Mutational Evolution in Relapsed Diffuse Large B-Cell Lymphoma

 ,
,  , ,
, ,  ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Selection
2.2. Pathology Review
2.3. Fluorescence in Situ Hybridization
2.4. DNA Isolation
2.5. Whole Exome Sequencing
2.6. Bioinformatics Approach
2.7. Genes Possibly Related to Therapy Resistance
3. Results
3.1. Patient Characteristics
3.2. Quality Control
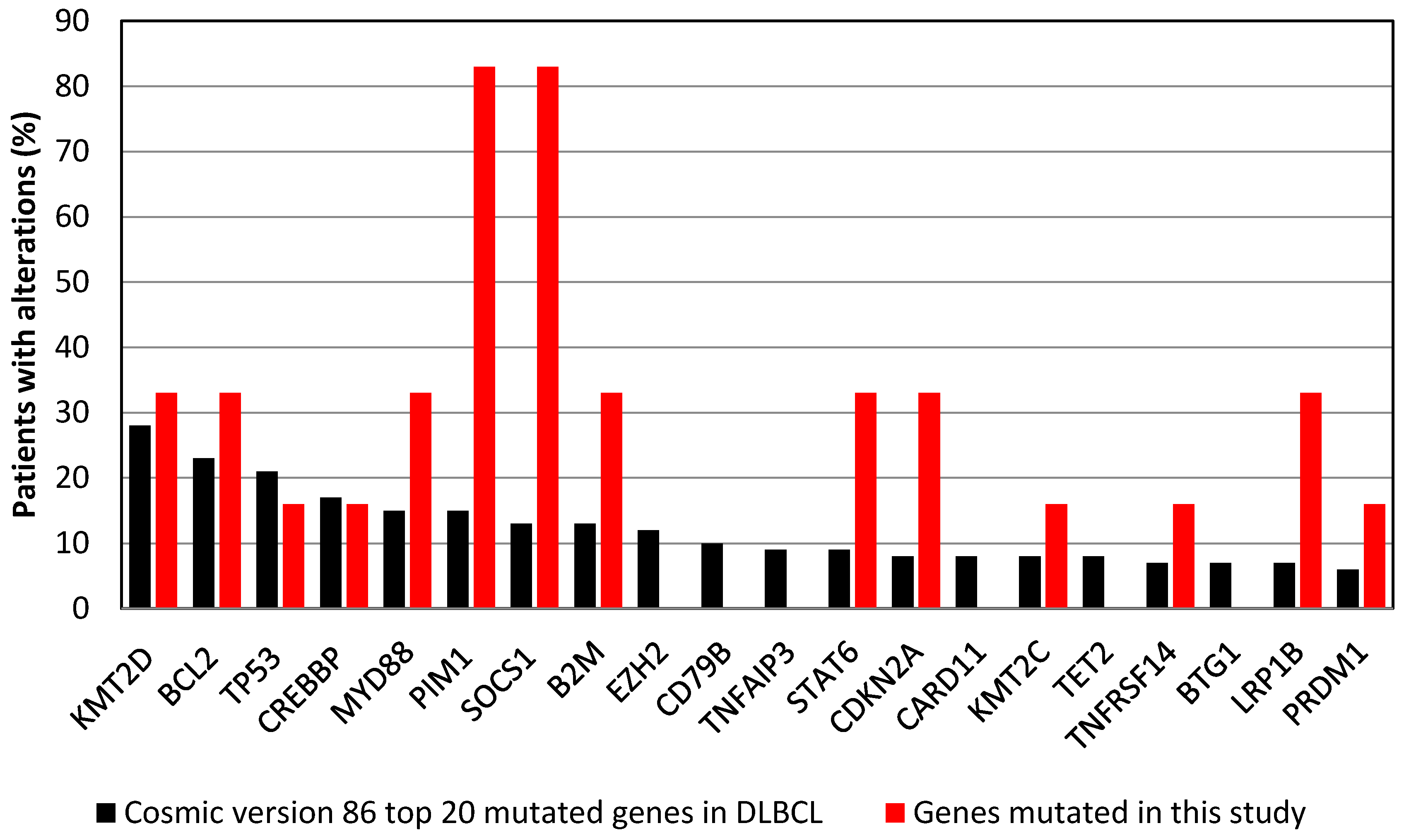
3.3. Commonly Mutated Genes
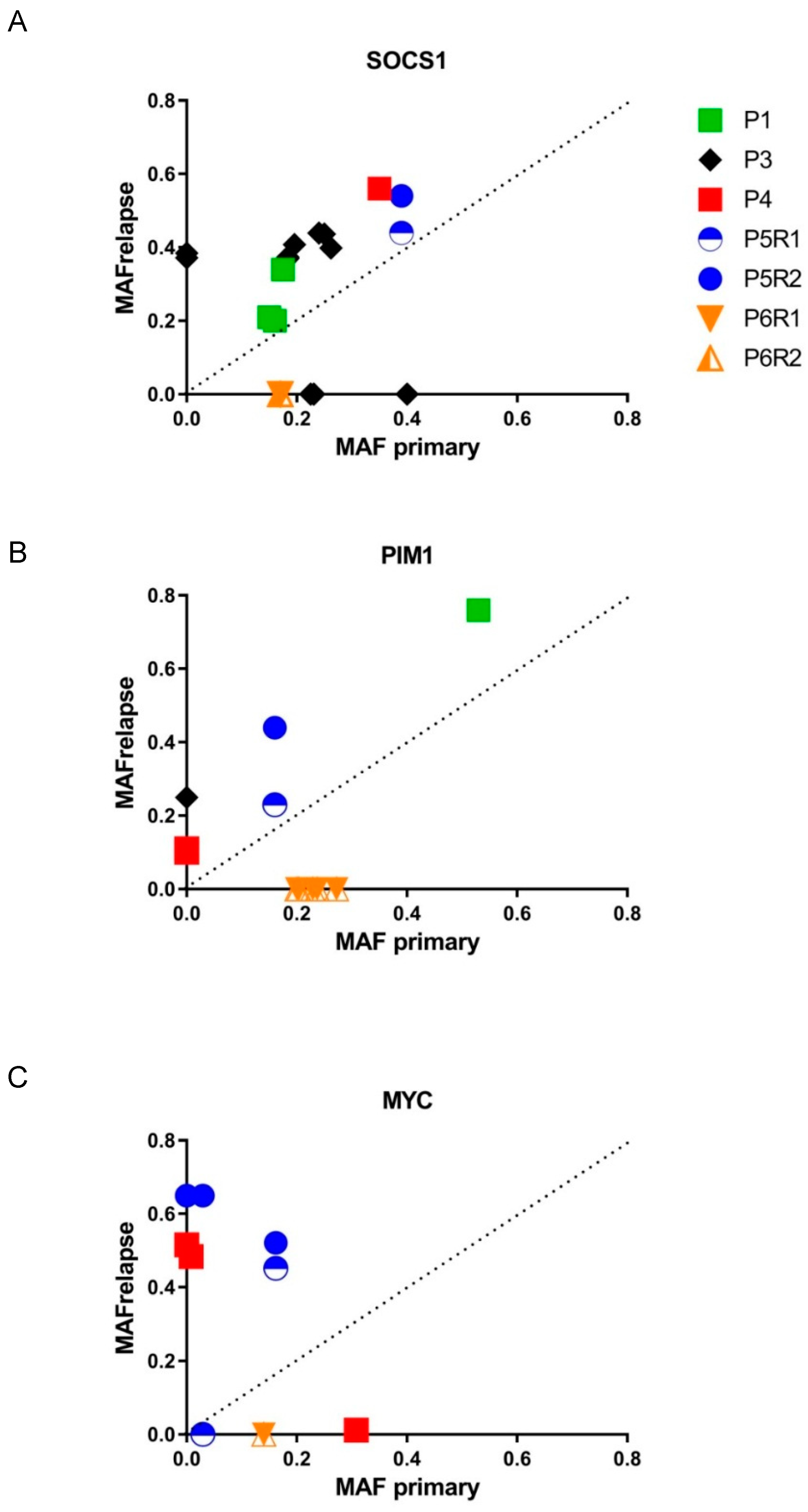
3.4. Mutational Evolution
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gascoyne, R.D.; Campo, E.; Jaffe, E.S. Diffuse large B-cell lymphoma, NOS. In WHO Classification of Tumours of Haematopoeitic and Lymphoid Tissues, rev. 4th ed.; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Eds.; International Agency for Research on Cancer: Lyon, France, 2017; pp. 291–297. [Google Scholar]
- Coiffier, B.; Lepage, E.; Briere, J.; Herbrecht, R.; Tilly, H.; Bouabdallah, R.; Morel, P.; Van Den Neste, E.; Salles, G.; Gaulard, P.; et al. CHOP Chemotherapy Plus Rituximab Compared with CHOP Alone in Elderly Patients with Diffuse Large-B-Cell Lymphoma. N. Engl. J. Med. 2002, 346, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Ziepert, M.; Hasenclever, D.; Kuhnt, E.; Glass, B.; Schmitz, N.; Pfreundschuh, M.; Loeffler, M. Standard International Prognostic Index Remains a Valid Predictor of Outcome for Patients with Aggressive CD20+ B-Cell Lymphoma in the Rituximab Era. J. Clin. Oncol. 2010, 28, 2373–2380. [Google Scholar] [CrossRef] [PubMed]
- Gisselbrecht, C.; Glass, B.; Mounier, N.; Singh Gill, D.; Linch, D.C.; Trneny, M.; Bosly, A.; Ketterer, N.; Shpilberg, O.; Hagberg, H.; et al. Salvage Regimens with Autologous Transplantation for Relapsed Large B-Cell Lymphoma in the Rituximab Era. J. Clin. Oncol. 2010, 28, 4184–4190. [Google Scholar] [CrossRef] [PubMed]
- Van Imhoff, G.W.; McMillan, A.; Matasar, M.J.; Radford, J.; Ardeshna, K.M.; Kuliczkowski, K.; Kim, W.; Hong, X.; Goerloev, J.S.; Davies, A.; et al. Ofatumumab Versus Rituximab Salvage Chemoimmunotherapy in Relapsed Or Refractory Diffuse Large B-Cell Lymphoma: The ORCHARRD Study. J. Clin. Oncol. 2017, 35, 544–551. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, A.A.; Eisen, M.B.; Davis, R.E.; Ma, C.; Lossos, I.S.; Rosenwald, A.; Boldrick, J.C.; Sabet, H.; Tran, T.; Yu, X.; et al. Distinct Types of Diffuse Large B-Cell Lymphoma Identified by Gene Expression Profiling. Nature 2000, 403, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.W.; Wright, G.W.; Williams, P.M.; Lih, C.J.; Walsh, W.; Jaffe, E.S.; Rosenwald, A.; Campo, E.; Chan, W.C.; Connors, J.M.; et al. Determining Cell-of-Origin Subtypes of Diffuse Large B-Cell Lymphoma using Gene Expression in Formalin-Fixed Paraffin-Embedded Tissue. Blood 2014, 123, 1214–1217. [Google Scholar] [CrossRef] [PubMed]
