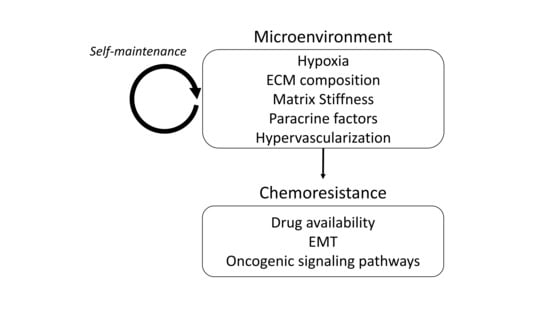
Chemoresistance and the Self-Maintaining Tumor Microenvironment


Abstract
{kind=link}
{kind=link}
{kind=link}
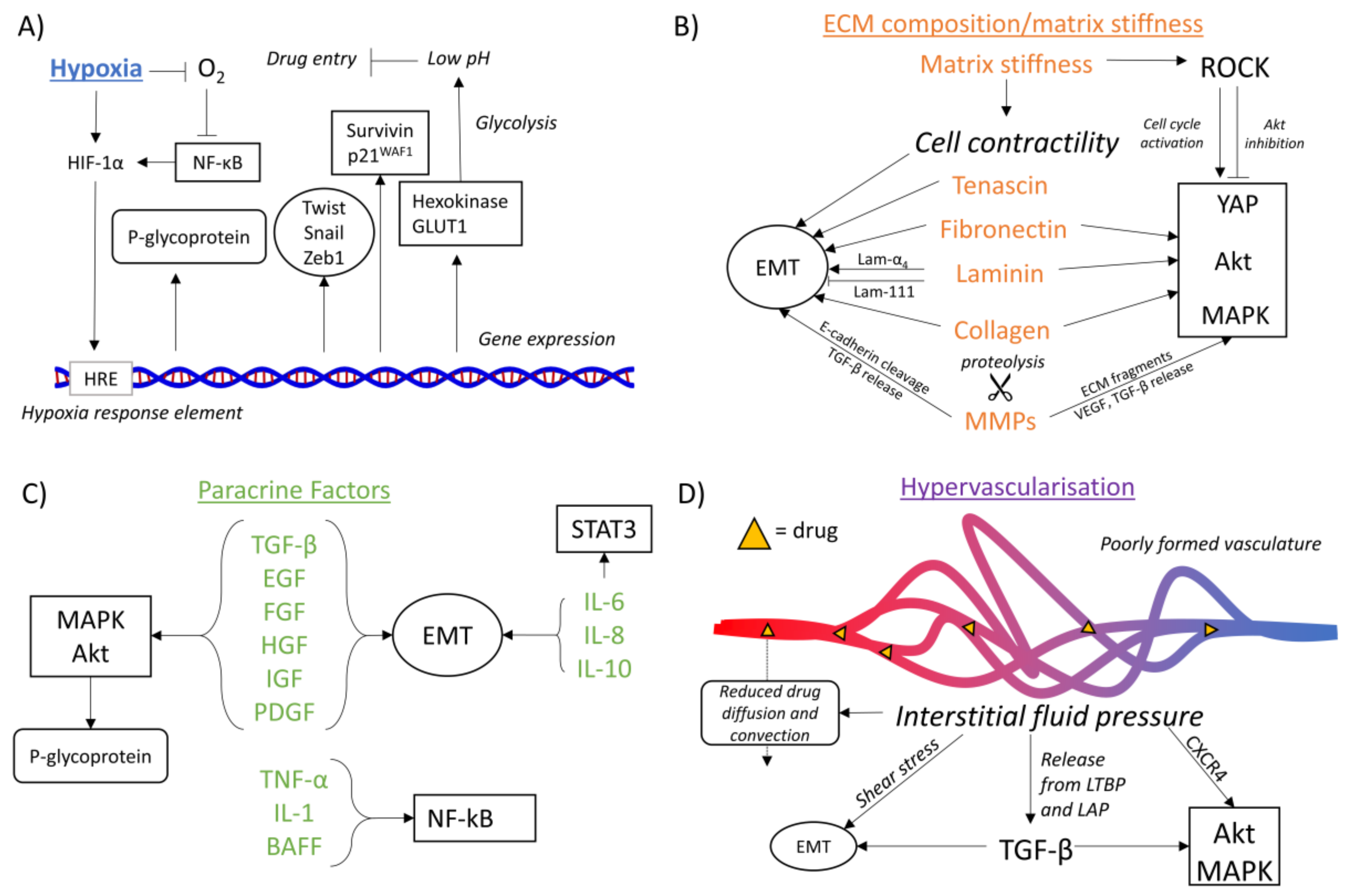{kind=link}
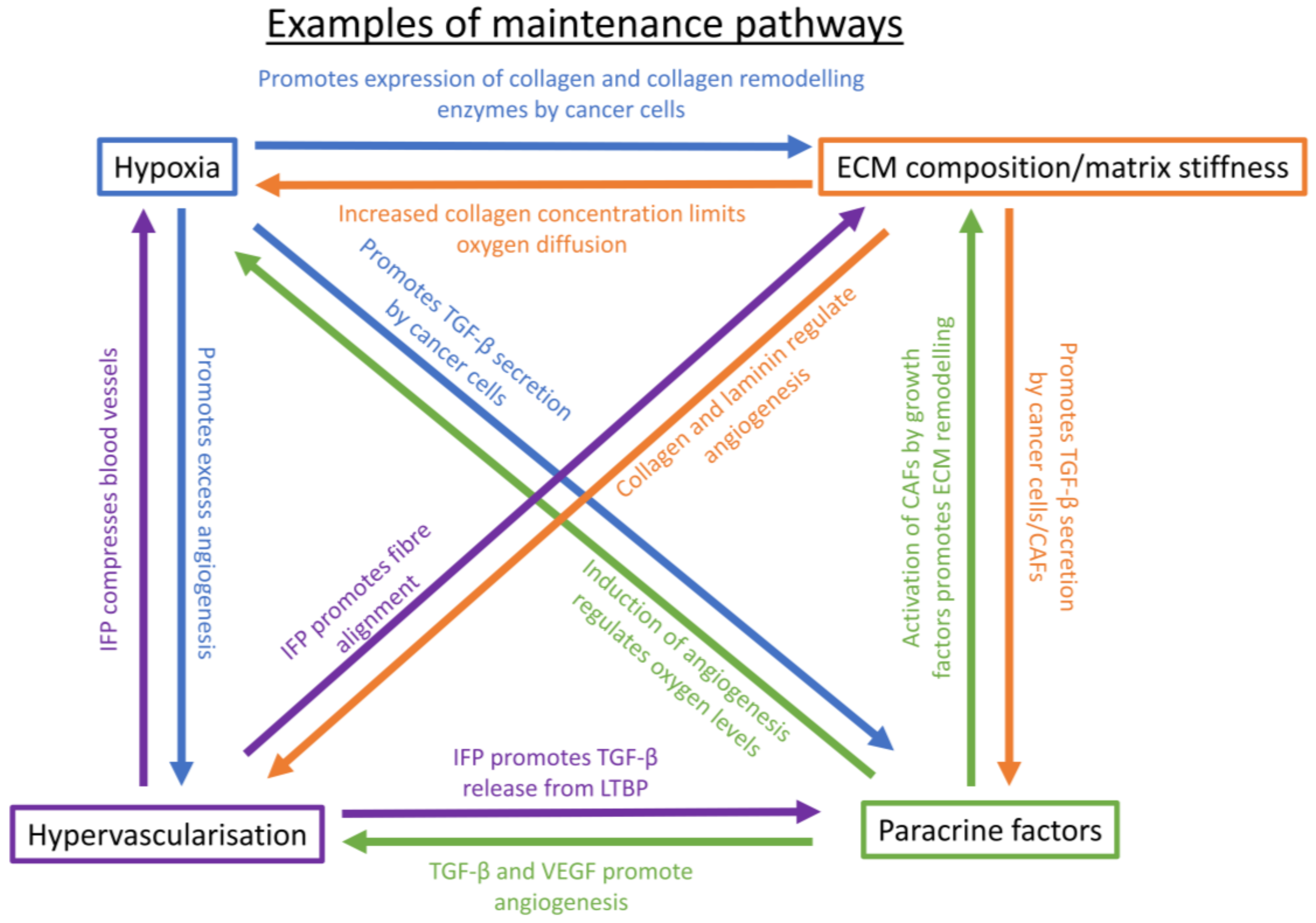{kind=link}
1. Introduction
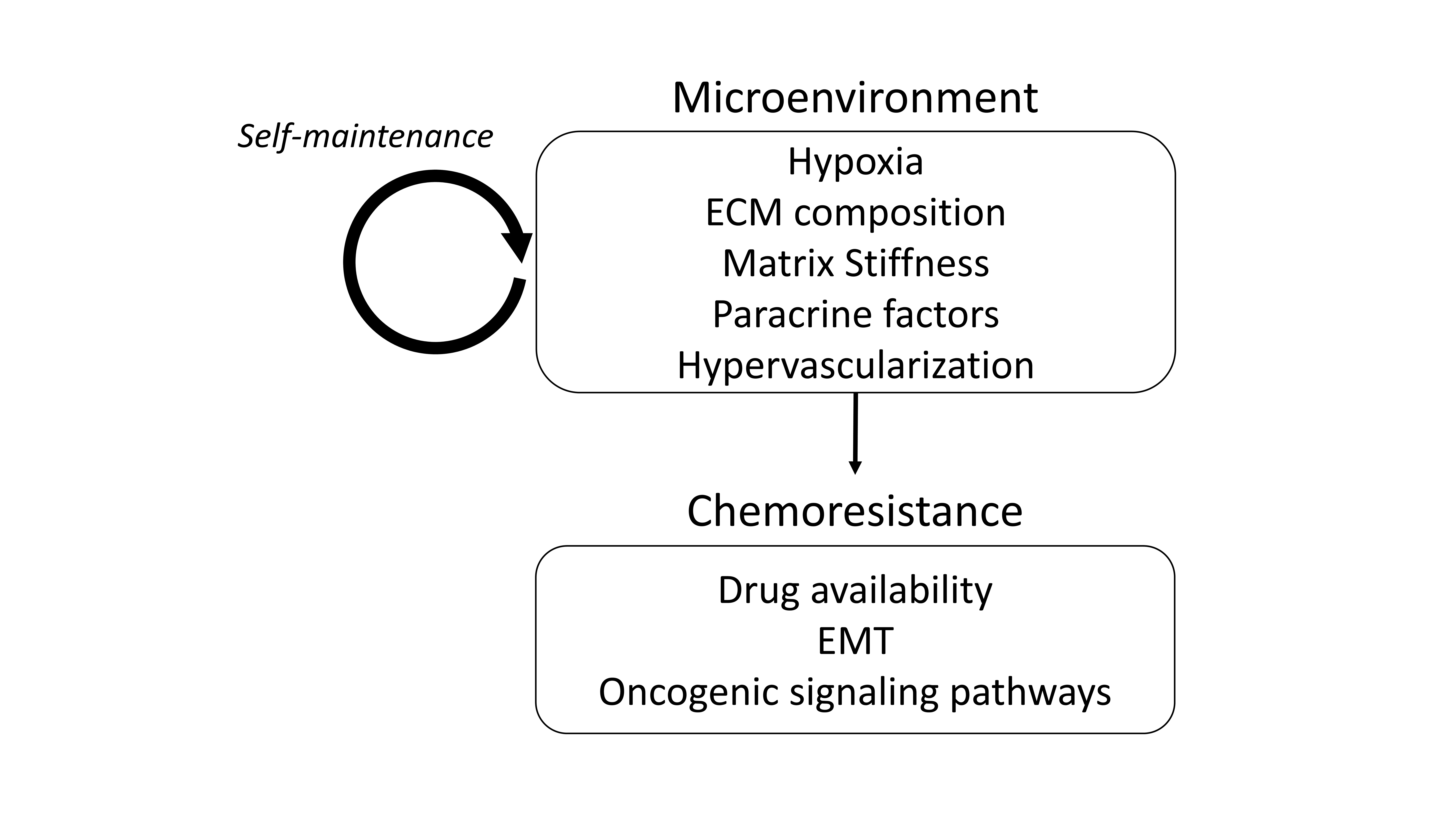
2. Mechanisms of Chemoresistance
2.1. Drug Availability
2.2. EMT
2.3. Oncogenic Signaling Pathways
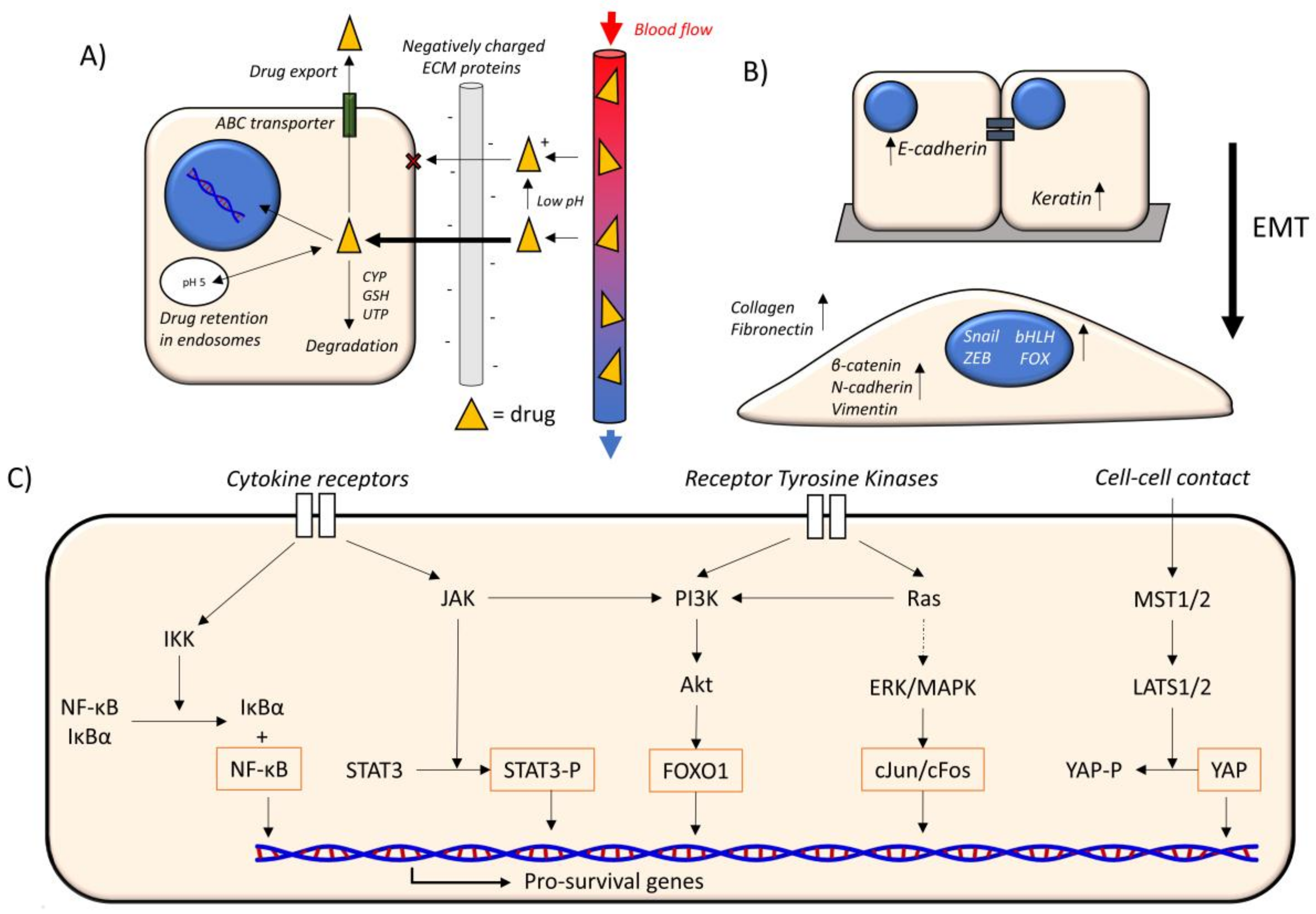
3. The Cancer Microenvironment
3.1. Hypoxia
3.2. ECM Composition
3.3. Matrix Stiffness
3.4. Paracrine Factors
3.5. Hypervascularization
4. Microenvironment-Mediated Chemoresistance
4.1. Hypoxia-Mediated Chemoresistance
4.1.1. Hypoxia and Drug Availability
4.1.2. Hypoxia and EMT
4.1.3. Hypoxia and Oncogenic Signaling Pathways
4.2. ECM Composition-Mediated Chemoresistance
4.2.1. ECM Composition and Drug Availability
4.2.2. ECM Composition and EMT
4.2.3. ECM Composition and Oncogenic Signaling Pathways
4.3. Matrix Stiffness-Mediated Chemoresistance
4.3.1. Matrix Stiffness and Drug Availability
4.3.2. Matrix Stiffness and EMT
4.3.3. Matrix Stiffness and Oncogenic Signaling Pathways
4.4. Paracrine Factor-Mediated Chemoresistance
4.4.1. Paracrine Factors and Drug Availability
4.4.2. Paracrine Factors and EMT
4.4.3. Paracrine Factors and Oncogenic Signaling Pathways
4.5. Hypervascularization-Mediated Chemoresistance
4.5.1. Hypervascularization and Drug Availability
4.5.2. Hypervascularization and EMT
4.5.3. Hypervascularization and Oncogenic Signaling Pathways
5. Maintenance of a Chemoresistance-Promoting Microenvironment
5.1. Hypoxia-Mediated Maintenance
5.2. ECM Composition and Matrix Stiffness-Mediated Maintenance
5.3. Paracrine Factor-Mediated Maintenance
5.4. Hypervascularization-Mediated Maintenance
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zheng, H. The molecular mechanisms of chemoresistance in cancers. Oncotarget 2017, 8, 59950–59964. [Google Scholar] [CrossRef] [PubMed]
- Senthebane, D.A.; Rowe, A.; Thomford, N.E.; Shipanga, H.; Munro, D.; Mazeedi, M.; Almazyadi, H.A.M.; Kallmeyer, K.; Dandara, C.; Pepper, M.S.; et al. The Role of Tumor Microenvironment in Chemoresistance: To Survive, Keep Your Enemies Closer. Int. J. Mol. Sci. 2017, 18, 1586. [Google Scholar] [CrossRef] [PubMed]
- Cox, T.R.; Erler, J.T. Remodeling and homeostasis of the extracellular matrix: Implications for fibrotic diseases and cancer. Dis. Model. Mech. 2011, 4, 165–178. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Nie, D.; Chakrabarty, S. Growth factors in tumor microenvironment. Front. Biosci. 2012, 15, 151–165. [Google Scholar] [CrossRef]
- Petrova, V.; Annicchiarico-Petruzzelli, M.; Melino, G.; Amelio, I. The hypoxic tumour microenvironment. Oncogenesis 2018, 7, 10. [Google Scholar] [CrossRef] [PubMed]
- Chorawala, M.; Oza, P.; Shah, G. Mechanisms of Anticancer Drugs Resistance: An Overview. Int. J. Pharm. Sci. Drug Res. 2012, 4, 1–9. [Google Scholar] [CrossRef]
- Dzobo, K.; Senthebane, D.A.; Rowe, A.; Thomford, N.E.; Mwapagha, L.M.; Al-Awwad, N.; Dandara, C.; Parker, M.I. Cancer Stem Cell Hypothesis for Therapeutic Innovation in Clinical Oncology? Taking the Root Out, Not Chopping the Leaf. OMICS 2016, 20, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J. Cancer stem cells and chemoresistance: The smartest survives the raid. Pharmacol. Ther. 2016, 160, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.; Saggar, J.K.; Yu, M.; Wang, M.; Tannock, I.F. Mechanisms of Drug Resistance Related to the Microenvironment of Solid Tumors and Possible Strategies to Inhibit Them. Cancer J. 2015, 21, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Stylianopoulos, T.; Poh, M.Z.; Insin, N.; Bawendi, M.G.; Fukumura, D.; Munn, L.L.; Jain, R.K. Diffusion of particles in the extracellular matrix: The effect of repulsive electrostatic interactions. Biophys. J. 2010, 99, 1342–1349. [Google Scholar] [CrossRef] [PubMed]
- Tannock, I.F.; Rotin, D. Acid pH in Tumors and Its Potential for Therapeutic Exploitation. Cancer Res. 1989, 49, 4973–4384. [Google Scholar]
- Michael, M.; Doherty, M.M. Tumoral drug metabolism: Overview and its implications for cancer therapy. J. Clin. Oncol. 2005, 23, 205–229. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.I.; Haber, M.; Henderson, M.J.; Norris, M.D. ABC transporters in cancer: More than just drug efflux pumps. Nat. Rev. Cancer 2010, 10, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Liang, X. Overcoming drug efflux-based multidrug resistance in cancer with nanotechnology. Chin. J. Cancer 2012, 31, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Krishna, R.; Mayer, L.D. Multidrug resistance (MDR) in cancer. Mechanisms, reversal using modulators of MDR and the role of MDR modulators in influencing the pharmacokinetics of anticancer drugs. Eur. J. Pharm. Sci. 2000, 11, 265–283. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Parri, M.; Chiarugi, P. Rac and Rho GTPases in cancer cell motility control. Cell Commun. Signal. 2010, 8, 23. [Google Scholar] [CrossRef] [PubMed]
- Wheelock, M.J.; Shintani, Y.; Maeda, M.; Fukumoto, Y.; Johnson, K.R. Cadherin switching. J. Cell Sci. 2008, 121, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Washington, M.K.; Crawford, H.C. Loss of FOXA1/2 is essential for the epithelial-to-mesenchymal transition in pancreatic cancer. Cancer Res. 2010, 70, 2115–2125. [Google Scholar] [CrossRef] [PubMed]
- Creighton, C.J.; Li, X.; Landis, M.; Dixon, J.M.; Neumeister, V.M.; Sjolund, A.; Rimm, D.L.; Wong, H.; Rodriguez, A.; Herschkowitz, J.I.; et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc. Natl. Acad. Sci. USA 2009, 106, 13820–13825. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.H.; Wang, C.H.; Huang, Z.J.; Liu, F.; Xu, C.W.; Li, X.L.; Chen, G.Q. miR-760 mediates chemoresistance through inhibition of epithelial mesenchymal transition in breast cancer cells. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 5002–5008. [Google Scholar] [PubMed]
- Li, J.; Liu, H.; Yu, J.; Yu, H. Chemoresistance to doxorubicin induces epithelial-mesenchymal transition via upregulation of transforming growth factor beta signaling in HCT116 colon cancer cells. Mol. Med. Rep. 2015, 12, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends. Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Asati, V.; Mahapatra, D.K.; Bharti, S.K. PI3K/Akt/mTOR and Ras/Raf/MEK/ERK signaling pathways inhibitors as anticancer agents: Structural and pharmacological perspectives. Eur. J. Med. Chem. 2016, 109, 314–341. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim. Biophys. Acta 2007, 1773, 1263–1284. [Google Scholar] [CrossRef] [PubMed]
- Peh, J.; Boudreau, M.W.; Smith, H.M.; Hergenrother, P.J. Overcoming Resistance to Targeted Anticancer Therapies through Small-Molecule-Mediated MEK Degradation. Cell Chem. Biol. 2018, 25, 996–1005.e4. [Google Scholar] [CrossRef] [PubMed]
- Godwin, P.; Baird, A.M.; Heavey, S.; Barr, M.P.; O’Byrne, K.J.; Gately, K. Targeting nuclear factor-kappa B to overcome resistance to chemotherapy. Front. Oncol. 2013, 3, 120. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ahmed, F.; Ali, S.; Philip, P.A.; Kucuk, O.; Sarkar, F.H. Inactivation of nuclear factor kappaB by soy isoflavone genistein contributes to increased apoptosis induced by chemotherapeutic agents in human cancer cells. Cancer Res. 2005, 65, 6934–6942. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, S.; Son, K.; Onda, S.; Schmidt, C.; Scrabas, G.; Okamoto, T.; Fujita, T.; Chiao, P.; Yanaga, K. Desensitization of NFκB for Overcoming Chemoresistance of Pancreatic Cancer Cells to TNF-α or Paclitaxel. Anticancer Res. 2012, 32, 4813–4822. [Google Scholar] [PubMed]
