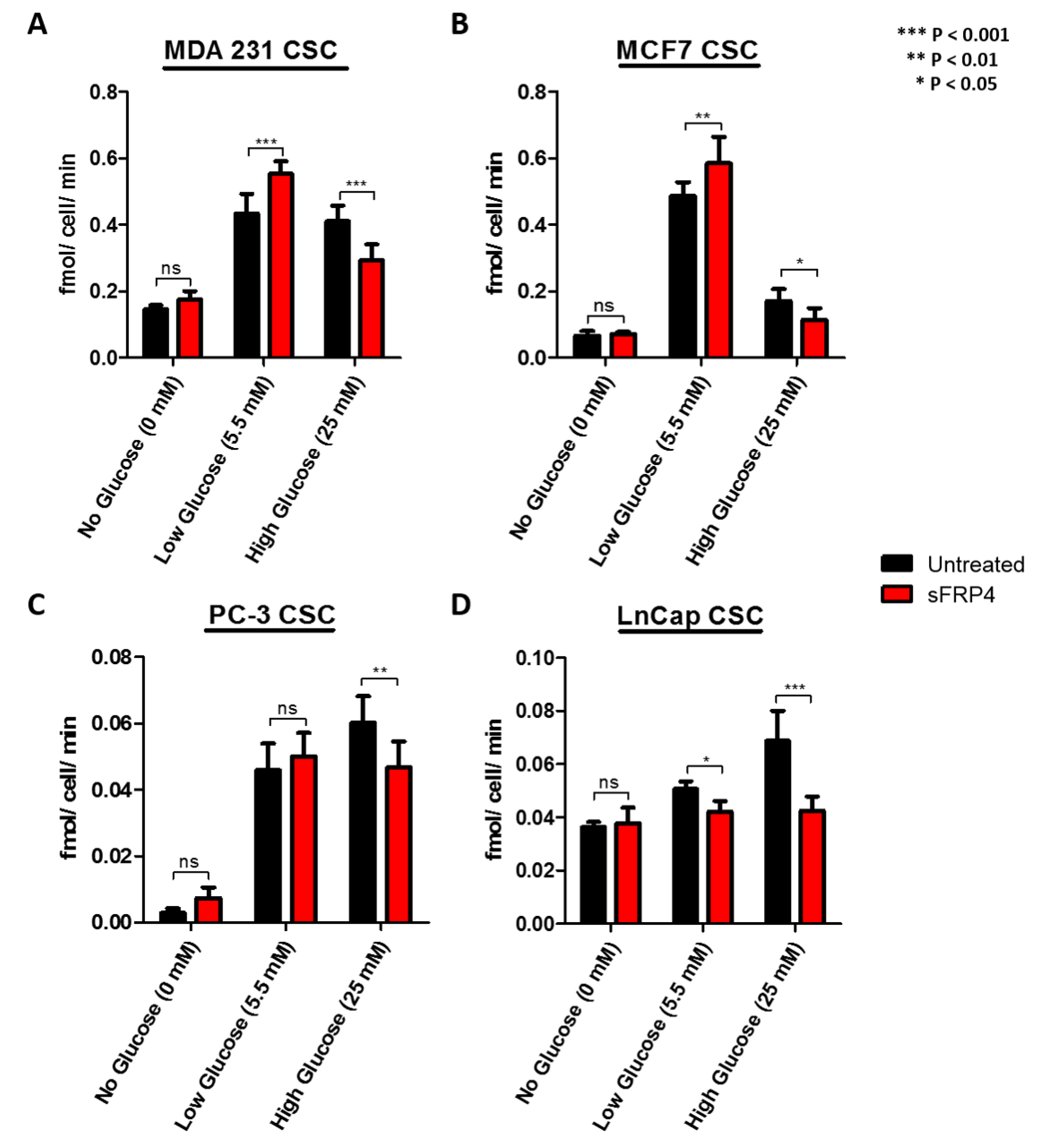
2.4. The Effect of Glucose and sFRP4 on Glucose-Uptake in CSCs
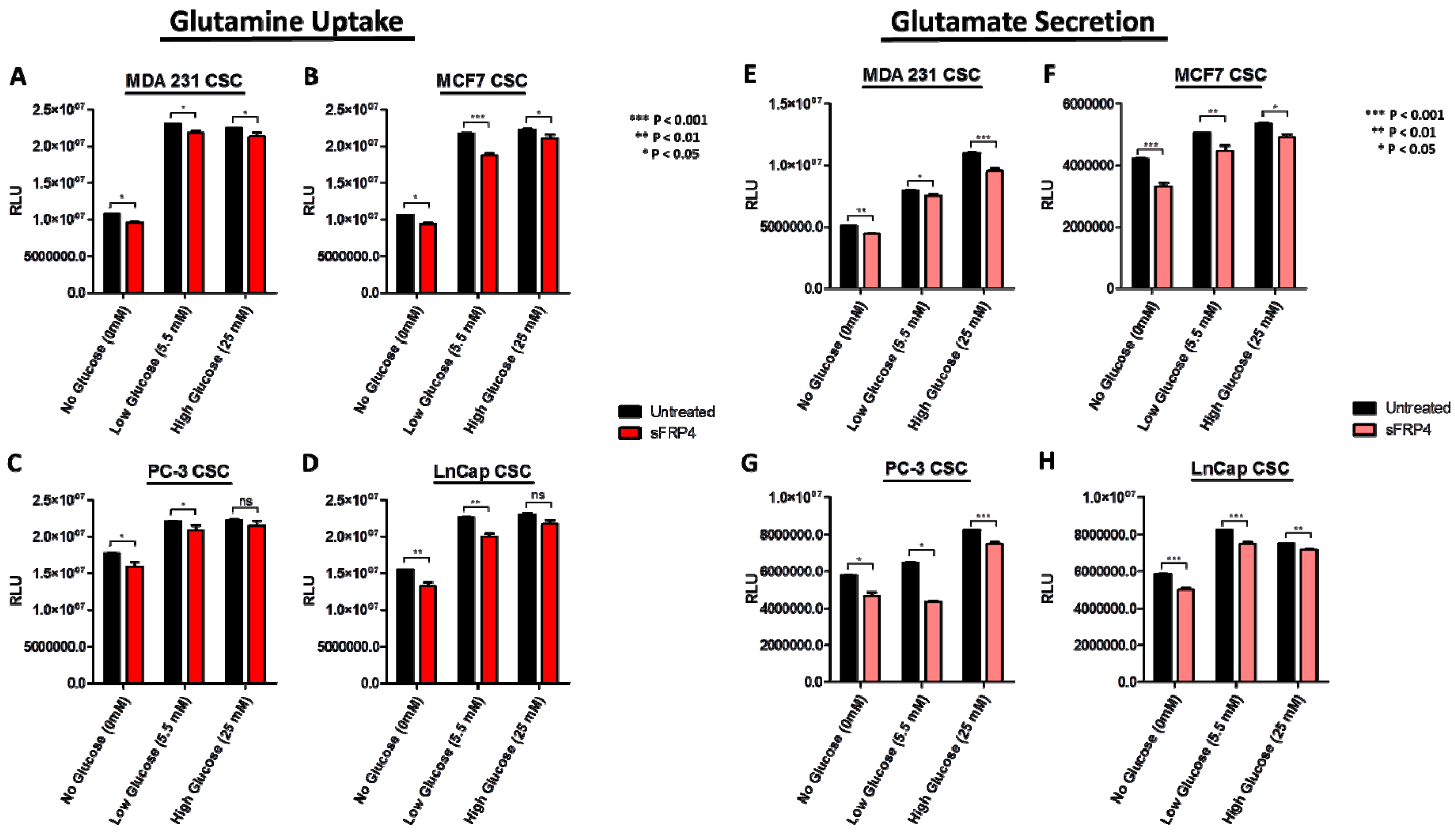
2.5. Changes in Extracellular Metabolites with sFRP4 Treatment of CSCs
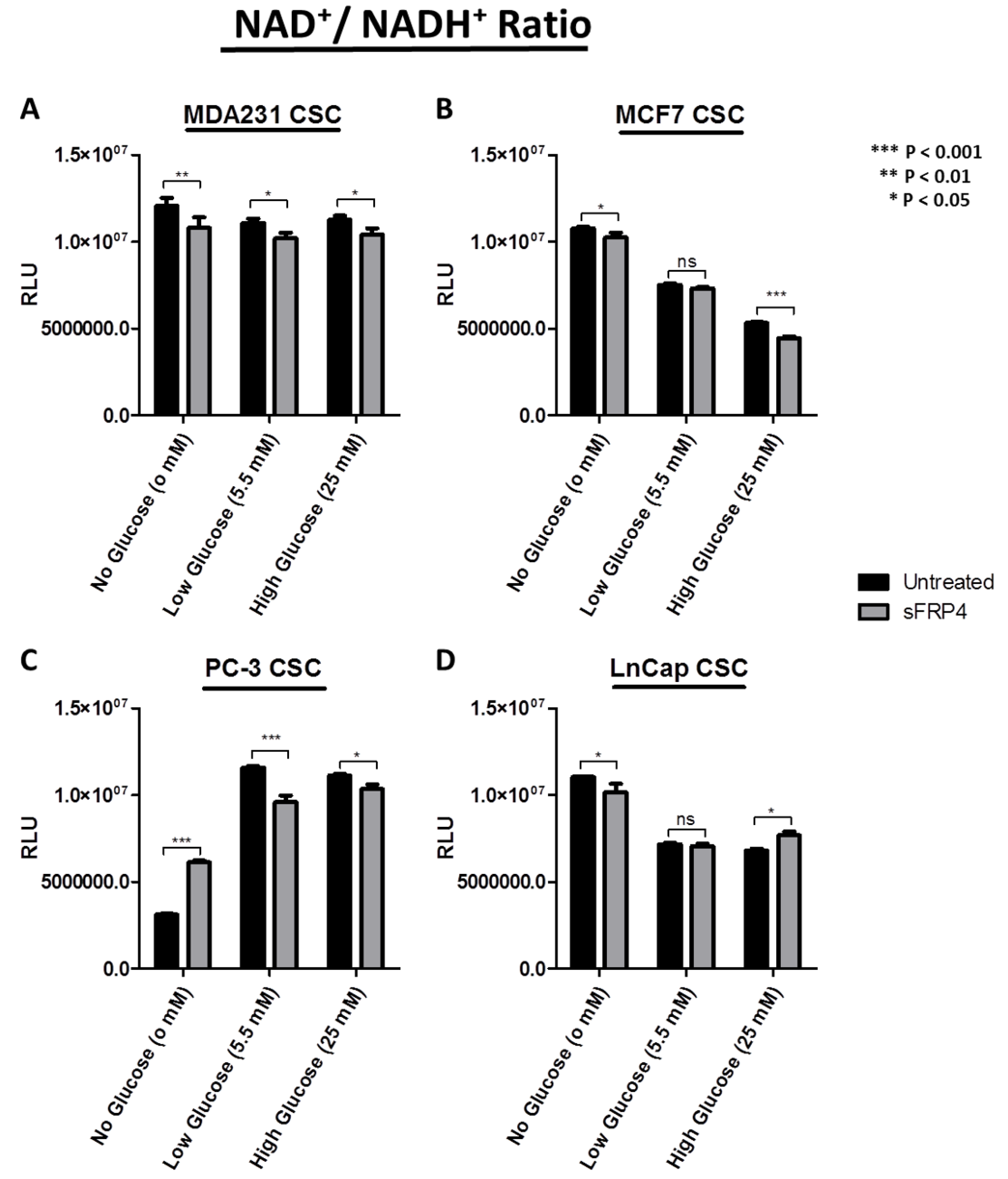
2.6. Changes in Redox Signature with sFRP4 Treatment of CSCs
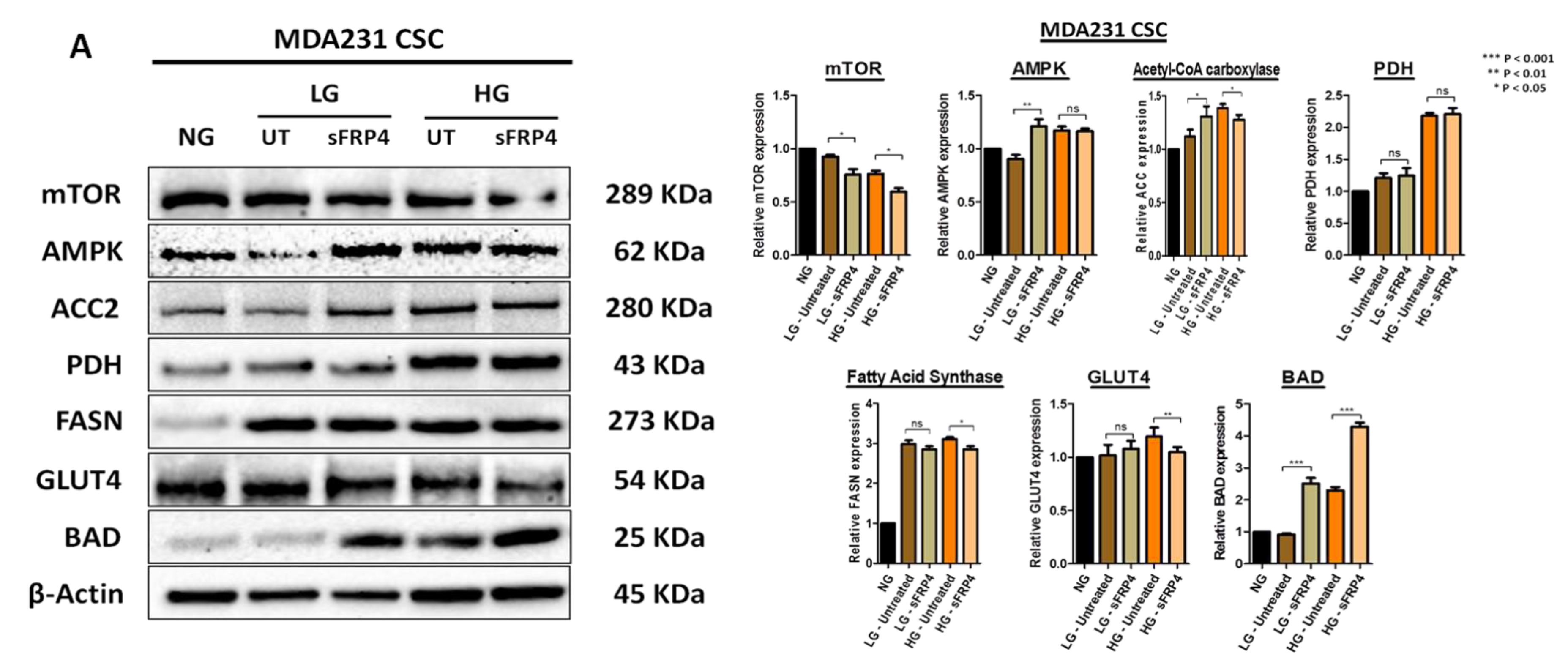
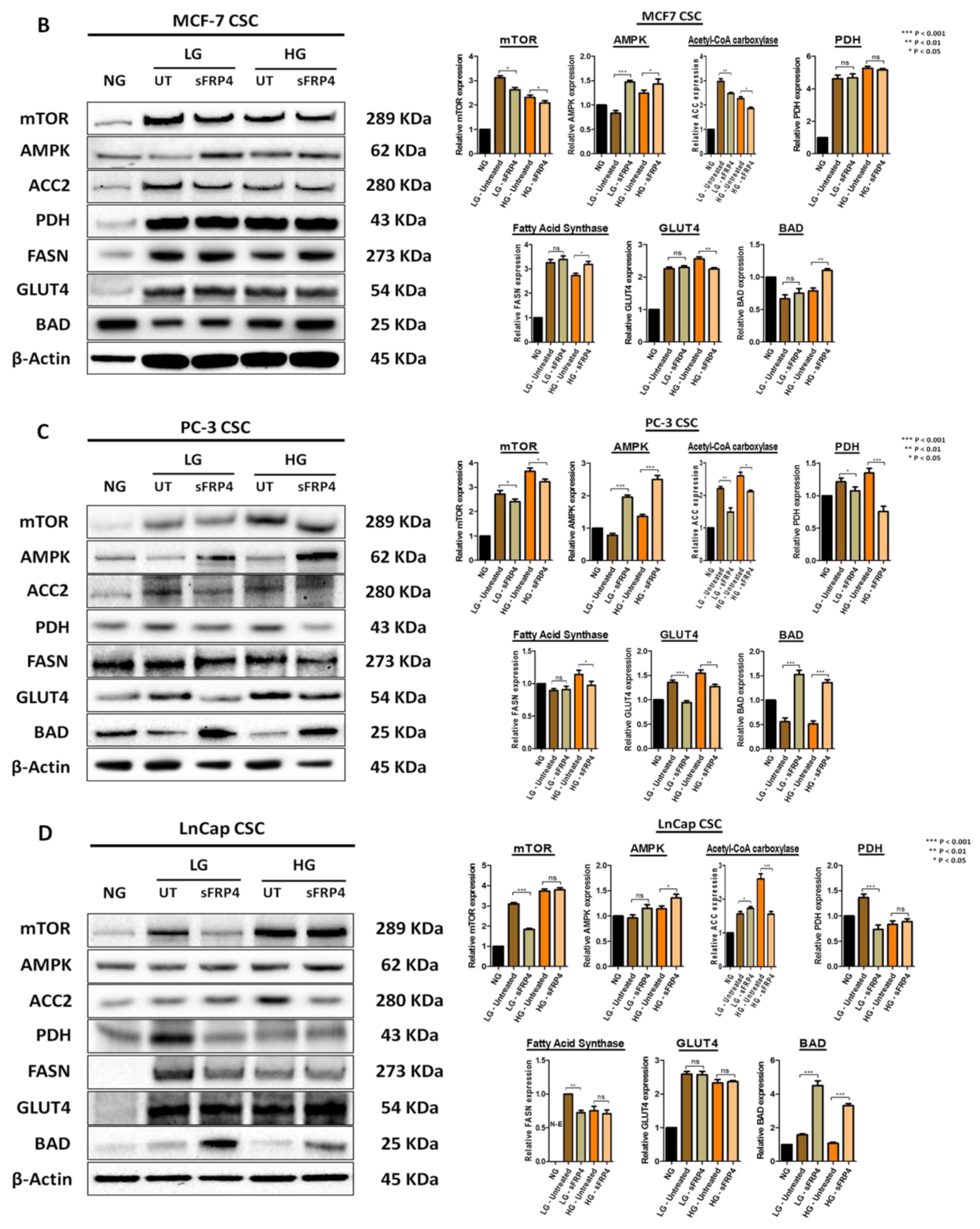
2.7. The Effect of sFRP4 on CSC Metabolism Target Proteins
3. Discussion
4. Materials and Methods
4.1. Cell Culture
Monolayer Cell Culture
4.2. Cancer Stem Cell Isolation
4.3. CSC Treatment
4.4. Viability Assay
4.5. Cell Surface Markers
4.6. The Cancer Genome Atlas Dataset
4.7. Glucose Uptake in CSCs
4.8. Detection of Extracellular Metabolites in CSC Medium
4.9. Detection of Qualitative NAD+/NADH in CSCs
4.10. Western Blotting
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Regenbrecht, C.R.; Lehrach, H.; Adjaye, J. Stemming cancer: Functional genomics of cancer stem cells in solid tumors. Stem Cell Rev. 2008, 4, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.M.; Ng, A.V.; Lam, S.; Hung, J.Y. Side population in human lung cancer cell lines and tumors is enriched with stem-like cancer cells. Cancer Res. 2007, 67, 4827–4833. [Google Scholar] [CrossRef] [PubMed]
- Dalerba, P.; Cho, R.W.; Clarke, M.F. Cancer stem cells: Models and concepts. Annu. Rev. Med. 2007, 58, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, D.; Becher, O.J.; Holland, E.C. Cancer stem cells and survival pathways. Cell Cycle 2008, 7, 1371–1378. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, A.; Kumar, S.; Arfuso, F.; Newsholme, P.; Dharmarajan, A. Secreted frizzled-related protein 4 (sFRP4) chemo-sensitizes cancer stem cells derived from human breast, prostate, and ovary tumor cell lines. Sci. Rep. 2017, 7, 2256. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Tan, W.; Wu, X.; Poustovoitov, M.; Strasner, A.; Li, W.; Borcherding, N.; Ghassemian, M.; Karin, M. A NIK-IKKα module expands ErbB2-induced tumor-initiating cells by stimulating nuclear export of p27/Kip1. Cancer Cell 2013, 23, 647–659. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.S.; Chandel, N.S. ROS links glucose metabolism to breast cancer stem cell and EMT phenotype. Cancer Cell 2013, 23, 265–267. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, Y.; Shingu, T.; Feng, L.; Chen, Z.; Ogasawara, M.; Keating, M.J.; Kondo, S.; Huang, P. Metabolic alterations in highly tumorigenic glioblastoma cells: Preference for hypoxia and high dependency on glycolysis. J. Biol. Chem. 2011, 286, 32843–32853. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Joven, J.; Cufi, S.; Corominas-Faja, B.; Oliveras-Ferraros, C.; Cuyas, E.; Martin-Castillo, B.; Lopez-Bonet, E.; Alarcon, T.; Vazquez-Martin, A. The Warburg effect version 2.0: Metabolic reprogramming of cancer stem cells. Cell Cycle (Georgetown, Tex.) 2013, 12, 1166–1179. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Wang, F.; Chen, G.; Zhang, H.; Feng, L.; Wang, L.; Colman, H.; Keating, M.J.; Li, X.; Xu, R.H.; et al. Effective elimination of cancer stem cells by a novel drug combination strategy. Stem Cells 2013, 31, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Bienz, M.; Clevers, H. Linking colorectal cancer to Wnt signaling. Cell 2000, 103, 311–320. [Google Scholar] [CrossRef]
- Longman, D.; Arfuso, F.; Viola, H.M.; Hool, L.C.; Dharmarajan, A.M. The role of the cysteine-rich domain and netrin-like domain of secreted frizzled-related protein 4 in angiogenesis inhibition in vitro. Oncol. Res. 2012, 20, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Bhuvanalakshmi, G.; Arfuso, F.; Millward, M.; Dharmarajan, A.; Warrier, S. Secreted frizzled-related protein 4 inhibits glioma stem-like cells by reversing epithelial to mesenchymal transition, inducing apoptosis and decreasing cancer stem cell properties. PLoS ONE 2015, 10, e0127517. [Google Scholar]
- Bodoor, K.; Haddad, Y.; Alkhateeb, A.; Al-Abbadi, A.; Dowairi, M.; Magableh, A.; Bsoul, N.; Ghabkari, A. DNA hypermethylation of cell cycle (p15 and p16) and apoptotic (p14, p53, DAPK and TMS1) genes in peripheral blood of leukemia patients. Asian Pac. J. Cancer Prev. 2014, 15, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Nakano, A.; Tsuji, D.; Miki, H.; Cui, Q.; El Sayed, S.M.; Ikegame, A.; Oda, A.; Amou, H.; Nakamura, S.; Harada, T.; et al. Glycolysis inhibition inactivates ABC transporters to restore drug sensitivity in malignant cells. PLoS ONE 2011, 6, e27222. [Google Scholar] [CrossRef] [PubMed]