- Offner, F.; Samoilova, O.; Osmanov, E.; Eom, H.S.; Topp, M.S.; Raposo, J.; Pavlov, V.; Ricci, D.; Chaturvedi, S.; Zhu, E.; et al. Frontline Rituximab, Cyclophosphamide, Doxorubicin, and Prednisone with Bortezomib (VR-CAP) Or Vincristine (R-CHOP) for Non-GCB DLBCL. Blood 2015, 126, 1893–1901. [Google Scholar] [CrossRef] [PubMed]
- Morin, R.D.; Mendez-Lago, M.; Mungall, A.J.; Goya, R.; Mungall, K.L.; Corbett, R.D.; Johnson, N.A.; Severson, T.M.; Chiu, R.; Field, M.; et al. Frequent Mutation of Histone-Modifying Genes in Non-Hodgkin Lymphoma. Nature 2011, 476, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Pasqualucci, L.; Trifonov, V.; Fabbri, G.; Ma, J.; Rossi, D.; Chiarenza, A.; Wells, V.A.; Grunn, A.; Messina, M.; Elliot, O.; et al. Analysis of the Coding Genome of Diffuse Large B-Cell Lymphoma. Nat. Genet. 2011, 43, 830–837. [Google Scholar] [CrossRef] [PubMed]
- Lohr, J.G.; Stojanov, P.; Lawrence, M.S.; Auclair, D.; Chapuy, B.; Sougnez, C.; Cruz-Gordillo, P.; Knoechel, B.; Asmann, Y.W.; Slager, S.L.; et al. Discovery and Prioritization of Somatic Mutations in Diffuse Large B-Cell Lymphoma (DLBCL) by Whole-Exome Sequencing. Proc. Natl. Acad. Sci. USA 2012, 109, 3879–3884. [Google Scholar] [CrossRef] [PubMed]
- Morin, R.D.; Mungall, K.; Pleasance, E.; Mungall, A.J.; Goya, R.; Huff, R.D.; Scott, D.W.; Ding, J.; Roth, A.; Chiu, R.; et al. Mutational and Structural Analysis of Diffuse Large B-Cell Lymphoma using Whole-Genome Sequencing. Blood 2013, 122, 1256–1265. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Grubor, V.; Love, C.L.; Banerjee, A.; Richards, K.L.; Mieczkowski, P.A.; Dunphy, C.; Choi, W.; Au, W.Y.; Srivastava, G.; et al. Genetic Heterogeneity of Diffuse Large B-Cell Lymphoma. Proc. Natl. Acad. Sci. USA 2013, 110, 1398–1403. [Google Scholar] [CrossRef] [PubMed]
- De Miranda, N.F.; Georgiou, K.; Chen, L.; Wu, C.; Gao, Z.; Zaravinos, A.; Lisboa, S.; Enblad, G.; Teixeira, M.R.; Zeng, Y.; et al. Exome Sequencing Reveals Novel Mutation Targets in Diffuse Large B-Cell Lymphomas Derived from Chinese Patients. Blood 2014, 124, 2544–2553. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Redmond, D.; Nie, K.; Eng, K.W.; Clozel, T.; Martin, P.; Tan, L.H.; Melnick, A.M.; Tam, W.; Elemento, O. Deep Sequencing Reveals Clonal Evolution Patterns and Mutation Events Associated with Relapse in B-Cell Lymphomas. Genome Biol. 2014, 15, 432. [Google Scholar] [PubMed]