- Zhao, Y.; Yang, X. The Hippo pathway in chemotherapeutic drug resistance. Int. J. Cancer 2015, 137, 2767–2773. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Lee, H.; Herrmann, A.; Buettner, R.; Jove, R. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat. Rev. Cancer 2014, 14, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Feng, J.; Hong, Z.; Chen, L.; Li, W.; Liao, S.; Wang, X.; Ji, T.; Wang, S.; Ma, D.; et al. Silencing of the STAT3 signaling pathway reverses the inherent and induced chemoresistance of human ovarian cancer cells. Biochem. Biophys. Res. Commun. 2013, 435, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Melton, D.W. Cisplatin regulates the MAPK kinase pathway to induce increased expression of DNA repair gene ERCC1 and increase melanoma chemoresistance. Oncogene 2012, 31, 2412–2422. [Google Scholar] [CrossRef] [PubMed]
- Hientz, K.; Mohr, A.; Bhakta-Guha, D.; Efferth, T. The role of p53 in cancer drug resistance and targeted chemotherapy. Oncotarget 2017, 8, 8921–8946. [Google Scholar] [CrossRef] [PubMed]
- Doktorova, H.; Hrabeta, J.; Khalil, M.A.; Eckschlager, T. Hypoxia-induced chemoresistance in cancer cells: The role of not only HIF-1. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech Repub. 2015, 159, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Muz, B.; de la Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Hewitson, K.S.; Schofield, C.J.; Ratcliffe, P.J. Hypoxia-Inducible Factor Prolyl-Hydroxylase: Purification and Assays of PHD2. Methods Enzymol. 2007, 435, 25–42. [Google Scholar] [CrossRef] [PubMed]
- Nurwidya, F.; Takahashi, F.; Minakata, K.; Murakami, A.; Takahashi, K. From tumor hypoxia to cancer progression: The implications of hypoxia-inducible factor-1 expression in cancers. Anat. Cell Biol. 2012, 45, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, R.; Pare, G.C.; Frederiksen, L.J.; Semenza, G.L.; Graham, C.H. Hypoxia-induced resistance to anticancer drugs is associated with decreased senescence and requires hypoxia-inducible factor-1 activity. Mol. Cancer Ther. 2008, 7, 1961–1973. [Google Scholar] [CrossRef] [PubMed]
- McMahon, S.; Charbonneau, M.; Grandmont, S.; Richard, D.E.; Dubois, C.M. Transforming growth factor beta1 induces hypoxia-inducible factor-1 stabilization through selective inhibition of PHD2 expression. J. Biol. Chem. 2006, 281, 24171–24181. [Google Scholar] [CrossRef] [PubMed]
- Adamaki, M. Cancer and the Cellular Response to Hypoxia. Pediatr. Ther. 2013, 03. [Google Scholar] [CrossRef]
- Xiong, G.-F.; Xu, R. Function of cancer cell-derived extracellular matrix in tumor progression. J. Cancer Metastasis Treat. 2016, 2, 357. [Google Scholar] [CrossRef]
- Ioachim, E.; Charchanti, A.; Briasoulis, E.; Karavasilis, V.; Tsanou, H.; Arvanitis, D.; Agnantis, N.; Pavlidis, N. Immunohistochemical expression of extracellular matrix components tenascin, fibronectin, collagen type IV and laminin in breast cancer: Their prognostic value and role in tumour invasion and progression. Eur. J. Cancer 2002, 38, 2362–2370. [Google Scholar] [CrossRef]
- Ruoslahti, E. RGD and other recognition sequences for integrins. Annu. Rev. Cell Dev. Biol. 1996, 12, 697–715. [Google Scholar] [CrossRef] [PubMed]
- Moreira, R.K. Hepatic stellate cells and liver fibrosis. Arch. Pathol. Lab. Med. 2007, 131, 1728–1734. [Google Scholar] [CrossRef] [PubMed]
- Heerboth, S.; Housman, G.; Leary, M.; Longacre, M.; Byler, S.; Lapinska, K.; Willbanks, A.; Sarkar, S. EMT and tumor metastasis. Clin. Transl. Med. 2015, 4, 6. [Google Scholar] [CrossRef] [PubMed]
- Krstic, J.; Santibanez, J.F. Transforming growth factor-beta and matrix metalloproteinases: Functional interactions in tumor stroma-infiltrating myeloid cells. Sci. World J. 2014, 2014, 521754. [Google Scholar] [CrossRef] [PubMed]
- Levental, K.R.; Yu, H.; Kass, L.; Lakins, J.N.; Egeblad, M.; Erler, J.T.; Fong, S.F.; Csiszar, K.; Giaccia, A.; Weninger, W.; et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 2009, 139, 891–906. [Google Scholar] [CrossRef] [PubMed]
- Mueller, S.; Sandrin, L. Liver stiffness: A novel parameter for the diagnosis of liver disease. Hepatic Med. Evid. Res. 2010, 2, 49–67. [Google Scholar] [CrossRef]
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Haining, A.W.M.; Rahikainen, R.; Cortes, E.; Lachowski, D.; Rice, A.; von Essen, M.; Hytonen, V.P.; Del Rio Hernandez, A. Mechanotransduction in talin through the interaction of the R8 domain with DLC1. PLoS Biol. 2018, 16, e2005599. [Google Scholar] [CrossRef] [PubMed]
- Lachowski, D.; Cortes, E.; Robinson, B.; Rice, A.; Rombouts, K.; Del Rio Hernandez, A.E. FAK controls the mechanical activation of YAP, a transcriptional regulator required for durotaxis. FASEB J. 2017, 32, 1099–1107. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Costanza, B.; Umelo, I.A.; Bellier, J.; Castronovo, V.; Turtoi, A. Stromal Modulators of TGF-beta in Cancer. J. Clin. Med. 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef] [PubMed]
- Ikushima, H.; Miyazono, K. TGFbeta signalling: A complex web in cancer progression. Nat. Rev. Cancer 2010, 10, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Edlund, S.; Bu, S.; Schuster, N.; Aspenstrom, P.; Heuchel, R.; Heldin, N.E.; Ten Dijke, P.; Heldin, C.H.; Landstrom, M. Transforming growth factor-beta1 (TGF-beta)-induced apoptosis of prostate cancer cells involves Smad7-dependent activation of p38 by TGF-beta-activated kinase 1 and mitogen-activated protein kinase kinase 3. Mol. Biol. Cell 2003, 14, 529–544. [Google Scholar] [CrossRef] [PubMed]
- Wikstrom, P.; Stattin, P.; Franck-Lissbrant, I.; Damber, J.; Bergh, A. Transforming Growth Factor B1 is associated with angiogenesis, metastasis, and poor clinical outcome in prostate cancer. Prostate 1998, 37, 19–29. [Google Scholar] [CrossRef]
- Adorno, M.; Cordenonsi, M.; Montagner, M.; Dupont, S.; Wong, C.; Hann, B.; Solari, A.; Bobisse, S.; Rondina, M.B.; Guzzardo, V.; et al. A Mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell 2009, 137, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Leight, J.; Wozniak, M.A.; Chen, S.; Lynch, M.; Chen, C. Matrix rigidity regulates a switch between TGF-β1–induced apoptosis and epithelial–mesenchymal transition. Mol. Biol. Cell 2012, 23, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Erdogan, B.; Webb, D.J. Cancer-associated fibroblasts modulate growth factor signaling and extracellular matrix remodeling to regulate tumor metastasis. Biochem. Soc. Trans. 2017, 45, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.; Charles, K.A.; Mantovani, A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell 2005, 7, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Landskron, G.; De la Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic inflammation and cytokines in the tumor microenvironment. J. Immunol. Res. 2014, 2014, 149185. [Google Scholar] [CrossRef] [PubMed]
- Vyas, D.; Laput, G.; Vyas, A.K. Chemotherapy-enhanced inflammation may lead to the failure of therapy and metastasis. Oncol Targets Ther. 2014, 7, 1015–1023. [Google Scholar] [CrossRef] [PubMed]