- Plaks, V.; Kong, N.; Werb, Z. The cancer stem cell niche: How essential is the niche in regulating stemness of tumor cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Warrier, S.; Balu, S.K.; Kumar, A.P.; Millward, M.; Dharmarajan, A. Wnt antagonist, secreted frizzled-related protein 4 (sFRP4), increases chemotherapeutic response of glioma stem-like cells. Oncol. Res. 2013, 21, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Warrier, S.; Bhuvanalakshmi, G.; Arfuso, F.; Rajan, G.; Millward, M.; Dharmarajan, A. Cancer stem-like cells from head and neck cancers are chemosensitized by the wnt antagonist, sFRP4, by inducing apoptosis, decreasing stemness, drug resistance and epithelial to mesenchymal transition. Cancer Gene Ther. 2014, 21, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.R.; Seo, J.H.; Chong, A.; Min, J.J.; Song, H.C.; Kim, Y.C.; Bom, H.S. Whole-body metabolic tumour volume of 18F-FDG PET/CT improves the prediction of prognosis in small cell lung cancer. Eur. J. Nucl. Med. Mol. Imaging 2012, 39, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Poulou, L.S.; Thanos, L.; Ziakas, P.D. Unifying the predictive value of pretransplant FDG PET in patients with lymphoma: A review and meta-analysis of published trials. Eur. J. Nucl. Med. Mol. Imaging 2010, 37, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Goerres, G.W.; Stupp, R.; Barghouth, G.; Hany, T.F.; Pestalozzi, B.; Dizendorf, E.; Schnyder, P.; Luthi, F.; von Schulthess, G.K.; Leyvraz, S. The value of PET, CT and in-line PET/CT in patients with gastrointestinal stromal tumours: Long-term outcome of treatment with imatinib mesylate. Eur. J. Nucl. Med. Mol. Imaging 2005, 32, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Kostakoglu, L.; Coleman, M.; Leonard, J.P.; Kuji, I.; Zoe, H.; Goldsmith, S.J. PET predicts prognosis after 1 cycle of chemotherapy in aggressive lymphoma and Hodgkin’s disease. J. Nucl. Med. 2002, 43, 1018–1027. [Google Scholar] [PubMed]
- Liu, P.P.; Liao, J.; Tang, Z.J.; Wu, W.J.; Yang, J.; Zeng, Z.L.; Hu, Y.; Wang, P.; Ju, H.Q.; Xu, R.H.; et al. Metabolic regulation of cancer cell side population by glucose through activation of the Akt pathway. Cell Death Differ. 2014, 21, 124–135. [Google Scholar] [CrossRef] [PubMed]
