Oncogenic Activation of Nrf2, Though as a Master Antioxidant Transcription Factor, Liberated by Specific Knockout of the Full-Length Nrf1α that Acts as a Dominant Tumor Repressor



Abstract
1. Introduction
2. Results
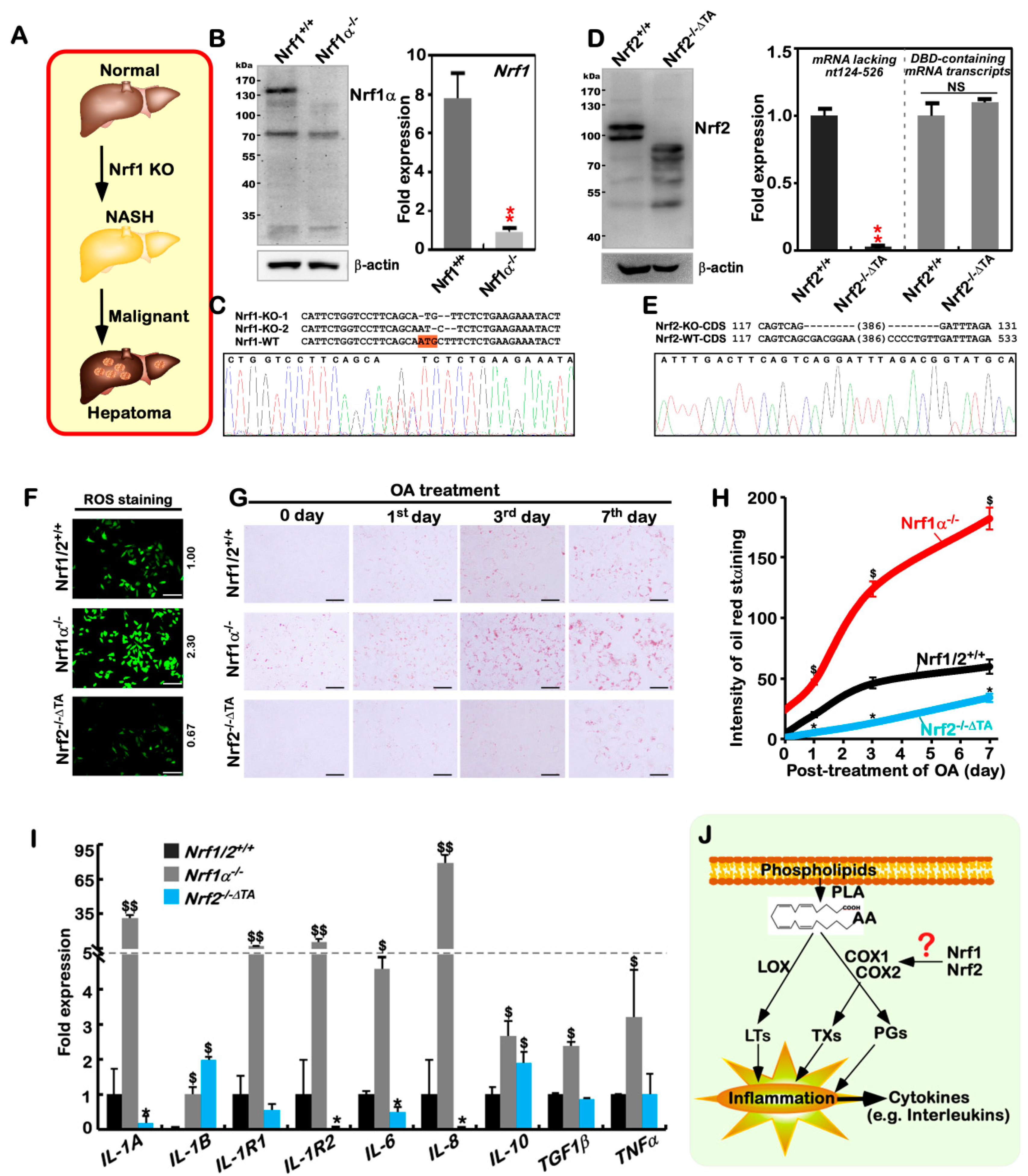
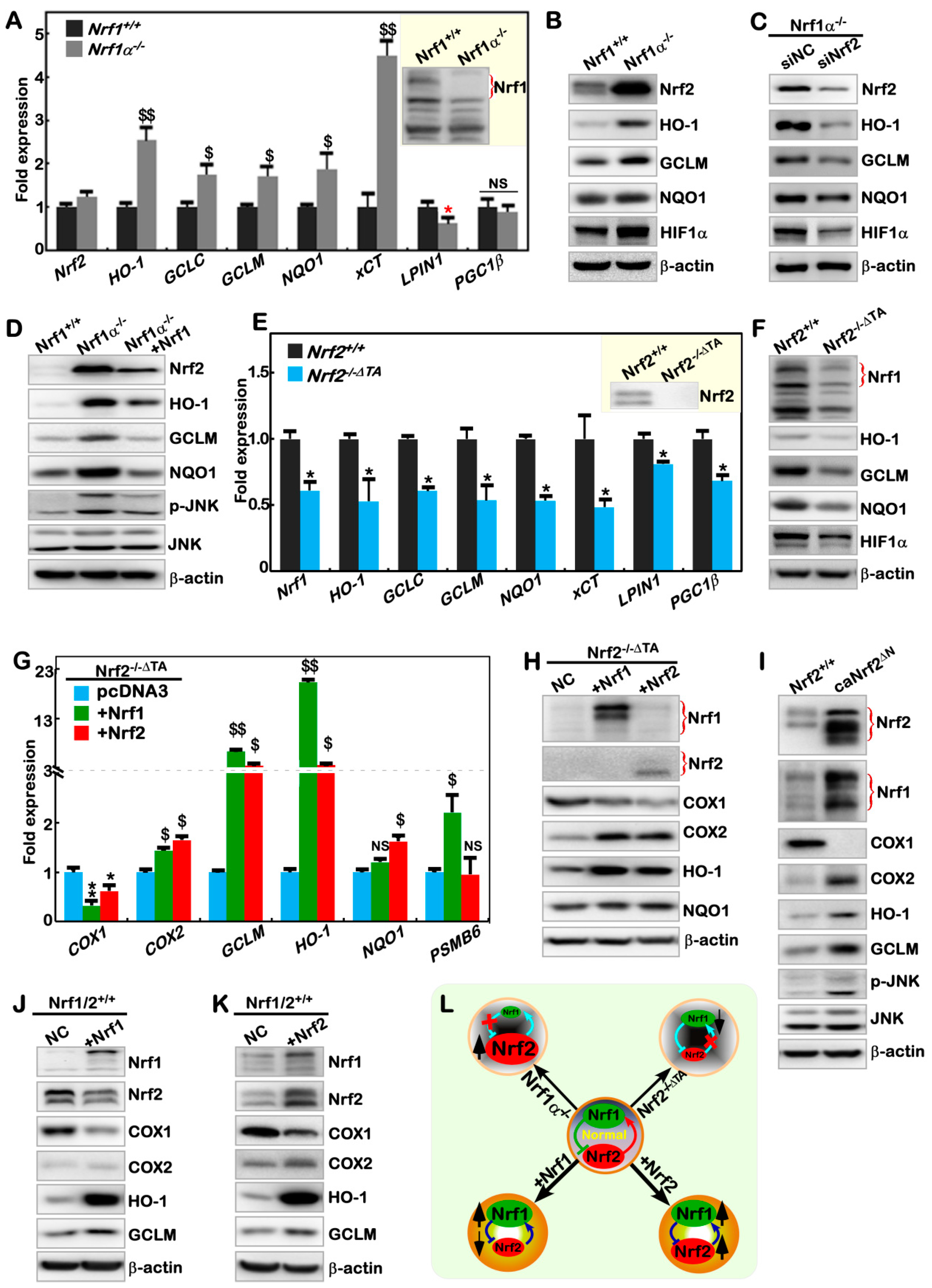
2.1. The Human Nrf1α−/−- and Nrf2−/−ΔTA -Driven Cell Models are Established
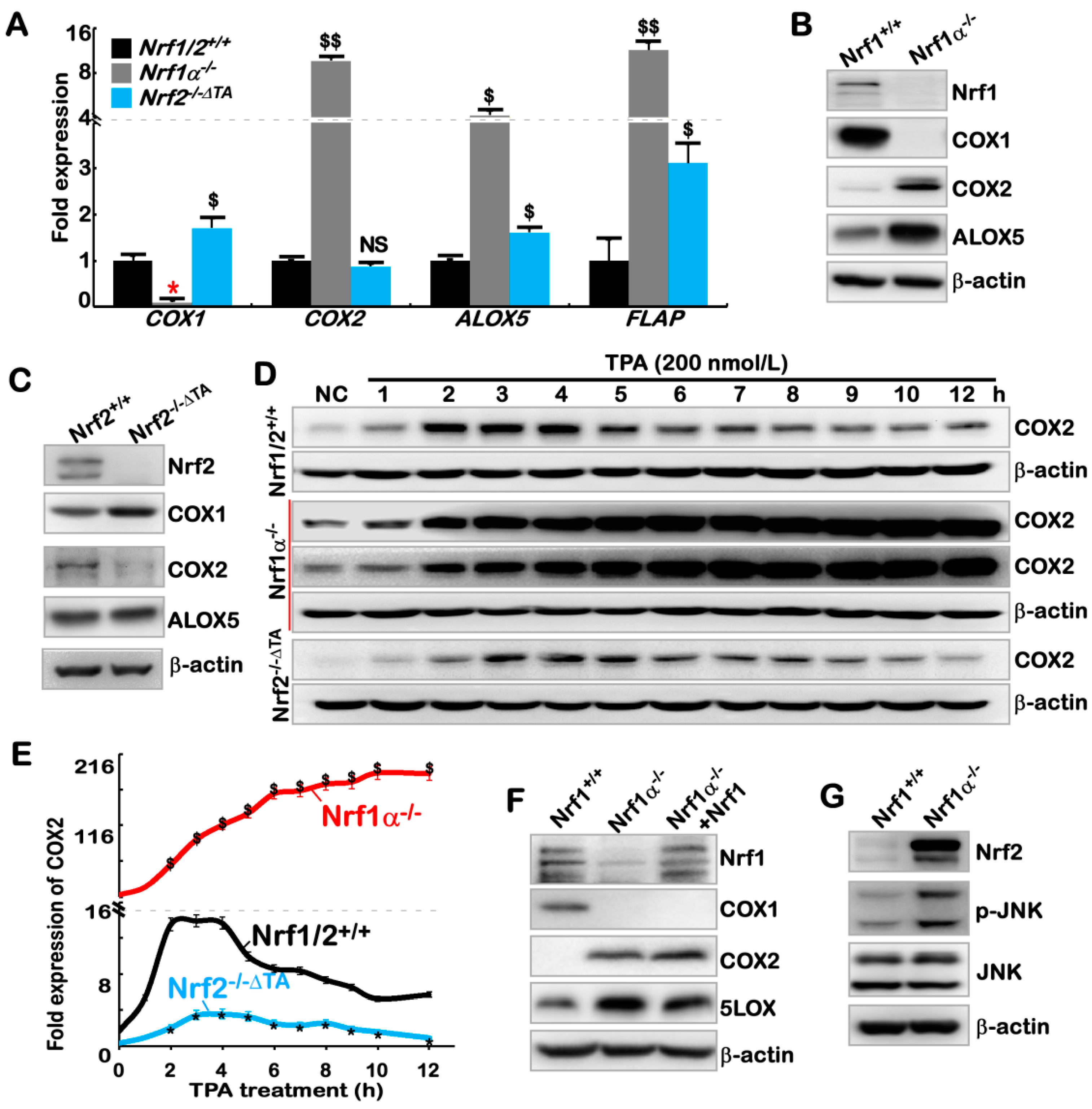
2.2. The Inflammation Marker COX2 Is Up-Regulated, while COX1 Is Down-Regulated, in Nrf1α−/− Cells
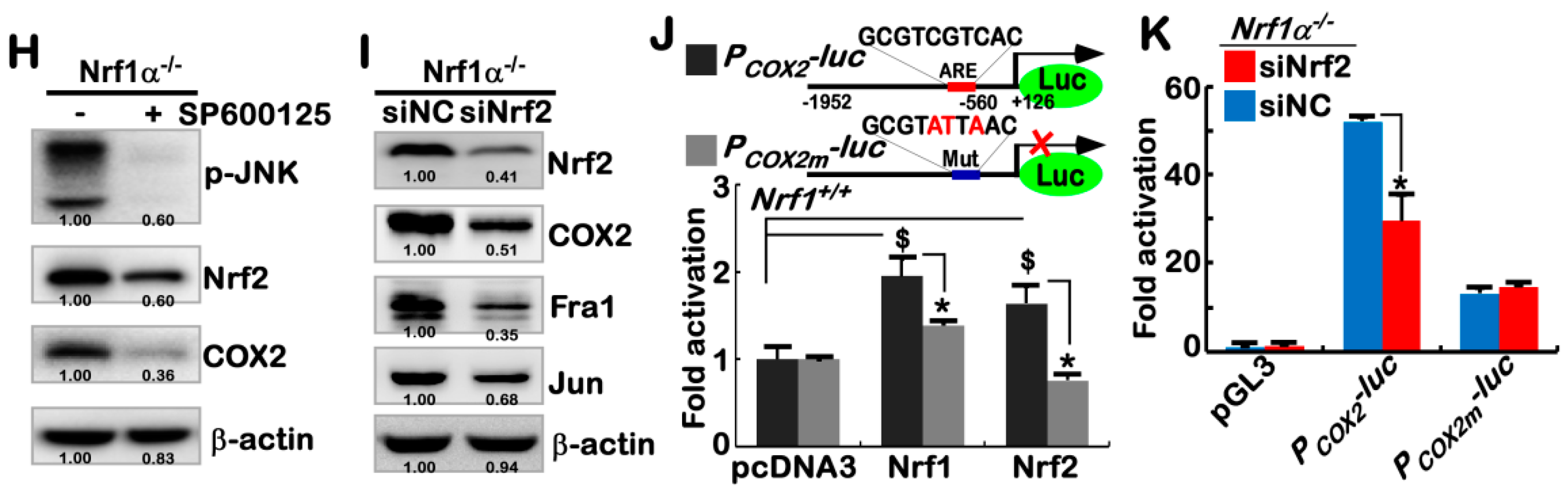
2.3. Hyper-Expression of COX2 Results from Increased Nrf2 and JNK-Mediated AP-1 in Nrf1α−/− Cells
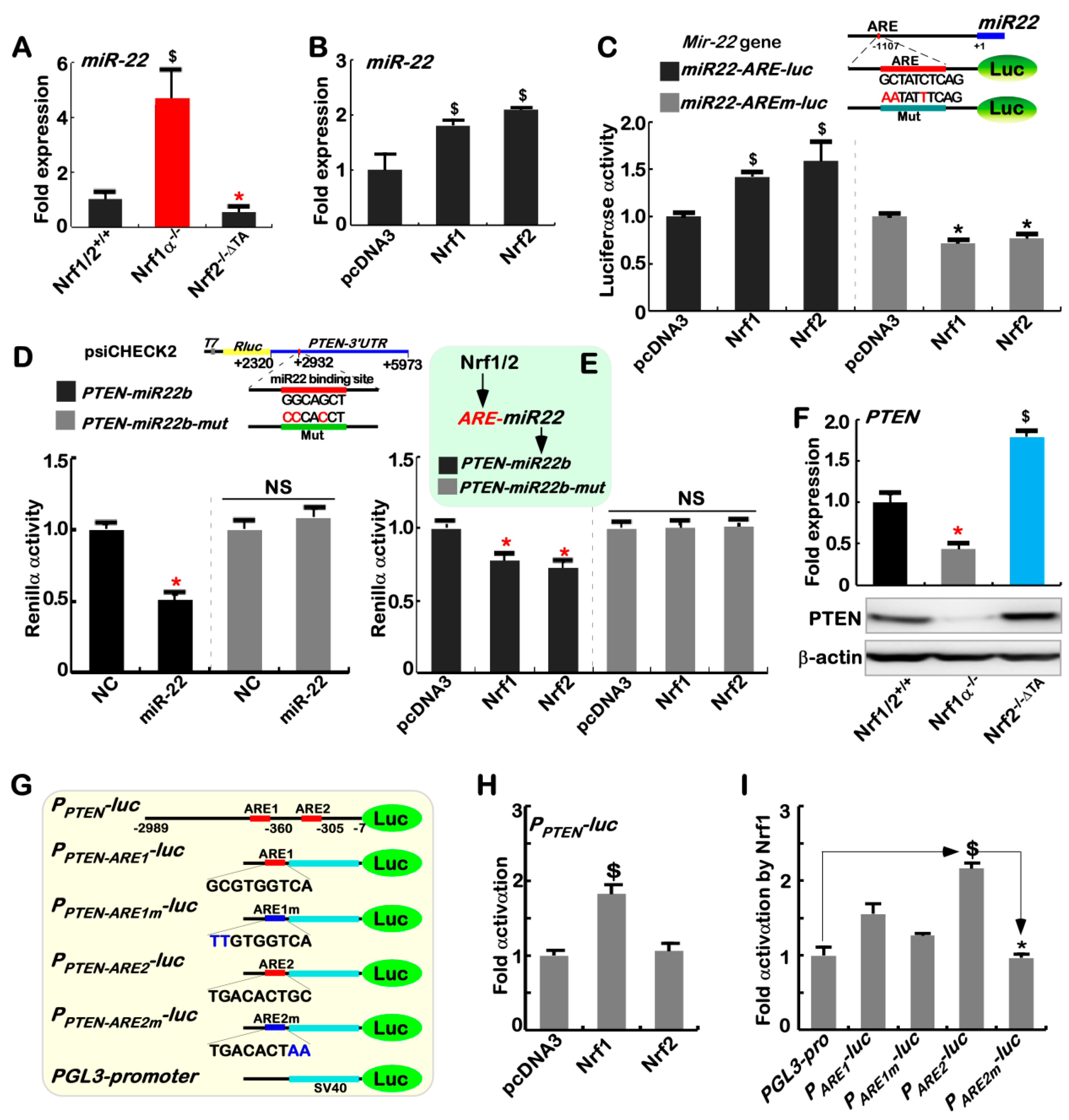
2.4. Nrf1α and Nrf2 Transactivate the ARE-Driven miR-22 Signaling to PTEN, but Not to COX1
2.5. Nrf1α and Nrf2 Have Mutual Inter-Regulatory Effects on Downstream Genes
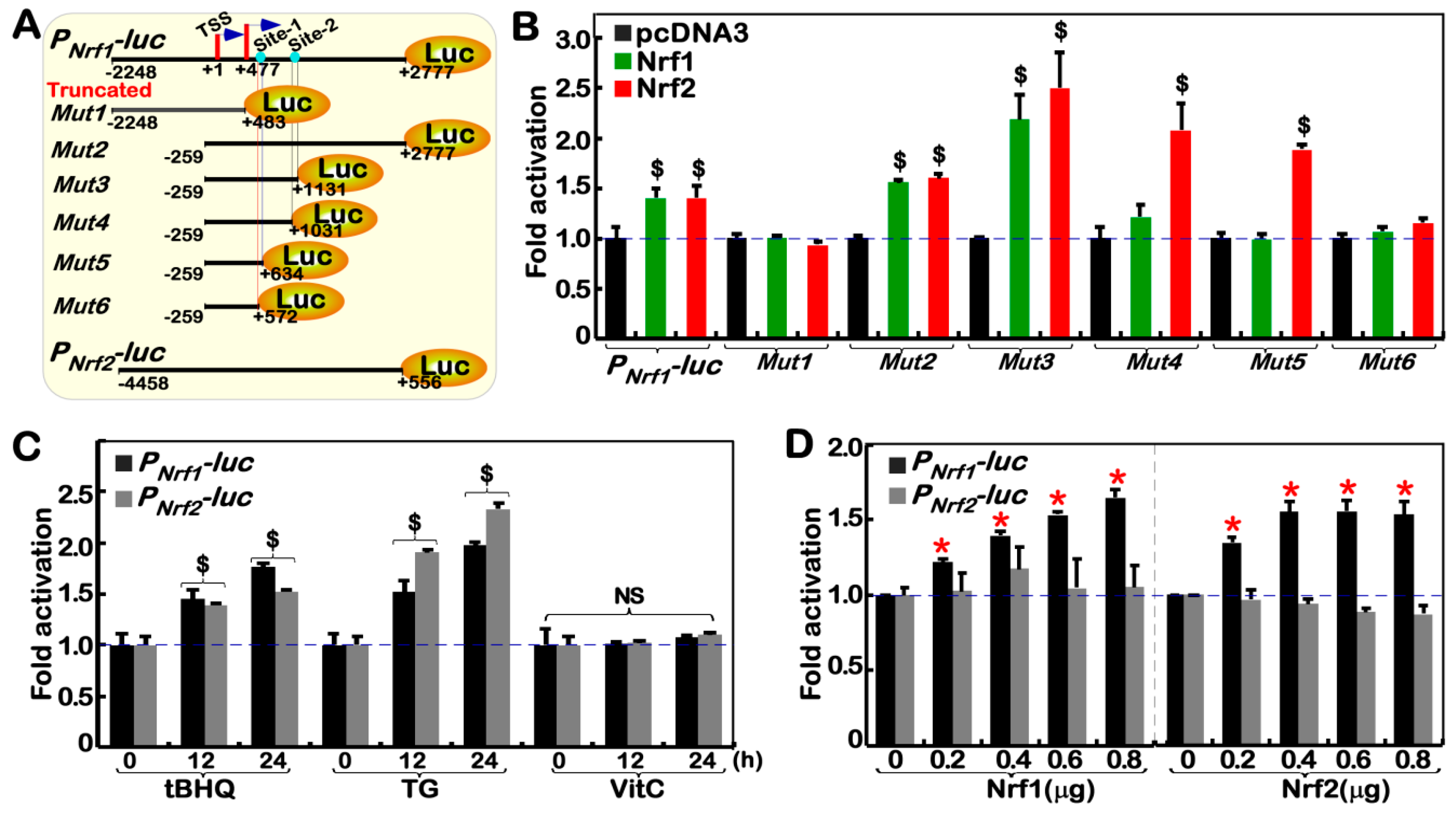
2.6. Nrf1α and Nrf2 Transactivate the Nrf1/Nfe2l1 Gene Promoter-Driven Reporter at Different Sites
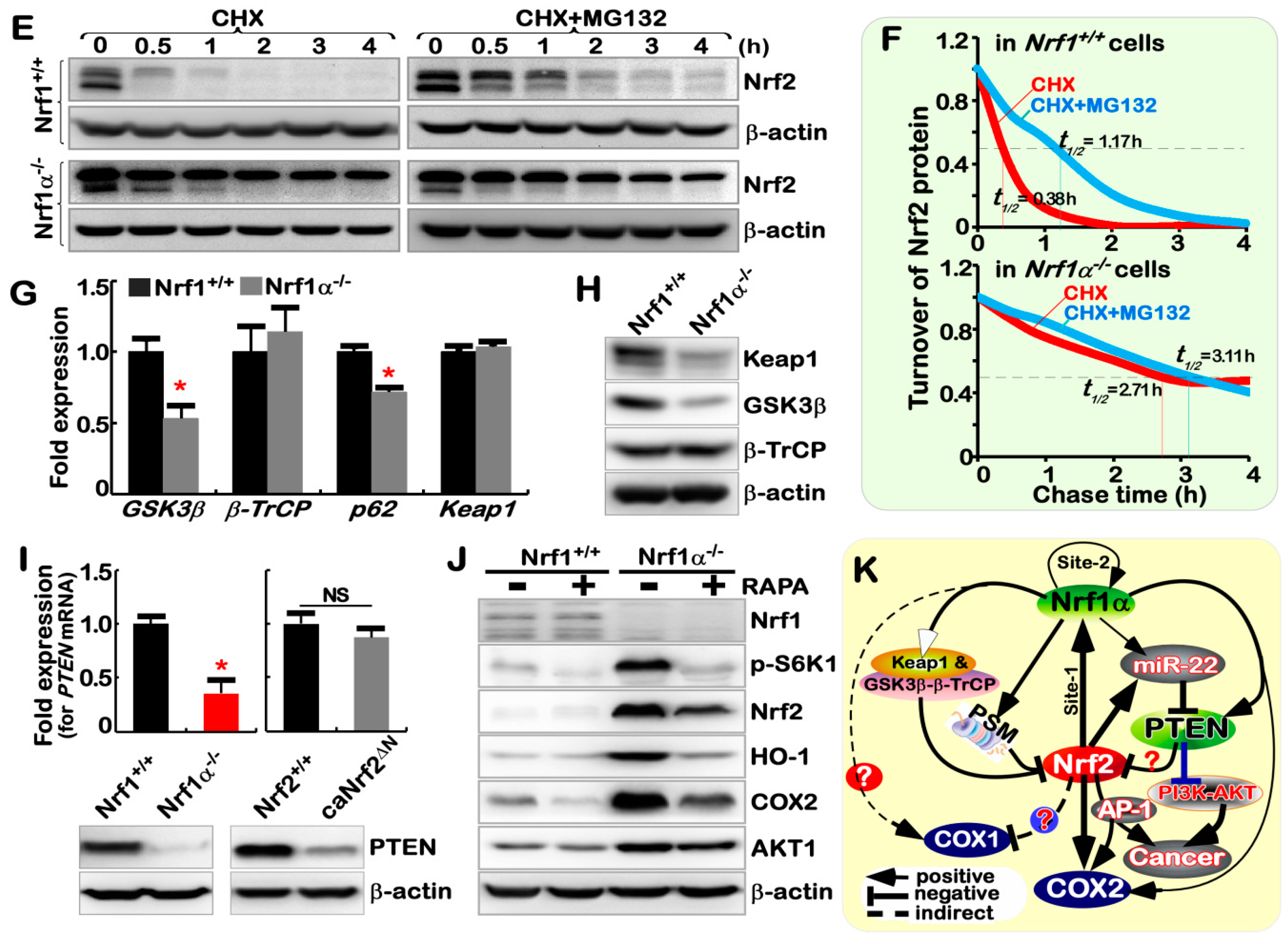
2.7. Nrf1α−/−-Leading Accumulation of Nrf2 Results from Decreased Keap1
2.8. Nrf1α and Nrf2 Exert Opposing and Unifying Roles in the Regulation of PTEN Signaling
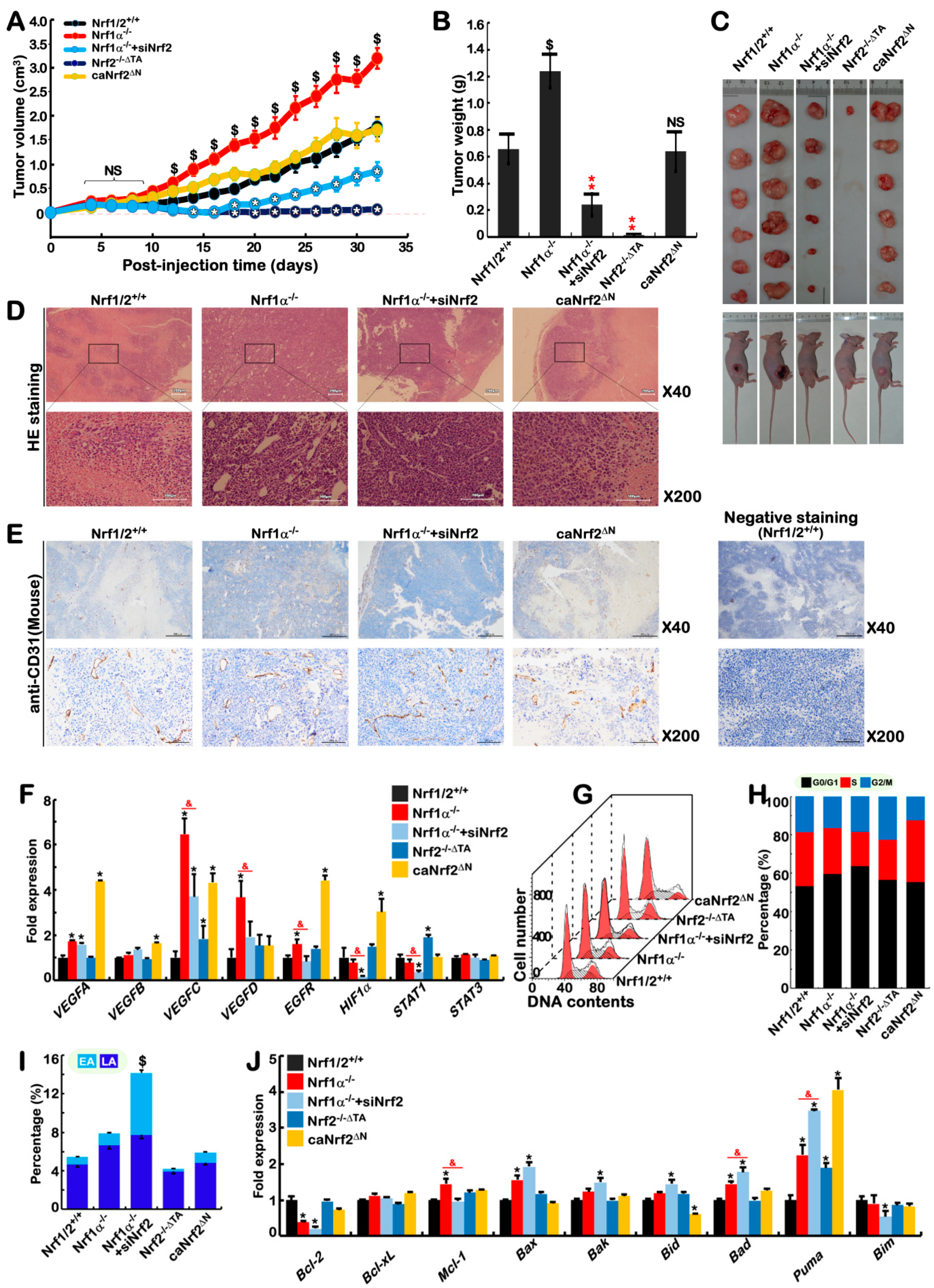
2.9. Blockage of Nrf1α+/+-Bearing or Nrf1α−/−-Derived Tumor Growth by Nrf2 Deficiency
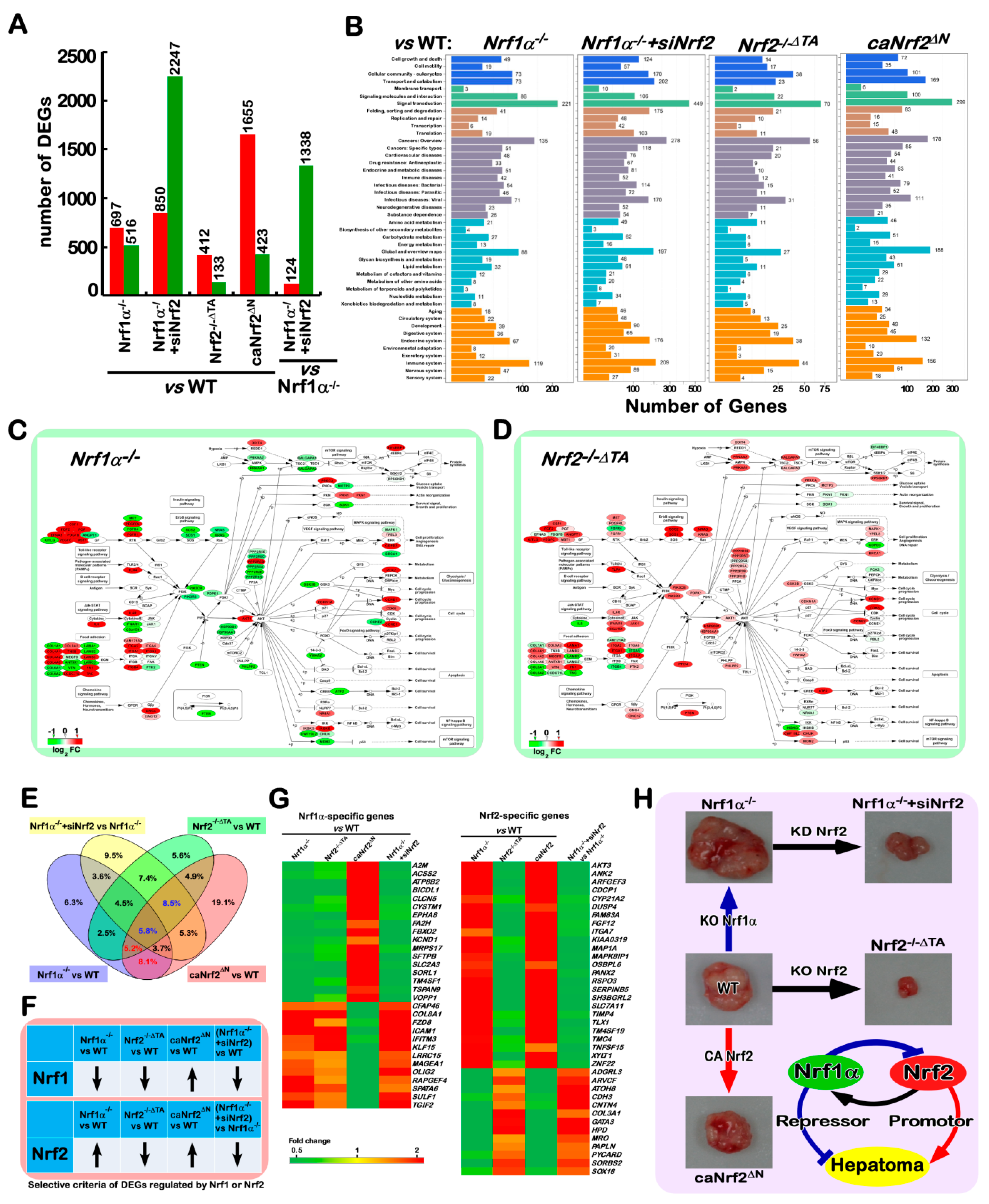
2.10. Different Subsets of Genes Are Finely Regulated by Nrf1α, Nrf2 Alone or Both
3. Discussion
4. Materials and Methods
4.1. Cell lines, Culture and Transfection
4.2. Expression Constructs and other Oligos Used for siRNA and miRNA
4.3. Subcutaneous Tumor Xenografts in Nude Mice
4.4. Histology and Immunohistochemistry
4.5. Immunocytochemistry and Confocal Microscopy
4.6. Subcellular Fractionation
4.7. Lipid Staining
4.8. Intracellular ROS staining
4.9. Luciferase Reporter Assay
4.10. Real-Time Quantitative PCR
4.11. Western Blotting
4.12. Flow Cytometry Analysis of Cell Cycle and Apoptosis
4.13. Key Resources Used for ’Wet Experiments’
4.14. The Genome-Wide Transcriptomic Analysis
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Corte, C.D.; Ferrari, F.; Villani, A.; Nobili, V. Epidemiology and natural history of NAFLD. J. Med. Biochem. 2015, 34, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Michelotti, G.A.; Machado, M.V.; Diehl, A.M. NAFLD, NASH and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Rowell, R.J.; Anstee, Q.M. An overview of the genetics, mechanisms and management of NAFLD and ALD. Clin. Med. 2015, 15 (Suppl. 6), s77–s82. [Google Scholar] [CrossRef]
- Wong, V.W.; Adams, L.A.; de Ledinghen, V.; Wong, G.L.; Sookoian, S. Noninvasive biomarkers in NAFLD and NASH - current progress and future promise. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 461–478. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Jump, D.B.; Tripathy, S.; Depner, C.M. Fatty acid-regulated transcription factors in the liver. Annu. Rev. Nutr. 2013, 33, 249–269. [Google Scholar] [CrossRef]
- Karagianni, P.; Talianidis, I. Transcription factor networks regulating hepatic fatty acid metabolism. Biochim. Biophys. Acta 2015, 1851, 2–8. [Google Scholar] [CrossRef]
- Nakayama, H.; Otabe, S.; Ueno, T.; Hirota, N.; Yuan, X.; Fukutani, T.; Hashinaga, T.; Wada, N.; Yamada, K. Transgenic mice expressing nuclear sterol regulatory element-binding protein 1c in adipose tissue exhibit liver histology similar to nonalcoholic steatohepatitis. Metabolism 2007, 56, 470–475. [Google Scholar] [CrossRef]
- Zhang, T.; Kho, D.H.; Wang, Y.; Harazono, Y.; Nakajima, K.; Xie, Y.; Raz, A. Gp78, an e3 ubiquitin ligase acts as a gatekeeper suppressing nonalcoholic steatohepatitis (NASH) and liver cancer. PLoS ONE 2015, 10, e0118448. [Google Scholar] [CrossRef]
- Yahagi, N.; Shimano, H.; Hasty, A.H.; Matsuzaka, T.; Ide, T.; Yoshikawa, T.; Amemiya-Kudo, M.; Tomita, S.; Okazaki, H.; Tamura, Y.; et al. Absence of sterol regulatory element-binding protein-1 (SREBP-1) ameliorates fatty livers but not obesity or insulin resistance in lep(ob)/lep(ob) mice. J. Biol. Chem. 2002, 277, 19353–19357. [Google Scholar] [CrossRef] [PubMed]
- Moon, Y.A.; Liang, G.; Xie, X.; Frank-Kamenetsky, M.; Fitzgerald, K.; Koteliansky, V.; Brown, M.S.; Goldstein, J.L.; Horton, J.D. The SCAP/SREBP pathway is essential for developing diabetic fatty liver and carbohydrate-induced hypertriglyceridemia in animals. Cell Metab. 2012, 15, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Horie, Y.; Suzuki, A.; Kataoka, E.; Sasaki, T.; Hamada, K.; Sasaki, J.; Mizuno, K.; Hasegawa, G.; Kishimoto, H.; Iizuka, M.; et al. Hepatocyte-specific PTEN deficiency results in steatohepatitis and hepatocellular carcinomas. J. Clin. Investig. 2004, 113, 1774–1783. [Google Scholar] [CrossRef] [PubMed]