- Tato-Costa, J.; Casimiro, S.; Pacheco, T.; Pires, R.; Fernandes, A.; Alho, I.; Pereira, P.; Costa, P.; Castelo, H.B.; Ferreira, J.; et al. Therapy-Induced Cellular Senescence Induces Epithelial-to-Mesenchymal Transition and Increases Invasiveness in Rectal Cancer. Clin. Colorectal. Cancer 2016, 15, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Yano, H.; Nishida, T.; Kamura, T.; Kojiro, M. Angiogenesis in Cancer. Vasc. Health Risk Manag. 2006, 2, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Weis, S.M.; Cheresh, D.A. Tumor angiogenesis: Molecular pathways and therapeutic targets. Nat. Med. 2011, 17, 1359–1370. [Google Scholar] [CrossRef] [PubMed]
- Siemann, D.W. The unique characteristics of tumor vasculature and preclinical evidence for its selective disruption by Tumor-Vascular Disrupting Agents. Cancer Treat. Rev. 2011, 37, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, S.; Allegrini, P.R.; Becquet, M.M.; McSheehy, P.M.J. Tumor Interstitial Fluid Pressure as an Early-Response Marker for Anticancer Therapeutics. Neoplasia 2009, 11, 874–881. [Google Scholar] [CrossRef] [PubMed]
- Crawford, Y.; Kasman, I.; Yu, L.; Zhong, C.; Wu, X.; Modrusan, Z.; Kaminker, J.; Ferrara, N. PDGF-C mediates the angiogenic and tumorigenic properties of fibroblasts associated with tumors refractory to anti-VEGF treatment. Cancer Cell 2009, 15, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.L.; Coomber, B.L.; Kerbel, R.S. A paradigm for therapy-induced microenvironmental changes in solid tumors leading to drug resistance. Differentiation 2002, 70, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Sriraman, S.K.; Aryasomayajula, B.; Torchilin, V.P. Barriers to drug delivery in solid tumors. Tissue Barriers 2014, 2, e29528. [Google Scholar] [CrossRef] [PubMed]
- Vukovic, V.; Tannock, I.F. Influence of low pH on cytotoxicity of paclitaxel, mitoxantrone and topotecan. Br. J. Cancer 1997, 75, 1167–1172. [Google Scholar] [CrossRef] [PubMed]
- Du Souich, P.; Fradette, C. The effect and clinical consequences of hypoxia on cytochrome P450, membrane carrier proteins activity and expression. Expert Opin. Drug Metab. Toxicol. 2011, 7, 1083–1100. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.A.; Roth, R.A.; LaPres, J.J. Hypoxia, drug therapy and toxicity. Pharmacol. Ther. 2007, 113, 229–246. [Google Scholar] [CrossRef] [PubMed]
- Comerford, K.; Wallace, T.; Karhausen, J.; Louis, N.; Montalto, M.; Colgan, S.P. Hypoxia-inducible Factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res. 2002, 62, 3387–3394. [Google Scholar] [PubMed]
- Wartenburg, M.; Ling, F.C.; Muschen, M.; Klein, F.; Acker, H.; Gassmann, M.; Petrat, K.; Putz, V.; Heschler, J.; Sauer, H. Regulation of the multidrug resistance transporter P-glycoprotein in multicellular tumor spheroids by hypoxia-inducible factor (HIF-1) and reactive oxygen species. FASEB J. 2003, 17, 503–505. [Google Scholar] [CrossRef] [PubMed]
- Kizaka-Kondo, S.; Inoue, M.; Harada, H.; Hiraoka, M. Tumour hypoxia: A target for selective cancer therapy. Cancer Sci. 2004, 94, 1021–1028. [Google Scholar] [CrossRef]
- Cheng, Z.X.; Sun, B.; Wang, S.J.; Gao, Y.; Zhang, Y.M.; Zhou, H.X.; Jia, G.; Wang, Y.W.; Kong, R.; Pan, S.H.; et al. Nuclear factor-kappaB-dependent epithelial to mesenchymal transition induced by HIF-1alpha activation in pancreatic cancer cells under hypoxic conditions. PLoS ONE 2011, 6, e23752. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.H.; Huang, C.; Feng, Z.Z.; Lv, X.H.; Qiu, Z.J. Hypoxia-induced snail expression through transcriptional regulation by HIF-1alpha in pancreatic cancer cells. Dig. Dis. Sci. 2013, 58, 3503–3515. [Google Scholar] [CrossRef] [PubMed]
- Yeo, C.D.; Kang, N.; Choi, S.Y.; Kim, B.N.; Park, C.K.; Kim, J.W.; Kim, Y.K.; Kim, S.J. The role of hypoxia on the acquisition of epithelial-mesenchymal transition and cancer stemness: A possible link to epigenetic regulation. Korean J. Intern. Med. 2017, 32, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.H.; Wu, M.Z.; Chiou, S.H.; Chen, P.M.; Chang, S.Y.; Liu, C.J.; Teng, S.C.; Wu, K.J. Direct regulation of TWIST by HIF-1alpha promotes metastasis. Nat. Cell Biol. 2008, 10, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Wang, J.; Li, J.; Post, M. Mouse snail is a target gene for HIF. Mol. Cancer Res. 2011, 9, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Shi, X.; Peng, Y.; Wu, M.; Zhang, P.; Xie, R.; Wu, Y.; Yan, Q.; Liu, S.; Wang, J. HIF-1alpha Promotes Epithelial-Mesenchymal Transition and Metastasis through Direct Regulation of ZEB1 in Colorectal Cancer. PLoS ONE 2015, 10, e0129603. [Google Scholar] [CrossRef]
- Lundgren, K.; Nordenskjold, B.; Landberg, G. Hypoxia, Snail and incomplete epithelial-mesenchymal transition in breast cancer. Br. J. Cancer 2009, 101, 1769–1781. [Google Scholar] [CrossRef] [PubMed]
- Okumura, Y.; Noda, T.; Eguchi, H.; Sakamoto, T.; Iwagami, Y.; Yamada, D.; Asaoka, T.; Wada, H.; Kawamoto, K.; Gotoh, K.; et al. Hypoxia-Induced PLOD2 is a Key Regulator in Epithelial-Mesenchymal Transition and Chemoresistance in Biliary Tract Cancer. Ann. Surg. Oncol. 2018, 25, 3728–3737. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.X.; Wang, D.W.; Liu, T.; Liu, W.X.; Xia, W.B.; Xu, J.; Zhang, Y.H.; Qu, Y.K.; Guo, L.Q.; Ding, L.; et al. Effects of the HIF-1alpha and NF-kappaB loop on epithelial-mesenchymal transition and chemoresistance induced by hypoxia in pancreatic cancer cells. Oncol. Rep. 2014, 31, 1891–1898. [Google Scholar] [CrossRef] [PubMed]
- Taylor, C.T.; Cummins, E.P. The role of NF-kappaB in hypoxia-induced gene expression. Ann. N. Y. Acad. Sci. 2009, 1177, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Sang, N.; Stiehl, D.P.; Bohensky, J.; Leshchinsky, I.; Srinivas, V.; Caro, J. MAPK signaling up-regulates the activity of hypoxia-inducible factors by its effects on p300. J. Biol. Chem. 2003, 278, 14013–14019. [Google Scholar] [CrossRef] [PubMed]
- Van Uden, P.; Kenneth, N.S.; Rocha, S. Regulation of hypoxia-inducible factor-1alpha by NF-kappaB. Biochem. J. 2008, 412, 477–484. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, K.; Fidler, I.J. Hypoxia increases resistance of human pancreatic cancer cells to apoptosis induced by gemcitabine. Clin. Cancer Res. 2004, 10, 2299–2306. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, Y.; Ma, Y.; Yang, L.; Wang, T.; Meng, X.; Zong, Z.; Sun, X.; Hua, X.; Li, H. Yes-associated protein (YAP) binds to HIF-1alpha and sustains HIF-1alpha protein stability to promote hepatocellular carcinoma cell glycolysis under hypoxic stress. J. Exp. Clin. Cancer Res. 2018, 37, 216. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Chen, Y.; Chen, L.; Cheng, H.; Mu, C.; Li, J.; Gao, R.; Zhou, C.; Cao, L.; Liu, J.; et al. Hypoxia regulates Hippo signalling through the SIAH2 ubiquitin E3 ligase. Nat. Cell Biol. 2014, 17, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Heddleston, J.M.; Li, Z.; Lathia, J.D.; Bao, S.; Hjelmeland, A.B.; Rich, J.N. Hypoxia inducible factors in cancer stem cells. Br. J. Cancer 2010, 102, 789–795. [Google Scholar] [CrossRef] [PubMed]
- Bao, B.; Azmi, A.S.; Ali, S.; Ahmad, A.; Li, Y.; Banerjee, S.; Kong, D.; Sarkar, F.H. The biological kinship of hypoxia with CSC and EMT and their relationship with deregulated expression of miRNAs and tumor aggressiveness. Biochim. Biophys. Acta 2012, 1826, 272–296. [Google Scholar] [CrossRef] [PubMed]