- Siegfried, Z.; Simon, I. DNA methylation and gene expression. Wiley Interdiscip. Rev. 2010, 2, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Vlashi, E.; Lagadec, C.; Vergnes, L.; Matsutani, T.; Masui, K.; Poulou, M.; Popescu, R.; Della Donna, L.; Evers, P.; Dekmezian, C.; et al. Metabolic state of glioma stem cells and nontumorigenic cells. Proc. Natl. Acad. Sci. USA 2011, 108, 16062–16067. [Google Scholar] [CrossRef] [PubMed]
- Ciavardelli, D.; Rossi, C.; Barcaroli, D.; Volpe, S.; Consalvo, A.; Zucchelli, M.; De Cola, A.; Scavo, E.; Carollo, R.; D’Agostino, D.; et al. Breast cancer stem cells rely on fermentative glycolysis and are sensitive to 2-deoxyglucose treatment. Cell Death Dis. 2014, 5, e1336. [Google Scholar] [CrossRef] [PubMed]
- Hammoudi, N.; Ahmed, K.B.; Garcia-Prieto, C.; Huang, P. Metabolic alterations in cancer cells and therapeutic implications. Chin. J. Cancer 2011, 30, 508–525. [Google Scholar] [CrossRef] [PubMed]
- Helmlinger, G.; Sckell, A.; Dellian, M.; Forbes, N.S.; Jain, R.K. Acid production in glycolysis-impaired tumors provides new insights into tumor metabolism. Clin. Cancer Res. 2002, 8, 1284–1291. [Google Scholar] [PubMed]
- Koochekpour, S.; Majumdar, S.; Azabdaftari, G.; Attwood, K.; Scioneaux, R.; Subramani, D.; Manhardt, C.; Lorusso, G.D.; Willard, S.S.; Thompson, H.; et al. Serum glutamate levels correlate with gleason score and glutamate blockade decreases proliferation, migration, and invasion and induces apoptosis in prostate cancer cells. Clin. Cancer Res. 2012, 18, 5888–5901. [Google Scholar] [CrossRef] [PubMed]
- Simpson, N.E.; Tryndyak, V.P.; Pogribna, M.; Beland, F.A.; Pogribny, I.P. Modifying metabolically sensitive histone marks by inhibiting glutamine metabolism affects gene expression and alters cancer cell phenotype. Epigenetics 2012, 7, 1413–1420. [Google Scholar] [CrossRef] [PubMed]
- Phang, J.M.; Liu, W.; Hancock, C. Bridging epigenetics and metabolism: Role of non-essential amino acids. Epigenetics 2013, 8, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.M.; Yu, Z.X.; Lin, H.; Fu, X.; Meadows, G.G. Selective amino acid restriction differentially affects the motility and directionality of du145 and PC3 prostate cancer cells. J. Cell. Physiol. 2008, 217, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.M.; Lin, H.; Liu, X.; Fang, W.; Meadows, G.G. Cell death of prostate cancer cells by specific amino acid restriction depends on alterations of glucose metabolism. J. Cell. Physiol. 2010, 224, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Maity, P. Effect of glutamine analogue-acivicin on tumor induced angiogenesis in Ehrlich ascites carcinoma. Indian J. Exp. Biol. 2005, 43, 407–413. [Google Scholar] [PubMed]