- Hollander, M.C.; Blumenthal, G.M.; Dennis, P.A. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat. Rev. Cancer 2011, 11, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Shimano, H.; Sato, R. SREBP-regulated lipid metabolism: Convergent physiology—Divergent pathophysiology. Nat. Rev. Endocrinol. 2017, 13, 710–730. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.R.; Chen, M.; Pandolfi, P.P. The functions and regulation of the PTEN tumour suppressor: New modes and prospects. Nat. Rev. Mol. Cell Biol. 2018, 19, 547–562. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Iwasaki, K.; Sugiyama, H.; Tsuji, Y. Role of the tumor suppressor PTEN in antioxidant responsive element-mediated transcription and associated histone modifications. Mol. Biol. Cell 2009, 20, 1606–1617. [Google Scholar] [CrossRef]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef]
- Rojo, A.I.; Rada, P.; Mendiola, M.; Ortega-Molina, A.; Wojdyla, K.; Rogowska-Wrzesinska, A.; Hardisson, D.; Serrano, M.; Cuadrado, A. The PTEN/Nrf2 axis promotes human carcinogenesis. Antioxid. Redox Signal. 2014, 21, 2498–2514. [Google Scholar] [CrossRef]
- Taguchi, K.; Hirano, I.; Itoh, T.; Tanaka, M.; Miyajima, A.; Suzuki, A.; Motohashi, H.; Yamamoto, M. Nrf2 enhances cholangiocyte expansion in PTEN-deficient livers. Mol. Cell. Biol. 2014, 34, 900–913. [Google Scholar] [CrossRef]
- Best, S.A.; De Souza, D.P.; Kersbergen, A.; Policheni, A.N.; Dayalan, S.; Tull, D.; Rathi, V.; Gray, D.H.; Ritchie, M.E.; McConville, M.J.; et al. Synergy between the Keap1/Nrf2 and PI3K pathways drives non-small-cell lung cancer with an altered immune microenvironment. Cell Metab. 2018, 27, 935–943. [Google Scholar] [CrossRef]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef]
- Wang, H.; Liu, X.; Long, M.; Huang, Y.; Zhang, L.; Zhang, R.; Zheng, Y.; Liao, X.; Wang, Y.; Liao, Q.; et al. Nrf2 activation by antioxidant antidiabetic agents accelerates tumor metastasis. Sci. Transl. Med. 2016, 8, 334ra351. [Google Scholar] [CrossRef] [PubMed]
- Schafer, M.; Dutsch, S.; Auf dem Keller, U.; Navid, F.; Schwarz, A.; Johnson, D.A.; Johnson, J.A.; Werner, S. Nrf2 establishes a glutathione-mediated gradient of UVB cytoprotection in the epidermis. Genes Dev. 2010, 24, 1045–1058. [Google Scholar] [CrossRef]
- Shanmugam, G.; Narasimhan, M.; Tamowski, S.; Darley-Usmar, V.; Rajasekaran, N.S. Constitutive activation of nrf2 induces a stable reductive state in the mouse myocardium. Redox Biol. 2017, 12, 937–945. [Google Scholar] [CrossRef]
- Auf dem Keller, U.; Huber, M.; Beyer, T.A.; Kumin, A.; Siemes, C.; Braun, S.; Bugnon, P.; Mitropoulos, V.; Johnson, D.A.; Johnson, J.A.; et al. Nrf transcription factors in keratinocytes are essential for skin tumor prevention but not for wound healing. Mol. Cell. Biol. 2006, 26, 3773–3784. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Chen, L.; Leung, L.; Yen, T.S.; Lee, C.; Chan, J.Y. Liver-specific inactivation of the Nrf1 gene in adult mouse leads to nonalcoholic steatohepatitis and hepatic neoplasia. Proc. Natl. Acad. Sci. USA 2005, 102, 4120–4125. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuji, M.; Katsuoka, F.; Kobayashi, A.; Aburatani, H.; Hayes, J.D.; Yamamoto, M. Nrf1 and Nrf2 play distinct roles in activation of antioxidant response element-dependent genes. J. Biol. Chem. 2008, 283, 33554–33562. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.Y.; Kwong, M.; Lu, R.H.; Chang, J.; Wang, B.; Yen, T.S.B.; Kan, Y.W. Targeted disruption of the ubiquitous cnc-bzip transcription factor, nrf-1, results in anemia and embryonic lethality in mice. Embo J. 1998, 17, 1779–1787. [Google Scholar] [CrossRef]
- Chen, L.Y.; Kwong, M.; Lu, R.H.; Ginzinger, D.; Lee, C.; Leung, L.; Chan, J.Y. Nrf1 is critical for redox balance and survival of liver cells during development. Mol. Cell. Biol. 2003, 23, 4673–4686. [Google Scholar] [CrossRef]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2 small maf heterodimer mediates the induction of Phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Zhang, Y.; Xiang, Y. Molecular and cellular basis for the unique functioning of Nrf1, an indispensable transcription factor for maintaining cell homoeostasis and organ integrity. Biochem. J. 2016, 473, 961–1000. [Google Scholar] [CrossRef] [PubMed]
- Bugno, M.; Daniel, M.; Chepelev, N.L.; Willmore, W.G. Changing gears in Nrf1 research, from mechanisms of regulation to its role in disease and prevention. Biochim. Biophys. Acta 2015, 1849, 1260–1276. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.S.; Lee, C.; Hu, T.; Nguyen, J.M.; Zhang, J.; Martin, M.V.; Vawter, M.P.; Huang, E.J.; Chan, J.Y. Loss of nuclear factor e2-related factor 1 in the brain leads to dysregulation of proteasome gene expression and neurodegeneration. Proc. Natl. Acad. Sci. USA 2011, 108, 8408–8413. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Tsukide, T.; Miyasaka, T.; Morita, T.; Mizoroki, T.; Saito, Y.; Ihara, Y.; Takashima, A.; Noguchi, N.; Fukamizu, A.; et al. Central nervous system-specific deletion of transcription factor Nrf1 causes progressive motor neuronal dysfunction. Genes Cells 2011, 16, 692–703. [Google Scholar] [CrossRef] [PubMed]