- Riddle, S.; Ahmad, A.; Ahmad, S.; Deeb, S.; Malkki, M.; Kelly Schneider, B.; Allen, C.; White, C. Hypoxia induces hexokinase II gene expression in human lung cell line A549. Am. J. Physiol. Lung Cell Mol. Physiol. 2000, 278, L407–L416. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, B.; Mohd Omar, M.F.; Soong, R. The Warburg effect and drug resistance. Br. J. Pharmacol. 2016, 173, 970–979. [Google Scholar] [CrossRef] [PubMed]
- Song, K.; Li, M.; Xu, X.-J.; Xuan, L.; Huang, G.-N.; Song, X.-L.; Liu, Q.-F. HIF-1α and GLUT1 Gene Expression is Associated with Chemoresistance of Acute Myeloid Leukemia. Asian Pac. J. Cancer Prev. 2014, 15, 1823–1829. [Google Scholar] [CrossRef] [PubMed]
- Schmid, T.; Zhou, J.; Brune, B. HIF-1 and p53: Communication of transcription factors under hypoxia. J. Cell Mol. Med. 2004, 8, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Obacz, J.; Pastorekova, S.; Vojtesek, B.; Hrstka, R. Cross-talk between HIF and p53 as mediators of molecular responses to physiological and genotoxic stresses. Mol. Cancer 2013, 12, 93. [Google Scholar] [CrossRef] [PubMed]
- Weniger, M.; Honselmann, K.C.; Liss, A.S. The Extracellular Matrix and Pancreatic Cancer: A Complex Relationship. Cancers 2018, 10, E316. [Google Scholar] [CrossRef] [PubMed]
- Netti, P.; Berk, D.; Swartz, M.; Godzinsky, A.; Jain, R.K. Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Res. 2000, 60, 2497–2503. [Google Scholar] [PubMed]
- Graff, B.A.; Vangberg, L.; Rofstad, E.K. Quantitative assessment of uptake and distribution of iron oxide particles (NC100150) in human melanoma xenografts by contrast-enhanced MRI. Magn. Reson. Med. 2004, 51, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Pun, S.H.; Tack, F.; Bellocq, N.C.; Cheng, J.; Grubbs, B.H.; Jensen, G.S.; Davis, M.E.; Brewster, M.; Janicot, M.; Janssens, B.; et al. Targeted delivery of RNA-cleaving DNA enzyme (DNAzyme) to tumor tissue by transferrin-modified, cyclodextrin-based particles. Cancer Biol. Ther. 2014, 3, 641–650. [Google Scholar] [CrossRef]
- Ricciardelli, C.; Ween, M.P.; Lokman, N.A.; Tan, I.A.; Pyragius, C.E.; Oehler, M.K. Chemotherapy-induced hyaluronan production: A novel chemoresistance mechanism in ovarian cancer. BMC Cancer 2013, 13, 476. [Google Scholar] [CrossRef] [PubMed]
- Bosnian, F.T.; Havenith, M.; Cleutjens, J.P.M. Basement Membranes in Cancer. Ultrastruct. Pathol. 1985, 8, 291–304. [Google Scholar] [CrossRef]
- Park, J.; Schwarzbauer, J.E. Mammary epithelial cell interactions with fibronectin stimulate epithelial-mesenchymal transition. Oncogene 2014, 33, 1649–1657. [Google Scholar] [CrossRef] [PubMed]
- Camara, J.; Jarai, G. Epithelial-mesenchymal transition in primary human bronchial epithelial cells is Smad-dependent and enhanced by fibronectin and TNF-alpha. Fibrogenes. Tissue Repair 2010, 3, 2. [Google Scholar] [CrossRef] [PubMed]
- Li, C.L.; Yang, D.; Cao, X.; Wang, F.; Hong, D.Y.; Wang, J.; Shen, X.C.; Chen, Y. Fibronectin induces epithelial-mesenchymal transition in human breast cancer MCF-7 cells via activation of calpain. Oncol. Lett. 2017, 13, 3889–3895. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.N.; Odero-Marah, V.A. The role of Snail in prostate cancer. Cell Adh. Migr. 2012, 6, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Koenig, A.; Mueller, C.; Hasel, C.; Adler, G.; Menke, A. Collagen type I induces disruption of E-cadherin-mediated cell-cell contacts and promotes proliferation of pancreatic carcinoma cells. Cancer Res. 2006, 66, 4662–4671. [Google Scholar] [CrossRef] [PubMed]
- Shintani, Y.; Maeda, M.; Chaika, N.; Johnson, K.R.; Wheelock, M.J. Collagen I promotes epithelial-to-mesenchymal transition in lung cancer cells via transforming growth factor-beta signaling. Am. J. Respir. Cell Mol. Biol. 2008, 38, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.K.; Lee, K.; Radisky, D.C.; Nelson, C.M. Extracellular matrix proteins regulate epithelial-mesenchymal transition in mammary epithelial cells. Differentiation 2013, 86, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Wondimu, Z.; Oikawa, Y.; Gentilcore, G.; Kiessling, R.; Egyhazi Brage, S.; Hansson, J.; Patarroyo, M. Laminins 411 and 421 differentially promote tumor cell migration via alpha6beta1 integrin and MCAM (CD146). Matrix Biol. 2014, 38, 69–83. [Google Scholar] [CrossRef] [PubMed]
- Nagaharu, K.; Zhang, X.; Yoshida, T.; Katoh, D.; Hanamura, N.; Kozuka, Y.; Ogawa, T.; Shiraishi, T.; Imanaka-Yoshida, K. Tenascin C induces epithelial-mesenchymal transition-like change accompanied by SRC activation and focal adhesion kinase phosphorylation in human breast cancer cells. Am. J. Pathol. 2011, 178, 754–763. [Google Scholar] [CrossRef] [PubMed]
- Noe, V.; Fingleton, B.; Jacobs, K.; Crawford, H.; Vermeulen, S.; Steelant, W.; Bruyneel, E.; Matrisian, L.; Mareel, M. Release of an invasion promoter E-cadherin fragment by matrilysin and stromelysin-1. J. Cell Sci. 2001, 114, 111–118. [Google Scholar] [PubMed]
- Radisky, D.C.; Levy, D.D.; Littlepage, L.E.; Liu, H.; Nelson, C.M.; Fata, J.E.; Leake, D.; Godden, E.L.; Albertson, D.G.; Angela Nieto, M.; et al. Rac1b and reactive oxygen species mediate MMP-3-induced EMT and genomic instability. Nature 2005, 436, 123–127. [Google Scholar] [CrossRef] [PubMed]
- Sever, R.; Brugge, J.S. Signal transduction in cancer. Cold Spring Harb Perspect. Med. 2015, 5, a006098. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Layseca, P.; Streuli, C.H. Signalling pathways linking integrins with cell cycle progression. Matrix Biol. 2014, 34, 144–153. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Kang, Y.M.; Kim, J.; Shin, N.; Hanks, S.; Song, W. The Integrin-coupled Signaling Adaptor p130Cas Suppresses Smad3 Function in Transforming Growth Factor-β Signaling. Mol. Biol. Cell 2008, 19, 2135–2146. [Google Scholar] [CrossRef] [PubMed]
- Senthebane, D.A.; Jonker, T.; Rowe, A.; Thomford, N.E.; Munro, D.; Dandara, C.; Wonkam, A.; Govender, D.; Calder, B.; Soares, N.C.; et al. The Role of Tumor Microenvironment in Chemoresistance: 3D Extracellular Matrices as Accomplices. Int. J. Mol. Sci. 2018, 19, 2861. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Ma, M.; Lin, X.H.; Liu, H.H.; Chen, J.; Chen, J.; Gao, D.M.; Cui, J.F.; Ren, Z.G.; Chen, R.X. Extracellular matrix collagen I promotes the tumor progression of residual hepatocellular carcinoma after heat treatment. BMC Cancer 2018, 18, 901. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Chang, T.T.; Huang, Y.; Chen, C.; Chou, C. COL11A1 confers chemoresistance on ovarian cancer cells through the activation of Akt/c/EBPβ pathway and PDK1 stabilization. Oncotarget 2015, 6, 23748–23763. [Google Scholar] [PubMed]
- Xie, J.W.; Haslam, S.Z. Extracellular matrix, Rac1 signaling, and estrogen-induced proliferation in MCF-7 breast cancer cells. Breast Cancer Res. Treat. 2008, 110, 257–268. [Google Scholar] [CrossRef] [PubMed]
- Slack-Davis, J.K.; Eblen, S.T.; Zecevic, M.; Boerner, S.A.; Tarcsafalvi, A.; Diaz, H.B.; Marshall, M.S.; Weber, M.J.; Parsons, J.T.; Catling, A.D. PAK1 phosphorylation of MEK1 regulates fibronectin-stimulated MAPK activation. J. Cell Biol. 2003, 162, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Yousif, N.G. Fibronectin promotes migration and invasion of ovarian cancer cells through up-regulation of FAK-PI3K/Akt pathway. Cell Biol. Int. 2014, 38, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.G.; Gumbiner, B.M. Adhesion to fibronectin regulates Hippo signaling via the FAK-Src-PI3K pathway. J. Cell Biol. 2015, 210, 503–515. [Google Scholar] [CrossRef] [PubMed]