- Willems, L.; Jacque, N.; Jacquel, A.; Neveux, N.; Maciel, T.T.; Lambert, M.; Schmitt, A.; Poulain, L.; Green, A.S.; Uzunov, M.; et al. Inhibiting glutamine uptake represents an attractive new strategy for treating acute myeloid leukemia. Blood 2013, 122, 3521–3532. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.; Xing, J.; Xiao, X.; Liou, A.K.; Gao, Y.; Yin, X.M.; Clark, R.S.; Graham, S.H.; Chen, J. Critical role of calpain I in mitochondrial release of apoptosis-inducing factor in ischemic neuronal injury. J. Neurosci. 2007, 27, 9278–9293. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Sheng, X.; Willson, A.K.; Roque, D.R.; Stine, J.E.; Guo, H.; Jones, H.M.; Zhou, C.; Bae-Jump, V.L. Glutamine promotes ovarian cancer cell proliferation through the mTOR/S6 pathway. Endocr. Relat. Cancer 2015, 22, 577–591. [Google Scholar] [CrossRef] [PubMed]
- Locasale, J.W.; Cantley, L.C. Metabolic flux and the regulation of mammalian cell growth. Cell Metab. 2011, 14, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Yuan, T.; Wu, Y.; Wang, Y.; Fan, T.W.; Miriyala, S.; Lin, Y.; Yao, J.; Shi, J.; Kang, T.; et al. Loss of FBP1 by snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell 2013, 23, 316–331. [Google Scholar] [CrossRef] [PubMed]
- Son, M.J.; Son, M.Y.; Seol, B.; Kim, M.J.; Yoo, C.H.; Han, M.K.; Cho, Y.S. Nicotinamide overcomes pluripotency deficits and reprogramming barriers. Stem Cells 2013, 31, 1121–1135. [Google Scholar] [CrossRef] [PubMed]
- Bonuccelli, G.; De Francesco, E.M.; de Boer, R.; Tanowitz, H.B.; Lisanti, M.P. Nadh autofluorescence, a new metabolic biomarker for cancer stem cells: Identification of vitamin C and cape as natural products targeting “stemness”. Oncotarget 2017, 8, 20667–20678. [Google Scholar] [CrossRef] [PubMed]
- Cuyas, E.; Corominas-Faja, B.; Menendez, J.A. The nutritional phenome of EMT-induced cancer stem-like cells. Oncotarget 2014, 5, 3970–3982. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Graham, P.H.; Hao, J.; Ni, J.; Bucci, J.; Cozzi, P.J.; Kearsley, J.H.; Li, Y. Acquisition of epithelial-mesenchymal transition and cancer stem cell phenotypes is associated with activation of the PI3K/Akt/mTOR pathway in prostate cancer radioresistance. Cell Death Dis. 2013, 4, e875. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wulfkuhle, J.; Zhang, H.; Gu, P.; Yang, Y.; Deng, J.; Margolick, J.B.; Liotta, L.A.; Petricoin, E., 3rd; Zhang, Y. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proc. Natl. Acad. Sci. USA 2007, 104, 16158–16163. [Google Scholar] [CrossRef] [PubMed]
- Douville, J.; Beaulieu, R.; Balicki, D. ALDH1 as a functional marker of cancer stem and progenitor cells. Stem Cells Dev. 2009, 18, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Huang, E.H.; Hynes, M.J.; Zhang, T.; Ginestier, C.; Dontu, G.; Appelman, H.; Fields, J.Z.; Wicha, M.S.; Boman, B.M. Aldehyde dehydrogenase 1 is a marker for normal and malignant human colonic stem cells (SC) and tracks SC overpopulation during colon tumorigenesis. Cancer Res. 2009, 69, 3382–3389. [Google Scholar] [CrossRef] [PubMed]
- Sunayama, J.; Matsuda, K.; Sato, A.; Tachibana, K.; Suzuki, K.; Narita, Y.; Shibui, S.; Sakurada, K.; Kayama, T.; Tomiyama, A.; et al. Crosstalk between the PI3K/mTOR and MEK/ERK pathways involved in the maintenance of self-renewal and tumorigenicity of glioblastoma stem-like cells. Stem Cells 2010, 28, 1930–1939. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.J.; Rho, J.K.; Kim, Y.M.; Jung, J.E.; Jin, Y.B.; Ko, Y.G.; Lee, J.S.; Lee, S.J.; Lee, J.C.; Park, M.J. Upregulation of CXCR4 is functionally crucial for maintenance of stemness in drug-resistant non-small cell lung cancer cells. Oncogene 2013, 32, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Song, C.W.; Lee, H.; Dings, R.P.; Williams, B.; Powers, J.; Santos, T.D.; Choi, B.H.; Park, H.J. Metformin kills and radiosensitizes cancer cells and preferentially kills cancer stem cells. Sci. Rep. 2012, 2, 362. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, A.; Deshpande, K.; Arfuso, F.; Newsholme, P.; Dharmarajan, A. Cancer stem cell metabolism: A potential target for cancer therapy. Mol. Cancer 2016, 15, 69. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef]
- Singh, R.; Cuervo, A.M. Lipophagy: Connecting autophagy and lipid metabolism. Int. J. Cell Biol. 2012, 2012, 282041. [Google Scholar] [CrossRef] [PubMed]