- Hirotsu, Y.; Higashi, C.; Fukutomi, T.; Katsuoka, F.; Tsujita, T.; Yagishita, Y.; Matsuyama, Y.; Motohashi, H.; Uruno, A.; Yamamoto, M. Transcription factor NF-E2-related factor 1 impairs glucose metabolism in mice. Genes Cells 2014, 19, 650–665. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Fu, J.; Xue, P.; Zhao, R.; Dong, J.; Liu, D.; Yamamoto, M.; Tong, Q.; Teng, W.; Qu, W.; et al. CNC-BZIP protein Nrf1-dependent regulation of glucose-stimulated insulin secretion. Antioxid. Redox Signal. 2015, 22, 819–831. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.G.; Ren, Y.G.; Li, S.J.; Hayes, J.D. Transcription factor Nrf1 is topologically repartitioned across membranes to enable target gene transactivation through its acidic glucose-responsive domains. PLoS ONE 2014, 9. [Google Scholar] [CrossRef]
- Widenmaier, S.B.; Snyder, N.A.; Nguyen, T.B.; Arduini, A.; Lee, G.Y.; Arruda, A.P.; Saksi, J.; Bartelt, A.; Hotamisligil, G.S. Nrf1 is an er membrane sensor that is central to cholesterol homeostasis. Cell 2017, 171, 1094–1109. [Google Scholar] [CrossRef]
- Bartelt, A.; Widenmaier, S.B.; Schlein, C.; Johann, K.; Goncalves, R.L.S.; Eguchi, K.; Fischer, A.W.; Parlakgul, G.; Snyder, N.A.; Nguyen, T.B.; et al. Brown adipose tissue thermogenic adaptation requires Nrf1-mediated proteasomal activity. Nat. Med. 2018, 24, 292–303. [Google Scholar] [CrossRef]
- Hou, Y.; Liu, Z.; Zuo, Z.; Gao, T.; Fu, J.; Wang, H.; Xu, Y.; Liu, D.; Yamamoto, M.; Zhu, B.; et al. Adipocyte-specific deficiency of Nfe2l1 disrupts plasticity of white adipose tissues and metabolic homeostasis in mice. Biochem. Biophys. Res. Commun. 2018. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lucocq, J.M.; Yamamoto, M.; Hayes, J.D. The NHB1 (N-terminal homology box 1) sequence in transcription factor Nrf1 is required to anchor it to the endoplasmic reticulum and also to enable its asparagine-glycosylation. Biochem. J. 2007, 408, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, S.; Xiang, Y.; Qiu, L.; Zhao, H.; Hayes, J.D. The selective post-translational processing of transcription factor Nrf1 yields distinct isoforms that dictate its ability to differentially regulate gene expression. Sci. Rep. 2015, 5, 12983. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, S.K.; den Besten, W.; Deshaies, R.J. p97-dependent retrotranslocation and proteolytic processing govern formation of active Nrf1 upon proteasome inhibition. Elife 2014, 3, e01856. [Google Scholar] [CrossRef] [PubMed]
- Sha, Z.; Goldberg, A.L. Proteasome-mediated processing of Nrf1 is essential for coordinate induction of all proteasome subunits and p97. Curr. Biol. 2014, 24, 1573–1583. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Halin, J.; Fan, Z.; Hu, S.; Wang, M.; Qiu, L.; Zhang, Z.; Mattjus, P.; Zhang, Y. Topovectorial mechanisms control the juxtamembrane proteolytic processing of Nrf1 to remove its N-terminal polypeptides during maturation of the cnc-bzip factor. Toxicol. Appl. Pharmacol. 2018, 360, 160–184. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Wang, M.; Hu, S.; Qiu, L.; Yang, F.; Zhang, Z.; Yu, S.; Pi, J.; Zhang, Y. Mechanisms controlling the multistage post-translational processing of endogenous Nrf1α/TCF11 proteins to yield distinct isoforms within the coupled positive and negative feedback circuits. Toxicol. Appl. Pharmacol. 2018, 360, 212–235. [Google Scholar] [CrossRef]
- Koizumi, S.; Irie, T.; Hirayama, S.; Sakurai, Y.; Yashiroda, H.; Naguro, I.; Ichijo, H.; Hamazaki, J.; Murata, S. The aspartyl protease DDI2 activates Nrf1 to compensate for proteasome dysfunction. Elife 2016, 5. [Google Scholar] [CrossRef]
- Zhang, Y.; Qiu, L.; Li, S.; Xiang, Y.; Chen, J.; Ren, Y. The C-terminal domain of Nrf1 negatively regulates the full-length CNC-BZIP factor and its shorter isoform LCR-F1/Nrf1β; both are also inhibited by the small dominant-negative Nrf1γ/δ isoforms that down-regulate are-battery gene expression. PLoS ONE 2014, 9, e0109159. [Google Scholar] [CrossRef]
- Ren, Y.G.; Qiu, L.; Lu, F.L.; Ru, X.F.; Li, S.J.; Xiang, Y.C.; Yu, S.W.; Zhang, Y.G. Talens-directed knockout of the full-length transcription factor Nrf1α that represses malignant behaviour of human hepatocellular carcinoma (HepG2) cells. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Tsujita, T.; Peirce, V.; Baird, L.; Matsuyama, Y.; Takaku, M.; Walsh, S.V.; Griffin, J.L.; Uruno, A.; Yamamoto, M.; Hayes, J.D. Transcription factor Nrf1 negatively regulates the cystine/glutamate transporter and lipid-metabolizing enzymes. Mol. Cell. Biol. 2014, 34, 3800–3816. [Google Scholar] [CrossRef] [PubMed]