- Weaver, V.M.; Lelievre, S.; Lakins, J.N.; Chrenek, M.; Jones, J.C.R.; Giancotti, F.; Werb, Z.; Bissell, M.J. β4 integrin-dependent formation of polarized three-dimensional architecture confers resistance to apoptosis in normal and malignant mammary epithelium. Cancer Cell 2002, 2, 205–216. [Google Scholar] [CrossRef]
- Gonzales, M.; Haan, K.; Baker, S.; Fitchmun, M.; Todorov, I.; Weitzman, S.; Jones, J.C.R. A Cell Signal Pathway Involving Laminin-5, α3β1 Integrin, and Mitogen-activated Protein Kinase Can Regulate Epithelial Cell Proliferation. Mol. Biol. Cell 1999, 10, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Underwood, T.J.; Hayden, A.L.; Derouet, M.; Garcia, E.; Noble, F.; White, M.J.; Thirdborough, S.; Mead, A.; Clemons, N.; Mellone, M.; et al. Cancer-associated fibroblasts predict poor outcome and promote periostin-dependent invasion in oesophageal adenocarcinoma. J. Pathol. 2015, 235, 466–477. [Google Scholar] [CrossRef] [PubMed]
- Mott, J.D.; Werb, Z. Regulation of matrix biology by matrix metalloproteinases. Curr. Opin. Cell Biol. 2004, 16, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Adamis, A.P. Ten years of anti-vascular endothelial growth factor therapy. Nat. Rev. Drug Discov. 2016, 15, 385–403. [Google Scholar] [CrossRef] [PubMed]
- Belotti, D.; Paganoni, P.; Manenti, L.; Garofalo, A.; Marchini, S.; Taraboletti, G.; Giavazzi, R. Matrix Metalloproteinases (MMP9 and MMP2) Induce the Release of Vascular Endothelial Growth Factor (VEGF) by Ovarian Carcinoma Cells: Implications for Ascites Formation. Cancer Res. 2003, 63, 5224–5229. [Google Scholar] [PubMed]
- Yu, Q.; Stamenovic, D. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000, 14, 163–176. [Google Scholar] [PubMed]
- Dallas, S.L.; Rosser, J.L.; Mundy, G.R.; Bonewald, L.F. Proteolysis of latent transforming growth factor-beta (TGF-beta)-binding protein-1 by osteoclasts. A cellular mechanism for release of TGF-beta from bone matrix. J. Biol. Chem. 2002, 277, 21352–21360. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, V.; Berglund, E.J.; Chen, D.X.; Kidambi, S. Substrate stiffness regulates primary hepatocyte functions. RSC Adv. 2015, 5, 80956–80966. [Google Scholar] [CrossRef]
- Katayama, K.; Noguchi, K.; Sugimoto, Y. Regulations of P-Glycoprotein/ABCB1/MDR1 in Human Cancer Cells. New J. Sci. 2014, 2014, 1–10. [Google Scholar] [CrossRef]
- Watabe, H.; Furuhama, T.; Tani-Ishii, N.; Mikuni-Takagaki, Y. Mechanotransduction activates alpha(5)beta(1) integrin and PI3K/Akt signaling pathways in mandibular osteoblasts. Exp. Cell Res. 2011, 317, 2642–2649. [Google Scholar] [CrossRef] [PubMed]
- Cha, B.; Geng, X.; Mahamud, M.R.; Fu, J.; Mukherjee, A.; Kim, Y.; Jho, E.H.; Kim, T.H.; Kahn, M.L.; Xia, L.; et al. Mechanotransduction activates canonical Wnt/beta-catenin signaling to promote lymphatic vascular patterning and the development of lymphatic and lymphovenous valves. Genes Dev. 2016, 30, 1454–1469. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, H.; Jiang, T.; Jin, K.; Luo, Z.; Shi, W.; Mei, H.; Wang, H.; Hu, Y.; Pang, Z.; et al. Cyclopamine treatment disrupts extracellular matrix and alleviates solid stress to improve nanomedicine delivery for pancreatic cancer. J. Drug Target 2018, 26, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, V.P.; Martin, J.D.; Liu, H.; Lacorre, D.A.; Jain, S.R.; Kozin, S.V.; Stylianopoulos, T.; Mousa, A.S.; Han, X.; Adstamongkonkul, P.; et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat. Commun. 2013, 4, 2516. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.C.; Fattet, L.; Tsai, J.H.; Guo, Y.; Pai, V.H.; Majeski, H.E.; Chen, A.C.; Sah, R.L.; Taylor, S.S.; Engler, A.J.; et al. Matrix stiffness drives epithelial-mesenchymal transition and tumour metastasis through a TWIST1-G3BP2 mechanotransduction pathway. Nat. Cell Biol. 2015, 17, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Chen, Q.; Lui, C.; Cichon, M.; Radisky, D.C.; Nelson, C.M. Matrix compliance regulates Rac1b localization, NADPH oxidase assembly, an epithelial-mesenchymal transition. Mol. Biol. Cell 2012, 23, 4097–4108. [Google Scholar] [CrossRef] [PubMed]
- Rice, A.J.; Cortes, E.; Lachowski, D.; Cheung, B.C.H.; Karim, S.A.; Morton, J.P.; Del Rio Hernandez, A. Matrix stiffness induces epithelial-mesenchymal transition and promotes chemoresistance in pancreatic cancer cells. Oncogenesis 2017, 6, e352. [Google Scholar] [CrossRef] [PubMed]
- Samuel, M.S.; Lopez, J.I.; McGhee, E.J.; Croft, D.R.; Strachan, D.; Timpson, P.; Munro, J.; Schroder, E.; Zhou, J.; Brunton, V.G.; et al. Actomyosin-mediated cellular tension drives increased tissue stiffness and beta-catenin activation to induce epidermal hyperplasia and tumor growth. Cancer Cell 2011, 19, 776–791. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Li, D.; Li, H.; Wang, L.; Tian, G.; Dong, Y. YAP overexpression promotes the epithelial-mesenchymal transition and chemoresistance in pancreatic cancer cells. Mol. Med. Rep. 2016, 13, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; He, K.; Hu, Y.; Guo, X.; Wang, D.; Shi, W.; Li, J.; Song, J. YAP modulates TGF-beta1-induced simultaneous apoptosis and EMT through upregulation of the EGF receptor. Sci. Rep. 2017, 7, 45523. [Google Scholar] [CrossRef] [PubMed]
- Mouw, J.K.; Yui, Y.; Damiano, L.; Bainer, R.O.; Lakins, J.N.; Acerbi, I.; Ou, G.; Wijekoon, A.C.; Levental, K.R.; Gilbert, P.M.; et al. Tissue mechanics modulate microRNA-dependent PTEN expression to regulate malignant progression. Nat. Med. 2014, 20, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Guan, L.; Li, S.; Jiang, Y.; Xiong, N.; Li, L.; Wu, C.; Zeng, H.; Liu, Y. Mechanosensitive caveolin-1 activation-induced PI3K/Akt/mTOR signaling pathway promotes breast cancer motility, invadopodia formation and metastasis in vivo. Oncotarget 2015, 7, 16227–16247. [Google Scholar]
- Provenzano, P.; Keely, P.J. Mechanical signaling through the cytoskeleton regulates cell proliferation by coordinated focal adhesion and Rho GTPase signaling. J. Cell Sci. 2011, 124, 1195–1205. [Google Scholar] [CrossRef] [PubMed]
- Paszek, M.J.; Zahir, N.; Johnson, K.R.; Lakins, J.N.; Rozenberg, G.I.; Gefen, A.; Reinhart-King, C.A.; Margulies, S.S.; Dembo, M.; Boettiger, D.; et al. Tensional homeostasis and the malignant phenotype. Cancer Cell 2005, 8, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Street, C.A.; Bryan, B.A. Rho Kinase Proteins—Pleiotropic Modulators of Cell Survival and Apoptosis. Anticancer Res. 2011, 31, 3645–3658. [Google Scholar] [PubMed]
- Ching, K.; Tenney, K.; Chen, J.; Morgan, E. Suppression of constitutive cytochrome P450 gene expressin by epidermal growth factor receptor ligands in cultured rat hepatocytes. Drug Metab. Dispos. 1996, 24, 542–546. [Google Scholar] [PubMed]
- Iber, H.; Morgan, E. Regulation of Hepatic Cytochrome P450 2C11 by Transforming Growth Factor-β, Hepatocyte Growth Factor, and Interleukin-11. Drug Metab. Dispos. 1998, 26, 1042–1044. [Google Scholar] [PubMed]
- Sukhai, M.; Piquette-Miller, M. Regulation of the Multidrug Resistance Genes by Stress Signals. J. Pharm. Pharmaceut. Sci. 2000, 3, 268–280. [Google Scholar]
- Chen, K.G.; Sikic, B.I. Molecular pathways: Regulation and therapeutic implications of multidrug resistance. Clin. Cancer Res. 2012, 18, 1863–1869. [Google Scholar] [CrossRef] [PubMed]