- Novak, A.J.; Asmann, Y.W.; Maurer, M.J.; Wang, C.; Slager, S.L.; Hodge, L.S.; Manske, M.; Price-Troska, T.; Yang, Z.Z.; Zimmermann, M.T.; et al. Whole-Exome Analysis Reveals Novel Somatic Genomic Alterations Associated with Outcome in Immunochemotherapy-Treated Diffuse Large B-Cell Lymphoma. Blood Cancer. J. 2015, 5, e346. [Google Scholar] [CrossRef] [PubMed]
- Wise, J.F.; Nakken, S.; Vodak, D.; Troen, G.; Lingjaerde, O.C.; Meza-Zepeda, L.A.; Myklebost, O.; Beiske, K.; Myklebust, J.H.; Hovig, E.; et al. Discovery of Recurrent Mutations Associated with Chemo-Immunotherapy Relapse in Diffuse Large B-Cell Lymphoma. Blood 2015, 26, 110. [Google Scholar]
- Morin, R.D.; Assouline, S.; Alcaide, M.; Mohajeri, A.; Johnston, R.L.; Chong, L.; Grewal, J.; Yu, S.; Fornika, D.; Bushell, K.; et al. Genetic Landscapes of Relapsed and Refractory Diffuse Large B-Cell Lymphomas. Clin. Cancer Res. 2016, 22, 2290–2300. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.; Zhang, J.; Davis, N.S.; Moffitt, A.B.; Love, C.L.; Waldrop, A.; Leppa, S.; Pasanen, A.; Meriranta, L.; Karjalainen-Lindsberg, M.L.; et al. Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell 2017, 171, 481–494. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, R.; Wright, G.W.; Huang, D.W.; Johnson, C.A.; Phelan, J.D.; Wang, J.Q.; Roulland, S.; Kasbekar, M.; Young, R.M.; Shaffer, A.L.; et al. Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2018, 378, 1396–1407. [Google Scholar] [CrossRef] [PubMed]
- Chapuy, B.; Stewart, C.; Dunford, A.J.; Kim, J.; Kamburov, A.; Redd, R.A.; Lawrence, M.S.; Roemer, M.G.M.; Li, A.J.; Ziepert, M.; et al. Molecular Subtypes of Diffuse Large B Cell Lymphoma are Associated with Distinct Pathogenic Mechanisms and Outcomes. Nat. Med. 2018, 24, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Moffitt, A.B.; Dave, S.S. Clinical Applications of the Genomic Landscape of Aggressive Non-Hodgkin Lymphoma. J. Clin. Oncol. 2017, 35, 955–962. [Google Scholar] [CrossRef] [PubMed]
- Younes, A.; Berry, D.A. From Drug Discovery to Biomarker-Driven Clinical Trials in Lymphoma. Nat. Rev. Clin. Oncol. 2012, 9, 643–653. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, S.; Caldas, C. The Implications of Clonal Genome Evolution for Cancer Medicine. N. Engl. J. Med. 2013, 368, 842–851. [Google Scholar] [CrossRef] [PubMed]