- Hirotsu, Y.; Hataya, N.; Katsuoka, F.; Yamamoto, M. NF-E2-related factor 1 (Nrf1) serves as a novel regulator of hepatic lipid metabolism through regulation of the lipin1 and PGC-1β genes. Mol. Cell. Biol. 2012, 32, 2760–2770. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.A.; Dubois, R.N. Colorectal cancer prevention and treatment by inhibition of cyclooxygenase-2. Nat. Rev. Cancer 2001, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Castellone, M.D.; Teramoto, H.; Williams, B.O.; Druey, K.M.; Gutkind, J.S. Prostaglandin E2 promotes colon cancer cell growth through a GS-axin-β-catenin signaling axis. Science 2005, 310, 1504–1510. [Google Scholar] [CrossRef] [PubMed]
- Chowdhry, S.; Nazmy, M.H.; Meakin, P.J.; Dinkova-Kostova, A.T.; Walsh, S.V.; Tsujita, T.; Dillon, J.F.; Ashford, M.L.; Hayes, J.D. Loss of Nrf2 markedly exacerbates nonalcoholic steatohepatitis. Free Radic. Biol. Med. 2010, 48, 357–371. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Dubois, R.N. The role of COX-2 in intestinal inflammation and colorectal cancer. Oncogene 2010, 29, 781–788. [Google Scholar] [CrossRef]
- Sherratt, P.J.; McLellan, L.I.; Hayes, J.D. Positive and negative regulation of prostaglandin E2 biosynthesis in human colorectal carcinoma cells by cancer chemopreventive agents. Biochem. Pharmacol. 2003, 66, 51–61. [Google Scholar] [CrossRef]
- Itoh, K.; Mochizuki, M.; Ishii, Y.; Ishii, T.; Shibata, T.; Kawamoto, Y.; Kelly, V.; Sekizawa, K.; Uchida, K.; Yamamoto, M. Transcription factor Nrf2 regulates inflammation by mediating the effect of 15-deoxy-δ(12,14)-prostaglandin J(2). Mol. Cell. Biol. 2004, 24, 36–45. [Google Scholar] [CrossRef]
- O’Banion, M.K. Cyclooxygenase-2: Molecular biology, pharmacology, and neurobiology. Crit. Rev. Neurobiol. 1999, 13, 45–82. [Google Scholar] [CrossRef]
- Tanabe, T.; Tohnai, N. Cyclooxygenase isozymes and their gene structures and expression. Prostaglandins Other Lipid Mediat. 2002, 68–69, 95–114. [Google Scholar] [CrossRef]
- Xu, K.; Shu, H.K. Transcription factor interactions mediate EGF-dependent COX-2 expression. Mol. Cancer Res. 2013, 11, 875–886. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, J.; Yang, X.; Han, X. Several transcription factors regulate COX-2 gene expression in pancreatic β-cells. Mol. Biol. Rep. 2007, 34, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Sharma-Walia, N.; George Paul, A.; Patel, K.; Chandran, K.; Ahmad, W.; Chandran, B. NFAT and CREB regulate kaposi’s sarcoma-associated herpesvirus-induced cyclooxygenase 2 (COX-2). J. Virol. 2010, 84, 12733–12753. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.J.; Wingerd, B.A.; Arakawa, T.; Smith, W.L. Cyclooxygenase-2 gene transcription in a macrophage model of inflammation. J. Immunol. 2006, 177, 8111–8122. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.; Garcia, G.E.; Crosby, K.; Inoue, H.; Thompson, I.M.; Troyer, D.A.; Kumar, A.P. Regulation of COX-2 by cyclic amp response element binding protein in prostate cancer: Potential role for nexrutine. Neoplasia 2007, 9, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Grall, F.T.; Prall, W.C.; Wei, W.; Gu, X.; Cho, J.Y.; Choy, B.K.; Zerbini, L.F.; Inan, M.S.; Goldring, S.R.; Gravallese, E.M.; et al. The ets transcription factor ese-1 mediates induction of the COX-2 gene by lps in monocytes. FEBS J. 2005, 272, 1676–1687. [Google Scholar] [CrossRef] [PubMed]
- Luedde, T.; Beraza, N.; Kotsikoris, V.; van Loo, G.; Nenci, A.; De Vos, R.; Roskams, T.; Trautwein, C.; Pasparakis, M. Deletion of NEMO/IKKγ in liver parenchymal cells causes steatohepatitis and hepatocellular carcinoma. Cancer Cell 2007, 11, 119–132. [Google Scholar] [CrossRef]
- Natarajan, K.; Singh, S.; Burke, T.R., Jr.; Grunberger, D.; Aggarwal, B.B. Caffeic acid phenethyl ester is a potent and specific inhibitor of activation of nuclear transcription factor NF-κB. Proc. Natl. Acad. Sci. USA 1996, 93, 9090–9095. [Google Scholar] [CrossRef]
- Shin, H.M.; Kim, M.H.; Kim, B.H.; Jung, S.H.; Kim, Y.S.; Park, H.J.; Hong, J.T.; Min, K.R.; Kim, Y. Inhibitory action of novel aromatic diamine compound on lipopolysaccharide-induced nuclear translocation of NF-κB without affecting ikappab degradation. FEBS Lett. 2004, 571, 50–54. [Google Scholar] [CrossRef]
- Tymianski, M.; Spigelman, I.; Zhang, L.; Carlen, P.L.; Tator, C.H.; Charlton, M.P.; Wallace, M.C. Mechanism of action and persistence of neuroprotection by cell-permeant Ca2+ chelators. J. Cereb. Blood Flow Metab. 1994, 14, 911–923. [Google Scholar] [CrossRef]
- Bennett, B.L.; Sasaki, D.T.; Murray, B.W.; O’Leary, E.C.; Sakata, S.T.; Xu, W.; Leisten, J.C.; Motiwala, A.; Pierce, S.; Satoh, Y.; et al. Sp600125, an anthrapyrazolone inhibitor of jun N-terminal kinase. Proc. Natl. Acad. Sci. USA 2001, 98, 13681–13686. [Google Scholar] [CrossRef] [PubMed]