- Bian, Y.; Yu, Y.; Wang, S.; Li, L. Up-regulation of fatty acid synthase induced by EGFR/ERK activation promotes tumor growth in pancreatic cancer. Biochem. Biophys. Res. Commun. 2015, 463, 612–617. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Wang, H.; Li, J.; Fang, X.; Pan, H.; Yuan, X.; Zhang, P. Up-regulated FASN expression promotes transcoelomic metastasis of ovarian cancer cell through epithelial-mesenchymal transition. Int. J. Mol. Sci. 2014, 15, 11539–11554. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Dong, L.; Wei, D.; Wang, X.; Zhang, S.; Li, H. Fatty acid synthase mediates the epithelial-mesenchymal transition of breast cancer cells. Int. J. Biol. Sci. 2014, 10, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Zaytseva, Y.Y.; Rychahou, P.G.; Gulhati, P.; Elliott, V.A.; Mustain, W.C.; O’Connor, K.; Morris, A.J.; Sunkara, M.; Weiss, H.L.; Lee, E.Y.; et al. Inhibition of fatty acid synthase attenuates CD44-associated signaling and reduces metastasis in colorectal cancer. Cancer Res. 2012, 72, 1504–1517. [Google Scholar] [CrossRef] [PubMed]
- Mullen, G.E.; Yet, L. Progress in the development of fatty acid synthase inhibitors as anticancer targets. Bioorg. Med. Chem. Lett. 2015, 25, 4363–4369. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.S.; Nemeria, N.S.; Furey, W.; Jordan, F. The pyruvate dehydrogenase complexes: Structure-based function and regulation. J. Biol. Chem. 2014, 289, 16615–16623. [Google Scholar] [CrossRef] [PubMed]
- Holness, M.J.; Sugden, M.C. Regulation of pyruvate dehydrogenase complex activity by reversible phosphorylation. Biochem. Soc. Trans. 2003, 31, 1143–1151. [Google Scholar] [CrossRef] [PubMed]
- Corominas-Faja, B.; Cuyas, E.; Gumuzio, J.; Bosch-Barrera, J.; Leis, O.; Martin, A.G.; Menendez, J.A. Chemical inhibition of acetyl-CoA carboxylase suppresses self-renewal growth of cancer stem cells. Oncotarget 2014, 5, 8306–8316. [Google Scholar] [CrossRef] [PubMed]
- Flier, J.S.; Mueckler, M.M.; Usher, P.; Lodish, H.F. Elevated levels of glucose transport and transporter messenger RNA are induced by ras or src oncogenes. Science 1987, 235, 1492–1495. [Google Scholar] [CrossRef] [PubMed]
- Sastry, K.S.; Al-Muftah, M.A.; Li, P.; Al-Kowari, M.K.; Wang, E.; Ismail Chouchane, A.; Kizhakayil, D.; Kulik, G.; Marincola, F.M.; Haoudi, A.; et al. Targeting proapoptotic protein bad inhibits survival and self-renewal of cancer stem cells. Cell Death Differ. 2014, 21, 1936–1949. [Google Scholar] [CrossRef] [PubMed]