- Ventura, R.A.; Martin-Subero, J.I.; Jones, M.; McParland, J.; Gesk, S.; Mason, D.Y.; Siebert, R. FISH Analysis for the Detection of Lymphoma-Associated Chromosomal Abnormalities in Routine Paraffin-Embedded Tissue. J. Mol. Diagn. 2006, 8, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Saber, A.; Hiltermann, T.J.N.; Kok, K.; Terpstra, M.M.; De Lange, K.; Timens, W.; Groen, H.J.M.; Van den Berg, A. Mutation Patterns in Small Cell and Non-Small Cell Lung Cancer Patients Suggest a Different Level of Heterogeneity between Primary and Metastatic Tumors. Carcinogenesis 2017, 38, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Van der Wekken, A.J.; Kuiper, J.L.; Saber, A.; Terpstra, M.M.; Wei, J.; Hiltermann, T.J.N.; Thunnissen, E.; Heideman, D.A.M.; Timens, W.; Schuuring, E.; et al. Overall Survival in EGFR Mutated Non-Small-Cell Lung Cancer Patients Treated with Afatinib After EGFR TKI and Resistant Mechanisms upon Disease Progression. PLoS ONE 2017, 12, e0182885. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and Accurate Long-Read Alignment with Burrows-Wheeler Transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Boomsma, D.I.; Wijmenga, C.; Slagboom, E.P.; Swertz, M.A.; Karssen, L.C.; Abdellaoui, A.; Ye, K.; Guryev, V.; Vermaat, M.; van Dijk, F.; et al. The Genome of the Netherlands: Design, and Project Goals. Eur. J. Hum. Genet. 2014, 22, 221–227. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing Next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The Variant Call Format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang le, L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A Program for Annotating and Predicting the Effects of Single Nucleotide Polymorphisms, SnpEff: SNPs in the Genome of Drosophila Melanogaster Strain w1118; Iso-2; Iso-3. Fly (Austin) 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jian, X.; Boerwinkle, E. dbNSFP V2.0: A Database of Human Non-Synonymous SNVs and their Functional Predictions and Annotations. Hum. Mutat. 2013, 34, E2393–E2402. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.A.; Beare, D.; Gunasekaran, P.; Leung, K.; Bindal, N.; Boutselakis, H.; Ding, M.; Bamford, S.; Cole, C.; Ward, S.; et al. COSMIC: Exploring the World’s Knowledge of Somatic Mutations in Human Cancer. Nucleic Acids Res. 2015, 43, D805–D811. [Google Scholar] [CrossRef] [PubMed]
- Khodabakhshi, A.H.; Morin, R.D.; Fejes, A.P.; Mungall, A.J.; Mungall, K.L.; Bolger-Munro, M.; Johnson, N.A.; Connors, J.M.; Gascoyne, R.D.; Marra, M.A.; et al. Recurrent Targets of Aberrant Somatic Hypermutation in Lymphoma. Oncotarget 2012, 3, 1308–1319. [Google Scholar] [CrossRef] [PubMed]