- Fanjul, A.; Dawson, M.I.; Hobbs, P.D.; Jong, L.; Cameron, J.F.; Harlev, E.; Graupner, G.; Lu, X.P.; Pfahl, M. A new class of retinoids with selective inhibition of AP-1 inhibits proliferation. Nature 1994, 372, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Ahn, C.B.; Je, J.Y.; Kim, Y.S.; Park, S.J.; Kim, B.I. Induction of Nrf2-mediated phase ii detoxifying/antioxidant enzymes in vitro by chitosan-caffeic acid against hydrogen peroxide-induced hepatotoxicity through JNK/ERK pathway. Mol. Cell Biochem. 2017, 424, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Bak, M.J.; Truong, V.L.; Ko, S.Y.; Nguyen, X.N.; Jun, M.; Hong, S.G.; Lee, J.W.; Jeong, W.S. Induction of nrf2/are-mediated cytoprotective genes by red ginseng oil through ASK1-MKK4/7-JNK and p38 MAPK signaling pathways in HepG2 cells. J. Ginseng Res. 2016, 40, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Tao, S.; Wang, S.; Moghaddam, S.J.; Ooi, A.; Chapman, E.; Wong, P.K.; Zhang, D.D. Oncogenic KRAS confers chemoresistance by upregulating Nrf2. Cancer Res. 2014, 74, 7430–7441. [Google Scholar] [CrossRef] [PubMed]
- Tan, G.; Shi, Y.; Wu, Z.H. MicroRNA-22 promotes cell survival upon uv radiation by repressing PTEN. Biochem. Biophys. Res. Commun. 2012, 417, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Huang, J.; Xiao, H.; Liang, Z. MicroRNA-22 is downregulated in clear cell renal cell carcinoma, and inhibits cell growth, migration and invasion by targeting pten. Mol. Med. Rep. 2016, 13, 4800–4806. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.J.; Morrow, J.D.; Roberts, L.J., 2nd; Marnett, L.J. Induction of prostaglandin endoperoxide synthase-1 (COX-1) in a human promonocytic cell line by treatment with the differentiating agent TPA. Adv. Exp. Med. Biol. 1997, 400A, 99–106. [Google Scholar]
- Leung, L.; Kwong, M.; Hou, S.; Lee, C.; Chan, J.Y. Deficiency of the Nrf1 and Nrf2 transcription factors results in early embryonic lethality and severe oxidative stress. J. Biol. Chem. 2003, 278, 48021–48029. [Google Scholar] [CrossRef]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The keap1-nrf2 system: A thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef]
- Radhakrishnan, S.K.; Lee, C.S.; Young, P.; Beskow, A.; Chan, J.Y.; Deshaies, R.J. Transcription factor Nrf1 mediates the proteasome recovery pathway after proteasome inhibition in mammalian cells. Mol. Cell 2010, 38, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Steffen, J.; Seeger, M.; Koch, A.; Kruger, E. Proteasomal degradation is transcriptionally controlled by TCF11 via an ERAD-dependent feedback loop. Mol. Cell 2010, 40, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Kang, M.I.; Watai, Y.; Tong, K.I.; Shibata, T.; Uchida, K.; Yamamoto, M. Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol. Cell. Biol. 2006, 26, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Pitha-Rowe, I.; Liby, K.; Royce, D.; Sporn, M. Synthetic triterpenoids attenuate cytotoxic retinal injury: Cross-talk between Nrf2 and PI3K/AKT signaling through inhibition of the lipid phosphatase PTEN. Investig. Ophthalmol. Vis. Sci. 2009, 50, 5339–5347. [Google Scholar] [CrossRef] [PubMed]
- Tebay, L.E.; Robertson, H.; Durant, S.T.; Vitale, S.R.; Penning, T.M.; Dinkova-Kostova, A.T.; Hayes, J.D. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues and energy status, and pathways through which it attenuates degenerative disease. Free Radic. Biol. Med. 2015, 88, 108–146. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.W.; Hong, H.L.; Luo, W.W.; Dai, C.M.; Chen, X.Y.; Wang, L.P.; Li, Q.; Li, Z.Q.; Liu, P.Q.; Li, Z.M. mTORC2 facilitates endothelial cell senescence by suppressing Nrf2 expression via the AKT/GSK-3β/C/EBPα signaling pathway. Acta Pharmacol. Sin. 2018, 39, 1837–1846. [Google Scholar] [CrossRef]
- Kwong, M.; Kan, Y.W.; Chan, J.Y. The CNC basic leucine zipper factor, Nrf1, is essential for cell survival in response to oxidative stress-inducing agents - role for Nrf1 in γ-GCS(l) and G(SS) expression in mouse fibroblasts. J. Biol. Chem. 1999, 274, 37491–37498. [Google Scholar] [CrossRef]
- Xu, X.D.; Song, X.W.; Li, Q.; Wang, G.K.; Jing, Q.; Qin, Y.W. Attenuation of microRNA-22 derepressed pten to effectively protect rat cardiomyocytes from hypertrophy. J. Cell Physiol. 2012, 227, 1391–1398. [Google Scholar] [CrossRef]
- Chetram, M.A.; Don-Salu-Hewage, A.S.; Hinton, C.V. Ros enhances CXCR4-mediated functions through inactivation of pten in prostate cancer cells. Biochem. Biophys. Res. Commun. 2011, 410, 195–200. [Google Scholar] [CrossRef]
- Kitagishi, Y.; Matsuda, S. Redox regulation of tumor suppressor pten in cancer and aging (review). Int. J. Mol. Med. 2013, 31, 511–515. [Google Scholar] [CrossRef]