- Hour, T.C.; Chung, S.D.; Kang, W.Y.; Lin, Y.C.; Chuang, S.J.; Huang, A.M.; Wu, W.J.; Huang, S.P.; Huang, C.Y.; Pu, Y.S. EGFR mediates docetaxel resistance in human castration-resistant prostate cancer through the Akt-dependent expression of ABCB1 (MDR1). Arch. Toxicol. 2015, 89, 591–605. [Google Scholar] [CrossRef] [PubMed]
- Colak, S.; Ten Dijke, P. Targeting TGF-beta Signaling in Cancer. Trends. Cancer 2017, 3, 56–71. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Billottet, C.; Tuefferd, M.; Gentien, D.; Rapinat, A.; Thiery, J.P.; Broet, P.; Jouanneau, J. Modulation of several waves of gene expression during FGF-1 induced epithelial-mesenchymal transition of carcinoma cells. J. Cell Biochem. 2008, 104, 826–839. [Google Scholar] [CrossRef] [PubMed]
- Lo, H.W.; Hsu, S.C.; Xia, W.; Cao, X.; Shih, J.Y.; Wei, Y.; Abbruzzese, J.L.; Hortobagyi, G.N.; Hung, M.C. Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelial-mesenchymal transition in cancer cells via up-regulation of TWIST gene expression. Cancer Res. 2007, 67, 9066–9076. [Google Scholar] [CrossRef] [PubMed]
- Grotegut, S.; von Schweinitz, D.; Christofori, G.; Lehembre, F. Hepatocyte growth factor induces cell scattering through MAPK/Egr-1-mediated upregulation of Snail. EMBO J. 2006, 25, 3534–3545. [Google Scholar] [CrossRef] [PubMed]
- Graham, T.R.; Zhau, H.E.; Odero-Marah, V.A.; Osunkoya, A.O.; Kimbro, K.S.; Tighiouart, M.; Liu, T.; Simons, J.W.; O’Regan, R.M. Insulin-like growth factor-I-dependent up-regulation of ZEB1 drives epithelial-to-mesenchymal transition in human prostate cancer cells. Cancer Res. 2008, 68, 2479–2488. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Litzenburger, B.C.; Cui, X.; Delgado, D.A.; Grabiner, B.C.; Lin, X.; Lewis, M.T.; Gottardis, M.M.; Wong, T.W.; Attar, R.M.; et al. Constitutively active type I insulin-like growth factor receptor causes transformation and xenograft growth of immortalized mammary epithelial cells and is accompanied by an epithelial-to-mesenchymal transition mediated by NF-kappaB and snail. Mol. Cell Biol. 2007, 27, 3165–3175. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Lin, C.; Liu, Z.R. P68 RNA helicase mediates PDGF-induced epithelial mesenchymal transition by displacing Axin from beta-catenin. Cell 2006, 127, 139–155. [Google Scholar] [CrossRef] [PubMed]
- Fisher, D.T.; Appenheimer, M.M.; Evans, S.S. The two faces of IL-6 in the tumor microenvironment. Semin. Immunol. 2014, 26, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, N.J.; Sasser, A.K.; Axel, A.E.; Vesuna, F.; Raman, V.; Ramirez, N.; Oberyszyn, T.M.; Hall, B.M. Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene 2009, 28, 2940–2947. [Google Scholar] [CrossRef] [PubMed]
- Fernando, R.I.; Castillo, M.D.; Litzinger, M.; Hamilton, D.H.; Palena, C. IL-8 signaling plays a critical role in the epithelial-mesenchymal transition of human carcinoma cells. Cancer Res. 2011, 71, 5296–5306. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; Zhang, C.; Li, A.; Wang, D.; Luo, Z.; Ping, Y.; Zhou, B.; Liu, S.; Li, H.; Yue, D.; et al. IL6 derived from cancer-associated fibroblasts promotes chemoresistance via CXCR7 in esophageal squamous cell carcinoma. Oncogene 2018, 37, 873–883. [Google Scholar] [CrossRef] [PubMed]
- Studebaker, A.W.; Storci, G.; Werbeck, J.L.; Sansone, P.; Sasser, A.K.; Tavolari, S.; Huang, T.; Chan, M.W.; Marini, F.C.; Rosol, T.J.; et al. Fibroblasts isolated from common sites of breast cancer metastasis enhance cancer cell growth rates and invasiveness in an interleukin-6-dependent manner. Cancer Res. 2008, 68, 9087–9095. [Google Scholar] [CrossRef] [PubMed]
- Sriuranpong, V.; Park, J.; Amornphimoltham, P.; Patel, V.; Nelkin, B.; Gutkind, J. Epidermal Growth Factor Receptor-independent Constitutive Activation of STAT3 in Head and Neck Squamous Cell Carcinoma Is Mediated by the Autocrine/Paracrine Stimulation of the Interleukin 6/gp130 Cytokine System. Cancer Res. 2003, 63, 2948–2956. [Google Scholar] [PubMed]
- Decker, T.; Kovarik, P. Serine phosphorylation of STATs. Oncogene 2000, 19, 2628–2637. [Google Scholar] [CrossRef] [PubMed]
- Karajannis, M.A.; Vincent, L.; Direnzo, R.; Shmelkov, S.V.; Zhang, F.; Feldman, E.J.; Bohlen, P.; Zhu, Z.; Sun, H.; Kussie, P.; et al. Activation of FGFR1beta signaling pathway promotes survival, migration and resistance to chemotherapy in acute myeloid leukemia cells. Leukemia 2006, 20, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Deying, W.; Feng, G.; Shumei, L.; Hui, Z.; Ming, L.; Hongqing, W. CAF-derived HGF promotes cell proliferation and drug resistance by up-regulating the c-Met/PI3K/Akt and GRP78 signalling in ovarian cancer cells. Biosci. Rep. 2017, 37, BSR20160470. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhou, B.P. TNF-alpha/NF-kappaB/Snail pathway in cancer cell migration and invasion. Br. J. Cancer 2010, 102, 639–644. [Google Scholar] [CrossRef] [PubMed]
- Lewis, A.M.; Varghese, S.; Xu, H.; Alexander, H.R. Interleukin-1 and cancer progression: The emerging role of interleukin-1 receptor antagonist as a novel therapeutic agent in cancer treatment. J. Transl. Med. 2006, 4, 48. [Google Scholar] [CrossRef] [PubMed]
- Hoesel, B.; Schmid, J. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12. [Google Scholar] [CrossRef] [PubMed]
- Hengeveld, P.J.; Kersten, M.J. B-cell activating factor in the pathophysiology of multiple myeloma: A target for therapy? Blood Cancer J. 2015, 5, e282. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Y.; Geng, L.; Yi, H.; Huo, W.; Talmon, G.; Kim, Y.C.; Wang, S.M.; Wang, J. Transforming Growth Factor beta Mediates Drug Resistance by Regulating the Expression of Pyruvate Dehydrogenase Kinase 4 in Colorectal Cancer. J. Biol. Chem. 2016, 291, 17405–17416. [Google Scholar] [CrossRef] [PubMed]
- Drubay, V.; Skrypek, N.; Cordiez, L.; Vasseur, R.; Schulz, C.; Boukrout, N.; Duchene, B.; Coppin, L.; Van Seuningen, I.; Jonckheere, N. TGF-βRII knock-down promotes tumor growth and chemoresistance to gemcitabine of pancreatic cancer cells via phosphorylation of STAT3. Cancers 2018, 10, 254. [Google Scholar] [CrossRef] [PubMed]
- Stylianopoulos, T.; Munn, L.L.; Jain, R.K. Reengineering the Physical Microenvironment of Tumors to Improve Drug Delivery and Efficacy: From Mathematical Modeling to Bench to Bedside. Trends. Cancer 2018, 4, 292–319. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Rubin, K.; Pietras, K.; Ostman, A. High interstitial fluid pressure—An obstacle in cancer therapy. Nat. Rev. Cancer 2004, 4, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef] [PubMed]
- Stéphanou, A.; McDougall, S.R.; Anderson, A.R.A.; Chaplain, M.A.J. Mathematical modelling of flow in 2D and 3D vascular networks: Applications to anti-angiogenic and chemotherapeutic drug strategies. Math. Comput. Model. 2005, 41, 1137–1156. [Google Scholar] [CrossRef]
- Primeau, A.J.; Rendon, A.; Hedley, D.; Lilge, L.; Tannock, I.F. The distribution of the anticancer drug Doxorubicin in relation to blood vessels in solid tumors. Clin. Cancer Res. 2005, 11, 8782–8788. [Google Scholar] [CrossRef] [PubMed]