- Szikriszt, B.; Poti, A.; Pipek, O.; Krzystanek, M.; Kanu, N.; Molnar, J.; Ribli, D.; Szeltner, Z.; Tusnady, G.E.; Csabai, I.; et al. A Comprehensive Survey of the Mutagenic Impact of Common Cancer Cytotoxics. Genome Biol. 2016, 17, 99. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Scherer, F.; Kurtz, D.M.; Newman, A.M.; Stehr, H.; Craig, A.F.; Esfahani, M.S.; Lovejoy, A.F.; Chabon, J.J.; Klass, D.M.; Liu, C.L.; et al. Distinct Biological Subtypes and Patterns of Genome Evolution in Lymphoma Revealed by Circulating Tumor DNA. Sci. Transl. Med. 2016, 8, 364ra155. [Google Scholar] [CrossRef] [PubMed]
- Landau, D.A.; Tausch, E.; Taylor-Weiner, A.N.; Stewart, C.; Reiter, J.G.; Bahlo, J.; Kluth, S.; Bozic, I.; Lawrence, M.; Bottcher, S.; et al. Mutations Driving CLL and their Evolution in Progression and Relapse. Nature 2015, 526, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, G.; Rasi, S.; Rossi, D.; Trifonov, V.; Khiabanian, H.; Ma, J.; Grunn, A.; Fangazio, M.; Capello, D.; Monti, S.; et al. Analysis of the Chronic Lymphocytic Leukemia Coding Genome: Role of NOTCH1 Mutational Activation. J. Exp. Med. 2011, 208, 1389–1401. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Kihira, S.; Liu, C.L.; Nair, R.V.; Salari, R.; Gentles, A.J.; Irish, J.; Stehr, H.; Vicente-Duenas, C.; Romero-Camarero, I.; et al. Mutations in Early Follicular Lymphoma Progenitors are Associated with Suppressed Antigen Presentation. Proc. Natl. Acad. Sci. USA 2015, 112, E1116–E1125. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Gentles, A.J.; Nair, R.V.; Irish, J.M.; Kihira, S.; Liu, C.L.; Kela, I.; Hopmans, E.S.; Myklebust, J.H.; Ji, H.; et al. Hierarchy in Somatic Mutations Arising during Genomic Evolution and Progression of Follicular Lymphoma. Blood 2013, 121, 1604–1611. [Google Scholar] [CrossRef] [PubMed]
- Bea, S.; Valdes-Mas, R.; Navarro, A.; Salaverria, I.; Martin-Garcia, D.; Jares, P.; Gine, E.; Pinyol, M.; Royo, C.; Nadeu, F.; et al. Landscape of Somatic Mutations and Clonal Evolution in Mantle Cell Lymphoma. Proc. Natl. Acad. Sci. USA 2013, 110, 18250–18255. [Google Scholar] [CrossRef] [PubMed]
- Spina, V.; Bruscaggin, A.; Cuccaro, A.; Martini, M.; Di Trani, M.; Forestieri, G.; Manzoni, M.; Condoluci, A.; Arribas, A.; Terzi-Di-Bergamo, L.; et al. Circulating Tumor DNA Reveals Genetics, Clonal Evolution, and Residual Disease in Classical Hodgkin Lymphoma. Blood 2018, 131, 2413–2425. [Google Scholar] [CrossRef] [PubMed]