- Kageyama, S.; Saito, T.; Obata, M.; Koide, R.H.; Ichimura, Y.; Komatsu, M. Negative regulation of the keap1-nrf2 pathway by a p62/SQSTM1 splicing variant. Mol. Cell. Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kwak, M.K.; Itoh, K.; Yamamoto, M.; Kensler, T.W. Enhanced expression of the transcription factor Nrf2 by cancer chemopreventive agents: Role of antioxidant response element-like sequences in the Nrf2 promoter. Mol. Cell. Biol. 2002, 22, 2883–2892. [Google Scholar] [CrossRef]
- Menegon, S.; Columbano, A.; Giordano, S. The dual roles of Nrf2 in cancer. Trends Mol. Med. 2016, 22, 578–593. [Google Scholar] [CrossRef] [PubMed]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. Nrf2 and the hallmarks of cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef]
- Wang, R.; Yu, Z.; Sunchu, B.; Shoaf, J.; Dang, I.; Zhao, S.; Caples, K.; Bradley, L.; Beaver, L.M.; Ho, E.; et al. Rapamycin inhibits the secretory phenotype of senescent cells by a Nrf2-independent mechanism. Aging Cell 2017, 16, 564–574. [Google Scholar] [CrossRef]
- Morton, C.L.; Houghton, P.J. Establishment of human tumor xenografts in immunodeficient mice. Nat. Protoc. 2007, 2, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.G.; Hayes, J.D. Identification of topological determinants in the N-terminal domain of transcription factor Nrf1 that control its orientation in the endoplasmic reticulum membrane. Biochem. J. 2010, 430, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microrna target sites in mammalian mRNAs. Elife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Landmead, B.; Salzberg, S.L. Hisat: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. Rsem: Accurate transcript quantification from RNA-seq data with or without a reference genome. Bmc Bioinformatics 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Tarazona, S.; Garcia-Alcalde, F.; Dopazo, J.; Ferrer, A.; Conesa, A. Differential expression in RNA-seq: A matter of depth. Genome Res. 2011, 21, 2213–2223. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. Kegg for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reagent or Resource | Source | Identifier |
|---|---|---|
| Antibodies | ||
| AKT1 | Abcam | ab32505 |
| ALOX5 | Sangon Biotech | D220061 |
| CD31 | Servicebio | GB11063-3 |
| COX1 | Sangon Biotech | D260197 |
| COX2 | Abcam | ab62331 |
| Fos | Abcam | ab134122 |
| Fra1 | Abcam | ab124722 |
| GCLM | Abcam | ab126704 |
| GSK3β | Sangon Biotech | D160468 |
| HIF1α | Abcam | ab51608 |
| Histone 3 | Bioss | bs-0349R |
| HO-1 | Abcam | ab52947 |
| JNK (Anti-JNK1+JNK2+JNK3) | Abcam | ab208035 |
| Jun | Proteintech | 10024-2-AP |
| KEAP1 | Sangon Biotech | D154142 |
| NQO1 | Abcam | ab80588 |
| Nrf1 | Zhang’s [97] | N/A |
| Nrf2 | Abcam | ab62352 |
| p-JNK (Anti-JNK1+JNK2+JNK3 (phospho T183+T183+T221)) | Abcam | ab124956 |
| p-S6K1( Anti-RPS6KB1(Phospho-Thr389/412)) | Sangon Biotech | D151473 |
| PTEN | Abcam | ab32199 |
| Ubiquitin | Cell Signaling Technology | 3933S |
| Alexa Fluor 488 - Conjugated Goat anti-rabbit IgG | ZSGB-BIO | ZF-0511 |
| α-Tubulin | Beyotime | AF0001 |
| β-actin | ZSGB-BIO | TA-09 |
| β-TrCP | Sangon Biotech | D154110 |
| Biological Samples: Cell Lines | ||
| HepG2 | Cell bank of the Chinese Academy of Sciences | TCHu72 |
| Nrf1α-/- | this paper | NA |
| Nrf2-/-ΔTA | this paper | NA |
| caNrf2ΔN | this paper | NA |
| HepG2Keap1-/- | this paper | NA |
| HL7702 | Cell bank of the Chinese Academy of Sciences | GNHu 6 |
| HL7702Nrf1α-/- | this paper | NA |
| MEF | courtesy of Akira Kobayashi | NA |
| MEFNrf1-/-(ΔDBD) | courtesy of Akira Kobayashi | NA |
| MEFNrf2-/-(ΔDBD) | courtesy of John D. Hayes | NA |
| MEFKeap1-/- | courtesy of John D. Hayes | NA |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 12-O-Tetradecanoylphorbol-13-acetate (TPA) | Beyotime | S1819 |
| BAPTA-Acetoxymethyl ester (BAPTA-AM) | Cayman Chemical | 15551 |
| Caffeic Acid Phenethyl Ester (CAPE) | Selleck | S7414 |
| cOmplete Tablets EASYpack | Roche | 4693116001 |
| cycloheximide (CHX) | Solarbio | C8030 |
| H-89 | Beyotime | S1643 |
| JSH-23 | Selleck | S7351 |
| MG132 | Sigma Aldrich | M7449 |
| oil red O | Sangon Biotech | A600395 |
| paraformaldehyde | Boster Biological Technology | AR1068 |
| PhosSTOP EASYpack | Roche | 4906845001 |
| Rapamycin (RAPA) | Sigma Aldrich | 37094 |
| sodium oleate | Solarbio | N/A |
| SP600125 | Selleck | S1460 |
| SR11302 | Cayman Chemical | 11302 |
| tert-Butylhydroquinone (tBHQ) | Sigma Aldrich | 112941 |
| Thapsigargin (TG) | Sangon Biotech | A616759 |
| Vitamin C (VC) | Sigma Aldrich | 33034 |
| Deposited Data | ||
| Oligonucleotides for siRNA or miRNA | ||
| siNrf2 FW | Sangon Biotech | GUAAGAAGCCAGAUGUUAAdTdT |
| siNrf2 REV | Sangon Biotech | UUAACAUCUGGCUUCUUACdTdT |
| siJUN FW | Sangon Biotech | GCAUGGACCUAACAUUCGAdTdT |