- Piotrowski-Daspit, A.S.; Tien, J.; Nelson, C.M. Interstitial fluid pressure regulates collective invasion in engineered human breast tumors via Snail, vimentin, and E-cadherin. Integr. Biol. 2016, 8, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Tchafa, A.M.; Ta, M.; Reginato, M.J.; Shieh, A.C. EMT Transition Alters Interstitial Fluid Flow-Induced Signaling in ERBB2-Positive Breast Cancer Cells. Mol. Cancer Res. 2015, 13, 755–764. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhou, F.Y.; Shen, Y.; Zhang, Y.; Yin, H.; Zeng, Y.; Liu, J.; Yan, Z.; Liu, X. Fluid shear stress induced epithelial-mesenchymal transition (EMT) in Hep-2 cells. Oncotarget 2016, 7, 32876–32892. [Google Scholar] [PubMed]
- Risvi, I.; Gurkan, U.; Tasoglu, S.; Alagic, N.; Celli, J.; Mensah, L.; Mai, Z.; Demirci, U.; Hasan, T. Flow induces epithelial-mesenchymal transition, cellular heterogeneity and biomarker modulation in 3D ovarian cancer nodules. Proc. Natl. Acad. Sci. USA 2013, 110, E1974–E1983. [Google Scholar] [CrossRef] [PubMed]
- Triantafillu, U.; Park, S.; Klaassen, N.; Raddatz, A.; Kim, Y. Fluid shear stress induces cancer stem cell-like phenotype in MCF7 breast cancer cell line without inducing epithelial to mesenchymal transition. Int. J. Oncol. 2017, 50, 993–1001. [Google Scholar] [CrossRef] [PubMed]
- Shields, J.D.; Fleury, M.E.; Yong, C.; Tomei, A.A.; Randolph, G.J.; Swartz, M.A. Autologous chemotaxis as a mechanism of tumor cell homing to lymphatics via interstitial flow and autocrine CCR7 signaling. Cancer Cell 2007, 11, 526–538. [Google Scholar] [CrossRef] [PubMed]
- Riol-Blanco, L.; Sanchez-Sanchez, N.; Torres, A.; Tejedor, A.; Narumiya, S.; Corbi, A.L.; Sanchez-Mateos, P.; Rodriguez-Fernandez, J.L. The Chemokine Receptor CCR7 Activates in Dendritic Cells Two Signaling Modules That Independently Regulate Chemotaxis and Migratory Speed. J. Immunol. 2005, 174, 4070–4080. [Google Scholar] [CrossRef] [PubMed]
- Munson, J.M.; Bellamkonda, R.V.; Swartz, M.A. Interstitial flow in a 3D microenvironment increases glioma invasion by a CXCR4-dependent mechanism. Cancer Res. 2013, 73, 1536–1546. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.D.; Bouchard, M.J.; Shieh, A.C. Interstitial Fluid Flow Increases Hepatocellular Carcinoma Cell Invasion through CXCR4/CXCL12 and MEK/ERK Signaling. PLoS ONE 2015, 10, e0142337. [Google Scholar] [CrossRef] [PubMed]
- Shieh, A.C.; Rozansky, H.A.; Hinz, B.; Swartz, M.A. Tumor cell invasion is promoted by interstitial flow-induced matrix priming by stromal fibroblasts. Cancer Res. 2011, 71, 790–800. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, M.; Guschel, M.; Bernd, A.; Bereiter-Hahn, J.; Kaufmann, R.; Tandi, C.; Wiig, H.; Kippenberger, S. Lowering of tumor interstitial fluid pressure reduces tumor cell proliferation in a xenograft tumor model. Neoplasia 2006, 8, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Polacheck, W.J.; German, A.E.; Mammoto, A.; Ingber, D.E.; Kamm, R.D. Mechanotransduction of fluid stresses governs 3D cell migration. Proc. Natl. Acad. Sci. USA 2014, 111, 2447–2452. [Google Scholar] [CrossRef] [PubMed]
- Santi, A.; Kugeratski, F.G.; Zanivan, S. Cancer Associated Fibroblasts: The Architects of Stroma Remodeling. Proteomics 2018, 18, e1700167. [Google Scholar] [CrossRef] [PubMed]
- Dzobo, K.; Senthebane, D.A.; Thomford, N.E.; Rowe, A.; Dandara, C.; Parker, M.I. Not Everyone Fits the Mold: Intratumor and Intertumor Heterogeneity and Innovative Cancer Drug Design and Development. OMICS 2018, 22, 17–34. [Google Scholar] [CrossRef] [PubMed]
- Gascard, P.; Tlsty, T.D. Carcinoma-associated fibroblasts: Orchestrating the composition of malignancy. Genes Dev. 2016, 30, 1002–1019. [Google Scholar] [CrossRef] [PubMed]
- Komohara, Y.; Jinushi, M.; Takeya, M. Clinical significance of macrophage heterogeneity in human malignant tumors. Cancer Sci. 2014, 105, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Gilkes, D.M.; Bajpai, S.; Chaturvedi, P.; Wirtz, D.; Semenza, G.L. Hypoxia-inducible factor 1 (HIF-1) promotes extracellular matrix remodeling under hypoxic conditions by inducing P4HA1, P4HA2, and PLOD2 expression in fibroblasts. J. Biol. Chem. 2013, 288, 10819–10829. [Google Scholar] [CrossRef] [PubMed]
- Sada, M.; Ohuchida, K.; Horioka, K.; Okumura, T.; Moriyama, T.; Miyasaka, Y.; Ohtsuka, T.; Mizumoto, K.; Oda, Y.; Nakamura, M. Hypoxic stellate cells of pancreatic cancer stroma regulate extracellular matrix fiber organization and cancer cell motility. Cancer Lett. 2016, 372, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.K.; Zhou, Y.; Cheng, C.; Zhong, S.; Wu, H.Q.; Wang, B.; Fan, P.; Xiong, J.X.; Yang, H.J.; Wu, H.S. Overexpression of membrane-type 2 matrix metalloproteinase induced by hypoxia-inducible factor-1alpha in pancreatic cancer: Implications for tumor progression and prognosis. Mol. Clin. Oncol. 2014, 2, 973–981. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.Y.; Jang, Y.S.; Min, S.Y.; Song, J.Y. Overexpression of MMP-9 and HIF-1alpha in Breast Cancer Cells under Hypoxic Conditions. J. Breast Cancer 2011, 14, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Kai, A.K.; Chan, L.K.; Lo, R.C.; Lee, J.M.; Wong, C.C.; Wong, J.C.; Ng, I.O. Down-regulation of TIMP2 by HIF-1alpha/miR-210/HIF-3alpha regulatory feedback circuit enhances cancer metastasis in hepatocellular carcinoma. Hepatology 2016, 64, 473–487. [Google Scholar] [CrossRef] [PubMed]
- Azimi, I.; Petersen, R.M.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. Hypoxia-induced reactive oxygen species mediate N-cadherin and SERPINE1 expression, EGFR signalling and motility in MDA-MB-468 breast cancer cells. Sci. Rep. 2017, 7, 15140. [Google Scholar] [CrossRef] [PubMed]
- Topalovski, M.; Hagopian, M.; Wang, M.; Brekken, R.A. Hypoxia and Transforming Growth Factor beta Cooperate to Induce Fibulin-5 Expression in Pancreatic Cancer. J. Biol. Chem. 2016, 291, 22244–22252. [Google Scholar] [CrossRef] [PubMed]
- Ju, J.A.; Godet, I.; Ye, I.C.; Byun, J.; Jayatilaka, H.; Lee, S.J.; Xiang, L.; Samanta, D.; Lee, M.H.; Wu, P.H.; et al. Hypoxia Selectively Enhances Integrin alpha5beta1 Receptor Expression in Breast Cancer to Promote Metastasis. Mol. Cancer Res. 2017, 15, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Koike, T.; Kimura, N.; Miyazaki, K.; Yabuta, T.; Kumamoto, K.; Takenoshita, S.; Chen, J.; Kobayashi, M.; Hosokawa, M.; Taniguchi, A.; et al. Hypoxia induces adhesion molecules on cancer cells: A missing link between Warburg effect and induction of selectin-ligand carbohydrates. Proc. Natl. Acad. Sci. USA 2004, 101, 8132–8137. [Google Scholar] [CrossRef] [PubMed]
- Taddei, M.L.; Cavallini, L.; Comito, G.; Giannoni, E.; Folini, M.; Marini, A.; Gandellini, P.; Morandi, A.; Pintus, G.; Raspollini, M.R.; et al. Senescent stroma promotes prostate cancer progression: The role of miR-210. Mol. Oncol. 2014, 8, 1729–1746. [Google Scholar] [CrossRef] [PubMed]
- Curran, C.S.; Keely, P.J. Breast tumor and stromal cell responses to TGF-beta and hypoxia in matrix deposition. Matrix Biol. 2013, 32, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Wang, L.F.; Wang, R.F. Role of cancer-associated fibroblasts in invasion and metastasis of gastric cancer. World J. Gastroenterol. 2015, 21, 9717–9726. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Guo, H.; Wu, H.; Tian, T.; Dong, D.; Zhang, Y.; Sui, Y.; Zhang, Y.; Zhao, D.; Wang, S.; et al. GPER in CAFs regulates hypoxia-driven breast cancer invasion in a CTGF-dependent manner. Oncol. Rep. 2015, 33, 1929–1937. [Google Scholar] [CrossRef] [PubMed]