- Al-Bazz, Y.O.; Underwood, J.C.; Brown, B.L.; Dobson, P.R. Prognostic significance of Akt, phospho-Akt and bad expression in primary breast cancer. Eur. J. Cancer 2009, 45, 694–704. [Google Scholar] [CrossRef] [PubMed]
- Condorelli, F.; Salomoni, P.; Cotteret, S.; Cesi, V.; Srinivasula, S.M.; Alnemri, E.S.; Calabretta, B. Caspase cleavage enhances the apoptosis-inducing effects of bad. Mol. Cell Biol. 2001, 21, 3025–3036. [Google Scholar] [CrossRef] [PubMed]
- Karsten, U.; Goletz, S. What makes cancer stem cell markers different? Springerplus 2013, 2, 301. [Google Scholar] [CrossRef] [PubMed]
- Collins, A.T.; Berry, P.A.; Hyde, C.; Stower, M.J.; Maitland, N.J. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005, 65, 10946–10951. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cbioportal. Sci. Signal. 2013, 6. [Google Scholar] [CrossRef] [PubMed]
- Valley, M.P.; Karassina, N.; Aoyama, N.; Carlson, C.; Cali, J.J.; Vidugiriene, J. A bioluminescent assay for measuring glucose uptake. Anal. Biochem. 2016, 505, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Leippe, D.; Sobol, M.; Vidugiris, G.; Cali, J.J.; Vidugiriene, J. Bioluminescent assays for glucose and glutamine metabolism: High-throughput screening for changes in extracellular and intracellular metabolites. SLAS Discov. 2017, 22, 366–377. [Google Scholar] [CrossRef] [PubMed]









© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deshmukh, A.; Arfuso, F.; Newsholme, P.; Dharmarajan, A. Regulation of Cancer Stem Cell Metabolism by Secreted Frizzled-Related Protein 4 (sFRP4). Cancers 2018, 10, 40. https://doi.org/10.3390/cancers10020040
Deshmukh A, Arfuso F, Newsholme P, Dharmarajan A. Regulation of Cancer Stem Cell Metabolism by Secreted Frizzled-Related Protein 4 (sFRP4). Cancers. 2018; 10(2):40. https://doi.org/10.3390/cancers10020040
Chicago/Turabian StyleDeshmukh, Abhijeet, Frank Arfuso, Philip Newsholme, and Arun Dharmarajan. 2018. "Regulation of Cancer Stem Cell Metabolism by Secreted Frizzled-Related Protein 4 (sFRP4)" Cancers 10, no. 2: 40. https://doi.org/10.3390/cancers10020040
APA StyleDeshmukh, A., Arfuso, F., Newsholme, P., & Dharmarajan, A. (2018). Regulation of Cancer Stem Cell Metabolism by Secreted Frizzled-Related Protein 4 (sFRP4). Cancers, 10(2), 40. https://doi.org/10.3390/cancers10020040