| siJUN REV | Sangon Biotech | UCGAAUGUUAGGUCCAUGCdTdT |
| siFra1 FW | Sangon Biotech | CAAACUGGAAGAUGAGAAAdTdT |
| siFra1 REV | Sangon Biotech | UUUCUCAUCUUCCAGUUUGdTdT |
| has-miR-22-3p FW | Sangon Biotech | AAGCUGCCAGUUGAAGAACUGU |
| has-miR-22-3p REV | Sangon Biotech | AGUUCUUCAACUGGCAGCUUUU |
| Normal control FW | Sangon Biotech | UUCUCCGAACGUGUCACGUdTdT |
| Normal control REV | Sangon Biotech | ACGUGACACGUUCGGAGAAdTdT |
| Oligonucleotides for qPCR | ||
| ALOX5 FW | Tsingke | GCTGCCCCAGCCAGATGGACTC |
| ALOX5 REV | Tsingke | CTGCTTGGTGTGGAAATGCTGA |
| COX1 FW | Tsingke | CGCCAGTGAATCCCTGTTGTT |
| COX1 REV | Tsingke | AAGGTGGCATTGACAAACTCC |
| COX2 FW | Tsingke | AAGTCCCTGAGCATCTACGGTTT |
| COX2 REV | Tsingke | GTTGTGTTCCCTCAGCCAGATT |
| FLAP FW | Tsingke | TCAGCGTGGTCCAGAATGG |
| FLAP REV | Tsingke | GCAAGTGTTCCGGTCCTCT |
| FOS FW | Tsingke | CACCGACCTGCCTGCAAGAT |
| FOS REV | Tsingke | GCTGGGAACAGGAAGTCATCAA |
| FOSB FW | Tsingke | GCTGCAAGATCCCCTACGAAG |
| FOSB REV | Tsingke | ACGAAGAAGTGTACGAAGGGTT |
| Fra1 FW | Tsingke | CCTGCCGCCCTGTACCTTGT |
| Fra1 REV | Tsingke | GTCTCCGCTGCTGCTGCTACTC |
| Fra2 FW | Tsingke | CACCATCAACGCCATCACGA |
| Fra2 REV | Tsingke | CGACGCTTCTCCTCCTCTTCAG |
| GCLC FW | Tsingke | TCAATGGGAAGGAAGGTGTGTT |
| GCLC REV | Tsingke | TTGTAGTCAGGATGGTTTGCGA |
| GCLM FW | Tsingke | GTGTGATGCCACCAGATTTGAC |
| GCLM REV | Tsingke | CACAATGACCGAATACCGCAGT |
| HO-1 FW | Tsingke | CAGAGCCTGGAAGACACCCTAA |
| HO-1 REV | Tsingke | AAACCACCCCAACCCTGCTAT |
| JUN FW | Tsingke | ATGGAAACGACCTTCTATGACGA |
| JUN REV | Tsingke | CGTTGCTGGACTGGATTATCA |
| JUNB FW | Tsingke | AGCCACCTCCCGTTTACACCAA |
| JUNB REV | Tsingke | ACGGTCTGCGGTTCCTCCTTGA |
| JUND FW | Tsingke | ATCGACATGGACACGCAGGAGC |
| JUND REV | Tsingke | GCTGTTGACGTGGCTGAGGACT |
| KEAP1 FW | Tsingke | AACAACTCGCCCGACGGCAACAC |
| KEAP1 REV | Tsingke | CATCCCGCTCTGGCTCATACCTC |
| LPIN1 FW | Tsingke | TGACCAATCGCCAACTCTGG |
| LPIN1 REV | Tsingke | TCAGCACCAAGATGTCGGCT |
| mir-22 FW | Tsingke | GCAAGCTGCCAGTTGAAG |
| mir-22 REV | Tsingke | GTGCAGGGTCCGAGGT |
| mir-22-RT | Tsingke | GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACACAGTT |
| NQO1 FW | Tsingke | AAGAAGAAAGGATGGGAGGTGG |
| NQO1 REV | Tsingke | GAACAGACTCGGCAGGATACTGA |
| Nrf1 FW | Tsingke | TGGAACAGCAGTGGCAAGATCTCA |
| Nrf1 REV | Tsingke | GGCACTGTACAGGATTTCACTTGC |
| Nrf2 FW | Tsingke | AATTGCCTGTAAGTCCTGGTCAT |
| Nrf2 REV | Tsingke | TCATTGAACTGCTCTTTGGACAT |
| Nrf2-/-ΔTA FW | Tsingke | CGACGGAAAGAGTATGAGCTGGA |
| Nrf2-/-ΔTA REV | Tsingke | ACTGGTTTCTGACTGGATGTGCT |
| PGC1βFW | Tsingke | TGGTGAGATTGAGGAGTGCGA |
| PGC1βREV | Tsingke | GCCTTGTCTGAGGTATTGAGGTATTC |
| PSMB6 FW | Tsingke | TCAAGAAGGAGGGCAGGTGT |
| PSMB6 REV | Tsingke | GTAAAGTGGCAACGGCGAA |
| PTEN FW | Tsingke | TTTGAAGACCATAACCCACCAC |
| PTEN REV | Tsingke | ATTACACCAGTTCGTCCCTTTC |
| β-actin FW | Tsingke | CATGTACGTTGCTATCCAGGC |
| β-actin REV | Tsingke | CTCCTTAATGTCACGCACGAT |
| Oligonucleotides for construct | ||
| COX1-LUC FW | Tsingke | GCCTCGGTACCCTGCCTGCTCTCTC |
| COX1-LUC REV | Tsingke | GATGAGAAGCTTACTACTCCTCAGACAGATC |
| COX1-UTR FW | Tsingke | GCAGGAAAGCAGCATTCTCGAGGGGAGAGCTTTGTGCTTGTC |
| COX1-UTR REV | Tsingke | CACTGATTAAAAGTCCCTCGCGGCCGCTAAAGTGCTTGTGTC |
| COX1-UTR-M FW | Tsingke | GTCTTGACTCATGTTTCTCATGAAGCTAATAAAATTCGC |
| COX1-UTR-M REV | Tsingke | AGCTTCATGAGAAACATGAGTCAAGACCTGGATG |
| COX2-LUC FW | Tsingke | CTACAAATTGAGGTACCTGGTGTAG |
| COX2-LUC REV | Tsingke | AATTGGAAGCTTACCGAGAGAACCTTC |
| COX2-LUC-M FW | Tsingke | GAGCAGATATACAGCCTATTAAGCGTATTAACTAAAACATAAAACATGTCAGCC |
| COX2-LUC-M REV | Tsingke | GGCTGACATGTTTTATGTTTTAGTTAATACGCTTAATAGGCTGTATATCTGCTC |
| FOS FW | Tsingke | GCTTTGCCTAAGCTTCACGATGATGTTCTCG |
| FOS REV | Tsingke | TTCCCTGAATTCTCACAGGGCCAGCAGCGTG |
| JUN FW | Tsingke | CACGTGAAGCTTCGGACTGTTCTATGACTGC |
| JUN REV | Tsingke | CGACGGTCTGAATTCAAAATGTTTGCAACTG |
| Keap1 sgRNA FW | Tsingke | AAACACCGTATGAGCCAGAGCGGGATG |
| Keap1 sgRNA REV | Tsingke | CTCTAAAACCATCCCGCTCTGGCTCATA |
| MIR-22-LUC FW | Tsingke | CAGTCCTCTGGGTTGAACAGAGCTATCTCAGACAGAGGAAGGTCGGACGGA |
| MIR-22-LUC REV | Tsingke | GATCTCCGTCCGACCTTCCTCTGTCTGAGATAGCTCTGTTCAACCCAGAGGACTGGTAC |
| MIR-22-LUC-M FW | Tsingke | CAGTCCTCTGGGTTGAACAGAAATATTTCAGACAGAGGAAGGTCGGACGGA |
| MIR-22-LUC-M REV | Tsingke | GATCTCCGTCCGACCTTCCTCTGTCTGAAATATTTCTGTTCAACCCAGAGGACTGGTAC |
| Nrf1 FW | Tsingke | CGGGGTACCATGCTTTCTCTGAAGAAATACTTAACGGAAGG |
| Nrf1 REV | Tsingke | GCTCTAGACACTTTCTCCGGTCCTTTGGCTTCC |
| Nrf1-LUC-#1 FW | Tsingke | CCTAGGCCTGCTAGCGCGACTGAGTTTGTCTCTACACCT |
| Nrf1-LUC-#1 REV | Tsingke | CTTCAGAGAAAAGCTTGCTGAAGGACCAGAATGTTTATGCT |
| Nrf1-LUC-#2 FW | Tsingke | CCTAGGCCTGCTAGCGCGACTGAGTTTGTCTCTACACCT |
| Nrf1-LUC-#2 REV | Tsingke | CGAACAAGTGAAGCTTCCCTGGCCTTGAC |
| Nrf1-LUC-#3 FW | Tsingke | CACCCAACGCGCTAGCCCACTAACATCG |
| Nrf1-LUC-#3 REV | Tsingke | CTTCAGAGAAAAGCTTGCTGAAGGACCAGAATGTTTATGCT |