- Noman, M.Z.; Desantis, G.; Janji, B.; Hasmim, M.; Karray, S.; Dessen, P.; Bronte, V.; Chouaib, S. PD-L1 is a novel direct target of HIF-1alpha, and its blockade under hypoxia enhanced MDSC-mediated T cell activation. J. Exp. Med. 2014, 211, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Raub, C.B.; Putnam, A.J.; Tromberg, B.J.; George, S.C. Predicting bulk mechanical properties of cellularized collagen gels using multiphoton microscopy. Acta Biomater. 2010, 6, 4657–4665. [Google Scholar] [CrossRef] [PubMed]
- Ungefroren, H.; Sebens, S.; Seidl, D.; Lehnert, H.; Hass, R. Interaction of tumor cells with the microenvironment. Cell Commun. Signal. 2011, 9, 18. [Google Scholar] [CrossRef] [PubMed]
- Januchowski, R.; Swierczewska, M.; Sterzynska, K.; Wojtowicz, K.; Nowicki, M.; Zabel, M. Increased Expression of Several Collagen Genes is Associated with Drug Resistance in Ovarian Cancer Cell Lines. J. Cancer 2016, 7, 1295–1310. [Google Scholar] [CrossRef] [PubMed]
- Calvo, F.; Ege, N.; Grande-Garcia, A.; Hooper, S.; Jenkins, R.P.; Chaudhry, S.I.; Harrington, K.; Williamson, P.; Moeendarbary, E.; Charras, G.; et al. Mechanotransduction and YAP-dependent matrix remodelling is required for the generation and maintenance of cancer-associated fibroblasts. Nat. Cell Biol. 2013, 15, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Ao, M.; Brewer, B.M.; Yang, L.; Franco Coronel, O.E.; Hayward, S.W.; Webb, D.J.; Li, D. Stretching fibroblasts remodels fibronectin and alters cancer cell migration. Sci. Rep. 2015, 5, 8334. [Google Scholar] [CrossRef] [PubMed]
- Lachowski, D.; Cortes, E.; Pink, D.; Chronopoulos, A.; Karim, S.A.; J, P.M.; Del Rio Hernandez, A.E. Substrate Rigidity Controls Activation and Durotaxis in Pancreatic Stellate Cells. Sci. Rep. 2017, 7, 2506. [Google Scholar] [CrossRef] [PubMed]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix metalloproteinases: Regulators of the tumor microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [PubMed]
- Trastour, C.; Benizri, E.; Ettore, F.; Ramaioli, A.; Chamorey, E.; Pouyssegur, J.; Berra, E. HIF-1alpha and CA IX staining in invasive breast carcinomas: Prognosis and treatment outcome. Int. J. Cancer 2007, 120, 1451–1458. [Google Scholar] [CrossRef] [PubMed]
- Colom, A.; Galgoczy, R.; Almendros, I.; Xaubet, A.; Farre, R.; Alcaraz, J. Oxygen diffusion and consumption in extracellular matrix gels: Implications for designing three-dimensional cultures. J. Biomed. Mater. Res. A 2014, 102, 2776–2784. [Google Scholar] [CrossRef] [PubMed]
- Surazynski, A.; Donald, S.P.; Cooper, S.K.; Whiteside, M.A.; Salnikow, K.; Liu, Y.; Phang, J.M. Extracellular matrix and HIF-1 signaling: The role of prolidase. Int. J. Cancer 2008, 122, 1435–1440. [Google Scholar] [CrossRef] [PubMed]
- Mongiat, M.; Andreuzzi, E.; Tarticchio, G.; Paulitti, A. Extracellular Matrix, a Hard Player in Angiogenesis. Int. J. Mol. Sci. 2016, 17, 1822. [Google Scholar] [CrossRef] [PubMed]
- Senger, D.R.; Davis, G.E. Angiogenesis. Cold Spring Harb Perspect. Biol. 2011, 3, a005090. [Google Scholar] [CrossRef] [PubMed]
- Bergers, G.; Brekken, R.; McMahon, G.; Vu, T.H.; Itoh, T.; Tamaki, K.; Tanzawa, K.; Thorpe, P.; Itohara, S.; Werb, Z.; et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat. Cell Biol. 2000, 2, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Voutouri, C.; Polydorou, C.; Papageorgis, P.; Gkretsi, V.; Stylianopoulos, T. Hyaluronan-Derived Swelling of Solid Tumors, the Contribution of Collagen and Cancer Cells, and Implications for Cancer Therapy. Neoplasia 2016, 18, 732–741. [Google Scholar] [CrossRef] [PubMed]
- De Veirman, K.; Rao, L.; De Bruyne, E.; Menu, E.; Van Valckenborgh, E.; Van Riet, I.; Frassanito, M.A.; Di Marzo, L.; Vacca, A.; Vanderkerken, K. Cancer associated fibroblasts and tumor growth: Focus on multiple myeloma. Cancers 2014, 6, 1363–1381. [Google Scholar] [CrossRef] [PubMed]
- Goumans, M.J.; Valdimarsdottir, G.; Itoh, S.; Lebrin, F.; Larsson, J.; Mummery, C.; Karlsson, S.; Ten Dijke, P. Activin receptor-like kinase (ALK)1 is an antagonistic mediator of lateral TGFbeta/ALK5 signaling. Mol. Cell 2003, 12, 817–828. [Google Scholar] [CrossRef]
- Hillen, F.; Griffioen, A.W. Tumour vascularization: Sprouting angiogenesis and beyond. Cancer Metastasis Rev. 2007, 26, 489–502. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N. VEGF and the quest for tumour angiogenesis factors. Nat. Rev. Cancer 2002, 2, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N. Pathways mediating VEGF-independent tumor angiogenesis. Cytokine Growth Factor Rev. 2010, 21, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Broders-Bondon, F.; Nguyen Ho-Bouldoires, T.H.; Fernandez-Sanchez, M.E.; Farge, E. Mechanotransduction in tumor progression: The dark side of the force. J. Cell Biol. 2018, 217, 1571–1587. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.P.; Hinz, B.; Swartz, M. Interstitial fluid flow induces myofibroblast differentiation and collagen alignment in vitro. J. Cell Sci. 2005, 118, 4731–4739. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Xie, X.; Wang, Z.; Hu, C.; Zheng, Q.; Wang, Y.; Chen, R.; Xue, T.; Chen, J.; Gao, D.; et al. Increasing matrix stiffness upregulates vascular endothelial growth factor expression in hepatocellular carcinoma cells mediated by integrin beta1. Biochem. Biophys. Res. Commun. 2014, 444, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Fleury, M.E.; Boardman, K.C.; Swartz, M.A. Autologous morphogen gradients by subtle interstitial flow and matrix interactions. Biophys. J. 2006, 91, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [PubMed]
- Mistry, I.N.; Thomas, M.; Calder, E.D.D.; Conway, S.J.; Hammond, E.M. Clinical Advances of Hypoxia-Activated Prodrugs in Combination With Radiation Therapy. Int. J. Radiat. Oncol. Biol. Phys 2017, 98, 1183–1196. [Google Scholar] [CrossRef] [PubMed]
- Koren, E.; Apte, A.; Jani, A.; Torchilin, V.P. Multifunctional PEGylated 2C5-immunoliposomes containing pH-sensitive bonds and TAT peptide for enhanced tumor cell internalization and cytotoxicity. J. Control. Release 2012, 160, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Chronopoulos, A.; Robinson, B.; Sarper, M.; Cortes, E.; Auernheimer, V.; Lachowski, D.; Attwood, S.; Garcia, R.; Ghassemi, S.; Fabry, B.; et al. ATRA mechanically reprograms pancreatic stellate cells to suppress matrix remodelling and inhibit cancer cell invasion. Nat. Commun. 2016, 7, 12630. [Google Scholar] [CrossRef] [PubMed]
- Cortes, E.; Lachowski, D.; Rice, A.; Chronopoulos, A.; Robinson, B.; Thorpe, S.; Lee, D.A.; Possamai, L.A.; Wang, H.; Pinato, D.J.; et al. RAR-beta is downregulated in HCC & cirrhosis and its expression inhibits myosin-driven activation and durotaxis in hepatic stellate cells. Hepatology 2018. [Google Scholar] [CrossRef]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yeldag, G.; Rice, A.; Del Río Hernández, A. Chemoresistance and the Self-Maintaining Tumor Microenvironment. Cancers 2018, 10, 471. https://doi.org/10.3390/cancers10120471
Yeldag G, Rice A, Del Río Hernández A. Chemoresistance and the Self-Maintaining Tumor Microenvironment. Cancers. 2018; 10(12):471. https://doi.org/10.3390/cancers10120471
Chicago/Turabian StyleYeldag, Gulcen, Alistair Rice, and Armando Del Río Hernández. 2018. "Chemoresistance and the Self-Maintaining Tumor Microenvironment" Cancers 10, no. 12: 471. https://doi.org/10.3390/cancers10120471
APA StyleYeldag, G., Rice, A., & Del Río Hernández, A. (2018). Chemoresistance and the Self-Maintaining Tumor Microenvironment. Cancers, 10(12), 471. https://doi.org/10.3390/cancers10120471