- Furman, R.R.; Cheng, S.; Lu, P.; Setty, M.; Perez, A.R.; Guo, A.; Racchumi, J.; Xu, G.; Wu, H.; Ma, J.; et al. Ibrutinib Resistance in Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2014, 370, 2352–2354. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, S.; Schaffer, M.; Deraedt, W.; Deraedt, W.; Davic, C.; Stepanchick, E.; Aquino, R.; Yuan, Z.; Kranenburg, B.; Avivi, I.; et al. Mutational Analysis of Patients with Primary Resistance to Single-Agent Ibrutinib in Relapsed Or Refractory Mantle Cell Lymphoma (MCL). Blood 2014, 124, 78. [Google Scholar]
- Rossi, D.; Diop, F.; Spaccarotella, E.; Monti, S.; Zanni, M.; Rasi, S.; Deambrogi, C.; Spina, V.; Bruscaggin, A.; Favini, C.; et al. Diffuse Large B-Cell Lymphoma Genotyping on the Liquid Biopsy. Blood 2017, 129, 1947–1957. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, D.M.; Scherer, F.; Jin, M.C.; Soo, J.; Craig, A.F.M.; Esfahani, M.S.; Chabon, J.J.; Stehr, H.; Liu, C.L.; Tibshirani, R.; et al. Circulating Tumor DNA Measurements as Early Outcome Predictors in Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2018, 36, 2845–2853. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.A.; Rooney, M.S.; Rajasagi, M.; Tiao, G.; Dixon, P.M.; Lawrence, M.S.; Stevens, J.; Lane, W.J.; Dellagatta, J.L.; Steelman, S.; et al. Comprehensive Analysis of Cancer-Associated Somatic Mutations in Class I HLA Genes. Nat. Biotechnol. 2015, 33, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Nijland, M.; Veenstra, R.N.; Visser, L.; Xu, C.; Kushekhar, K.; van Imhoff, G.W.; Kluin, P.M.; van den Berg, A.; Diepstra, A. HLA Dependent Immune Escape Mechanisms in B-Cell Lymphomas: Implications for Immune Checkpoint Inhibitor Therapy? Oncoimmunology 2017, 6, e1295202. [Google Scholar] [CrossRef] [PubMed]
- Schif, B.; Lennerz, J.K.; Kohler, C.W.; Bentink, S.; Kreuz, M.; Melzner, I.; Ritz, O.; Trumper, L.; Loeffler, M.; Spang, R.; et al. SOCS1 Mutation Subtypes Predict Divergent Outcomes in Diffuse Large B-Cell Lymphoma (DLBCL) Patients. Oncotarget 2013, 4, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Kuo, H.P.; Ezell, S.A.; Hsieh, S.; Schweighofer, K.J.; Cheung, L.W.; Wu, S.; Apatira, M.; Sirisawad, M.; Eckert, K.; Liang, Y.; et al. The Role of PIM1 in the Ibrutinib-Resistant ABC Subtype of Diffuse Large B-Cell Lymphoma. Am. J. Cancer. Res. 2016, 6, 2489–2501. [Google Scholar] [PubMed]