| Nrf1-LUC-#4 FW | Tsingke | CACCCAACGCGCTAGCCCACTAACATCG |
| Nrf1-LUC-#4 REV | Tsingke | ACTGCACTCAAGCTTGGGCAACAAGAGCAA |
| Nrf1-LUC-#5 FW | Tsingke | CACCCAACGCGCTAGCCCACTAACATCG |
| Nrf1-LUC-#5 REV | Tsingke | CTACTAAGCTTGACTATTCCGTCCA |
| Nrf1-LUC-#6 FW | Tsingke | CACCCAACGCGCTAGCCCACTAACATCG |
| Nrf1-LUC-#6 REV | Tsingke | GTTCAAGCTTCCGGACAAAGTC |
| Nrf1-LUC-#7 FW | Tsingke | CACCCAACGCGCTAGCCCACTAACATCG |
| Nrf1-LUC-#7 REV | Tsingke | CTGGTAAGCTTCTGCCCGGATAC |
| Nrf2 FW | Tsingke | GAGCCCGGTACCACGGTCCACAGCTC |
| Nrf2 REV | Tsingke | AAAACTAGCTCGAGAAAGGTCAAATCCTCCT |
| Nrf2 sgRNA-1 FW | Tsingke | AAACACCGTATTTGACTTCAGTCAGCGA |
| Nrf2 sgRNA-1 REV | Tsingke | CTCTAAAACTCGCTGACTGAAGTCAAATA |
| Nrf2 sgRNA-2 FW | Tsingke | AAACACCGTGCATACCGTCTAAATCAAC |
| Nrf2 sgRNA-2 REV | Tsingke | CTCTAAAACGTTGATTTAGACGGTATGCA |
| Nrf2 sgRNA-3 FW | Tsingke | AAACACCGTGGATTTGATTGACATACTT |
| Nrf2 sgRNA-3 REV | Tsingke | CTCTAAAACAAGTATGTCAATCAAATCCA |
| Nrf2-LUC FW | Tsingke | CCAGGAGTTTGGTACCAGCCTGGGCAACATAGTGA |
| Nrf2-LUC REV | Tsingke | CCAGCTCCAAGTAGATCTTGATGAGCTGTGGA |
| PTEN-LUC FW | Tsingke | GGTACTTGGAGGCTGGTACCATATTCTAGCAC |
| PTEN-LUC REV | Tsingke | CGGGAGATCTGAGGGCAGGGCAGGGCA |
| PTEN-LUC-M1 FW | Tsingke | GAGCATTGTTTTCACCTGGTCCTTTTCACCTGTGCACAGGTAACCTCAG |
| PTEN-LUC-M1 REV | Tsingke | GTGCGTTGAGCAGTGTCACTGACTCGAGTCTGAGGTTACCTGTG |
| PTEN-LUC-M2 FW | Tsingke | GAGCAGCGTGGTCACCTGGTCCTTTTCACCTGTGCACAGGTAACCTCAG |
| PTEN-LUC-M2 REV | Tsingke | GTGCGTTGATTAGTTTCACTGACTCGAGTCTGAGGTTACCTGTG |
| PTEN-UTR FW | Tsingke | ATGGCAATAGGACCTCGAGTCAGATTACCAGTTATAGGAACAATTCTC |
| PTEN-UTR REV | Tsingke | CTTTCCATATGCTGGCGGCCGCGTACAGAATAATAGACAAAAGC |
| PTEN-UTR-M FW | Tsingke | CATATTGGTGCTAGAAAAAAGGAAGTGAATCTGTATTGGGGTACAG |
| PTEN-UTR-M REV | Tsingke | TGTACCCCAATACAGATTCACTTCCTTTTTTCTAGCACCAATATGCT |
| Recombinant DNA | ||
| pARE-luc | Zhang’s [97] | N/A |
| pcDNA3.1 | invitrogen | V79020 |
| pGL3-Basic | Promega | VQP0121 |
| pGL3-promoter | Promega | VQP0124 |
| pRL-TK | Promega | VQP0126 |
| psiCHECK2 | Promega | C8021 |
| Software and Algorithms | ||
| Canvas X | Canvas GFX, Inc. | https://www.canvasgfx.com/ |
| Chromas 2.4.1 | Technelysium Pty Ltd. | http://technelysium.com.au/wp/chromas/ |
| cytoscape | [98] | http://www.cytoscape.org/ |
| Excel | Microsoft | https://www.microsoft.com/ |
| FlowJo 7.6.5. | FlowJo | https://www.flowjo.com/ |
| KEGG | Kanehisa Laboratories | https://www.kegg.jp/ |
| Primer Premier 5 | PREMIER Biosoft International | https://www.PremierBiosoft.com/ |
| Targetscan 7.2 | [99] | http://www.targetscan.org/vert_72/ |
| Venny 2.1.0 | BioinfoGP, CNB-CSIC | http://bioinfogp.cnb.csic.es/tools/venny/index.html |
| Others | ||
| Cas9/gRNA Construct Kit | v-solid | VK001 |
| KeyGEN DAPI staining kit | KeyGEN BioTECH | KGA215 |
| DAB kit | Boster Biological Technology | AR1022 |
| Dual-luciferase reporter assay system | Promega | E1910 |
| FastTALETM TALEN Assembly Kit | SIDANSAI | 2801 |
| GoTaq® qPCR Master Mix | Promega | A6001 |
| Hematoxylin and Eosin Staining Kit | Beyotime | C0105 |
| Lenti-Pac HIV Expression Packaging Kit | Gene Copoeia | HPK-LvTR |
| Lipofectamine® 3000 Transfection Kit | Invitrogen | L3000-015 |
| Nuclei Isolation Kit | Sigma | NUC101-1KT |
| Reactive Oxygen Species Assay Kit | Beyotime | S0033 |
| Revert Aid First Strand Synthesis Kit | Thermo | K1622 |
| RNAsimple Total RNA Kit | Tiangen Biotech | DP419 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiu, L.; Wang, M.; Hu, S.; Ru, X.; Ren, Y.; Zhang, Z.; Yu, S.; Zhang, Y. Oncogenic Activation of Nrf2, Though as a Master Antioxidant Transcription Factor, Liberated by Specific Knockout of the Full-Length Nrf1α that Acts as a Dominant Tumor Repressor. Cancers 2018, 10, 520. https://doi.org/10.3390/cancers10120520
Qiu L, Wang M, Hu S, Ru X, Ren Y, Zhang Z, Yu S, Zhang Y. Oncogenic Activation of Nrf2, Though as a Master Antioxidant Transcription Factor, Liberated by Specific Knockout of the Full-Length Nrf1α that Acts as a Dominant Tumor Repressor. Cancers. 2018; 10(12):520. https://doi.org/10.3390/cancers10120520
Chicago/Turabian StyleQiu, Lu, Meng Wang, Shaofan Hu, Xufang Ru, Yonggang Ren, Zhengwen Zhang, Siwang Yu, and Yiguo Zhang. 2018. "Oncogenic Activation of Nrf2, Though as a Master Antioxidant Transcription Factor, Liberated by Specific Knockout of the Full-Length Nrf1α that Acts as a Dominant Tumor Repressor" Cancers 10, no. 12: 520. https://doi.org/10.3390/cancers10120520
APA StyleQiu, L., Wang, M., Hu, S., Ru, X., Ren, Y., Zhang, Z., Yu, S., & Zhang, Y. (2018). Oncogenic Activation of Nrf2, Though as a Master Antioxidant Transcription Factor, Liberated by Specific Knockout of the Full-Length Nrf1α that Acts as a Dominant Tumor Repressor. Cancers, 10(12), 520. https://doi.org/10.3390/cancers10120520