- Brault, L.; Menter, T.; Obermann, E.C.; Knapp, S.; Thommen, S.; Schwaller, J.; Tzankov, A. PIM Kinases are Progression Markers and Emerging Therapeutic Targets in Diffuse Large B-Cell Lymphoma. Br. J. Cancer 2012, 107, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Do, H.; Dobrovic, A. Sequence Artifacts in DNA from Formalin-Fixed Tissues: Causes and Strategies for Minimization. Clin. Chem. 2015, 61, 64–71. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Patient Characteristics | Clinicopathological Characteristics | Outcome | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | M/F | Age | Stage | IPI | Morphology | COO | Aberrant IHC | FISH | Primary Biopsy | Relapse Biopsy 1 | Relapse Biopsy 2 | End-of-Treatment | PFS (Months) | OS (Months) |
| 1 | F | 53 | 4 | 3 | DLBCL | GCB | n.a. | Inconclusive | Jejunum | Lymph node | - | CR | 7 | 101 |
| 2 | M | 45 | 2 | 2 | DLBCL | ABC | n.a. | MYC– | Lymph node | Lymph node | - | CR | 17 | 56 † |
| 3 | M | 65 | 3 | 2 | DLBCL | GCB | n.a. | MYC– | Soft tissue | Soft tissue | - | PR | 7 | 14 † |
| 4 | F | 57 | 3 | 1 | DLBCL | ABC | CD20– | MYC– | Lymph node | Lymph node | - | PD | 5 | 8 † |
| 5 | F | 57 | 4 | 4 | HGBCL MYC+/BCL6+ # | Unclassified | CD5+ | MYC+ BCL6+ | Lymph node A | Lymph node B * | Lymph node C * | n.a. | 14 | 36 † |
| 6 | M | 79 | 1 | 2 | DLBCL | GCB | n.a. | MYC– | Soft palate | Skin site A ** | Skin site B ** | CR | 55 | 55 † |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nijland, M.; Seitz, A.; Terpstra, M.; Van Imhoff, G.W.; Kluin, P.M.; Van Meerten, T.; Atayar, Ç.; Van Kempen, L.C.; Diepstra, A.; Kok, K.; et al. Mutational Evolution in Relapsed Diffuse Large B-Cell Lymphoma. Cancers 2018, 10, 459. https://doi.org/10.3390/cancers10110459
Nijland M, Seitz A, Terpstra M, Van Imhoff GW, Kluin PM, Van Meerten T, Atayar Ç, Van Kempen LC, Diepstra A, Kok K, et al. Mutational Evolution in Relapsed Diffuse Large B-Cell Lymphoma. Cancers. 2018; 10(11):459. https://doi.org/10.3390/cancers10110459
Chicago/Turabian StyleNijland, Marcel, Annika Seitz, Martijn Terpstra, Gustaaf W. Van Imhoff, Philip M Kluin, Tom Van Meerten, Çiğdem Atayar, Léon C. Van Kempen, Arjan Diepstra, Klaas Kok, and et al. 2018. "Mutational Evolution in Relapsed Diffuse Large B-Cell Lymphoma" Cancers 10, no. 11: 459. https://doi.org/10.3390/cancers10110459
APA StyleNijland, M., Seitz, A., Terpstra, M., Van Imhoff, G. W., Kluin, P. M., Van Meerten, T., Atayar, Ç., Van Kempen, L. C., Diepstra, A., Kok, K., & Van den Berg, A. (2018). Mutational Evolution in Relapsed Diffuse Large B-Cell Lymphoma. Cancers, 10(11), 459. https://doi.org/10.3390/cancers10110459