Non-Coding Variants in BRCA1 and BRCA2 Genes: Potential Impact on Breast and Ovarian Cancer Predisposition

 , , and
, , and
Abstract
1. Hereditary Breast and Ovarian Cancer (HBOC) Syndrome
2. Germline Cancer-Associated Variants in the Regulatory Regions
3. Regulatory Regions in BRCA1 and BRCA2 Genes
4. Methods to Assess the Pathogenicity of BRCA1/2 Non-Coding Variants
4.1. In Silico Tools and Genetic Data
4.2. In Vitro Studies
4.2.1. Assays to Measure Splicing
4.2.2. Assays to Measure Interaction between Enhancers and Promoters
4.2.3. Assays to Measure Gene Expression and Protein Function (Functional Assays)
4.2.4. Assays to Investigate the Underlying Mechanism of Variant Impact
4.3. Tumor Features
5. Impact of BRCA1/2 Non-Coding Variants on Breast and Ovarian Cancer Predisposition
6. Clinical Practice Recommendations for Non-Coding Variants’ Carriers
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Wooster, R.; Bignell, G.; Lancaster, J.; Swift, S.; Seal, S.; Mangion, J.; Collins, N.; Gregory, S.; Gumbs, C.; Micklem, G. Identification of the breast cancer susceptibility gene BRCA2. Nature 1995, 378, 789–792. [Google Scholar] [CrossRef] [PubMed]
- Miki, Y.; Swensen, J.; Shattuck-Eidens, D.; Futreal, P.A.; Harshman, K.; Tavtigian, S.; Liu, Q.; Cochran, C.; Bennett, L.M.; Ding, W. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994, 266, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.-A.; Mooij, T.M.; Roos-Blom, M.-J.; Jervis, S.; van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 2017, 317, 2402–2416. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, A.; Pharoah, P.D.P.; Narod, S.; Risch, H.A.; Eyfjord, J.E.; Hopper, J.L.; Loman, N.; Olsson, H.; Johannsson, O.; Borg, A.; et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: A combined analysis of 22 studies. Am. J. Hum. Genet. 2003, 72, 1117–1130. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Parmigiani, G. Meta-analysis of BRCA1 and BRCA2 penetrance. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2007, 25, 1329–1333. [Google Scholar] [CrossRef] [PubMed]
- Robson, M.; Im, S.-A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Tutt, A.; Tovey, H.; Cheang, M.C.U.; Kernaghan, S.; Kilburn, L.; Gazinska, P.; Owen, J.; Abraham, J.; Barrett, S.; Barrett-Lee, P.; et al. Carboplatin in BRCA1/2-mutated and triple-negative breast cancer BRCAness subgroups: the TNT Trial. Nat. Med. 2018, 24, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib Maintenance Therapy in Platinum-Sensitive Relapsed Ovarian Cancer. N. Engl. J. Med. 2012, 366, 1382–1392. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.L.; Oza, A.M.; Lorusso, D.; Aghajanian, C.; Oaknin, A.; Dean, A.; Colombo, N.; Weberpals, J.I.; Clamp, A.; Scambia, G.; et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1949–1961. [Google Scholar] [CrossRef]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.-G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Michailidou, K.; Hall, P.; Gonzalez-Neira, A.; Ghoussaini, M.; Dennis, J.; Milne, R.L.; Schmidt, M.K.; Chang-Claude, J.; Bojesen, S.E.; Bolla, M.K.; et al. Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat. Genet. 2013, 45, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Michailidou, K.; Lindström, S.; Dennis, J.; Beesley, J.; Hui, S.; Kar, S.; Lemaçon, A.; Soucy, P.; Glubb, D.; Rostamianfar, A.; et al. Association analysis identifies 65 new breast cancer risk loci. Nature 2017, 551, 92–94. [Google Scholar] [CrossRef] [PubMed]
- Brzovic, P.S.; Rajagopal, P.; Hoyt, D.W.; King, M.C.; Klevit, R.E. Structure of a BRCA1-BARD1 heterodimeric RING-RING complex. Nat. Struct. Biol. 2001, 8, 833–837. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.S.; Green, R.; Glover, J.N. Crystal structure of the BRCT repeat region from the breast cancer-associated protein BRCA1. Nat. Struct. Biol. 2001, 8, 838–842. [Google Scholar] [CrossRef] [PubMed]
- Varma, A.K.; Brown, R.S.; Birrane, G.; Ladias, J.A.A. Structural basis for cell cycle checkpoint control by the BRCA1-CtIP complex. Biochemistry 2005, 44, 10941–10946. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Paul, A.; Su, D.; Mehmood, S.; Foo, T.K.; Ochi, T.; Bunting, E.L.; Xia, B.; Robinson, C.V.; Wang, B.; et al. Structure of BRCA1-BRCT/Abraxas Complex Reveals Phosphorylation-Dependent BRCT Dimerization at DNA Damage Sites. Mol. Cell. 2016, 61, 434–448. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.S.; Baldeyron, C.; Carreira, A. Molding BRCA2 function through its interacting partners. Cell. Cycle. 2015, 14, 3389–3395. [Google Scholar] [CrossRef] [PubMed]
- von Nicolai, C.; Ehlén, Å.; Martin, C.; Zhang, X.; Carreira, A. A second DNA binding site in human BRCA2 promotes homologous recombination. Nat. Commun. 2016, 7, 12813. [Google Scholar] [CrossRef] [PubMed]
- Caputo, S.; Benboudjema, L.; Sinilnikova, O.; Rouleau, E.; Béroud, C.; Lidereau, R. Description and analysis of genetic variants in French hereditary breast and ovarian cancer families recorded in the UMD-BRCA1/BRCA2 databases. Nucleic. Acids. Res. 2012, 40, D992–D1002. [Google Scholar] [CrossRef] [PubMed]
- Castéra, L.; Harter, V.; Muller, E.; Krieger, S.; Goardon, N.; Ricou, A.; Rousselin, A.; Paimparay, G.; Legros, A.; Bruet, O.; et al. Landscape of pathogenic variations in a panel of 34 genes and cancer risk estimation from 5131 HBOC families. Genet. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kondrashova, O.; Topp, M.; Nesic, K.; Lieschke, E.; Ho, G.-Y.; Harrell, M.I.; Zapparoli, G.V.; Hadley, A.; Holian, R.; Boehm, E.; et al. Methylation of all BRCA1 copies predicts response to the PARP inhibitor rucaparib in ovarian carcinoma. Nat. Commun. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.R.; Roberts, N.; McGowan, S.; Hay, D.; Giannoulatou, E.; Lynch, M.; De Gobbi, M.; Taylor, S.; Gibbons, R.; Higgs, D.R. Analysis of hundreds of cis-regulatory landscapes at high resolution in a single, high-throughput experiment. Nat. Genet. 2014, 46, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Heidari, N.; Phanstiel, D.H.; He, C.; Grubert, F.; Jahanbani, F.; Kasowski, M.; Zhang, M.Q.; Snyder, M.P. Genome-wide map of regulatory interactions in the human genome. Genome Res. 2014, 24, 1905–1917. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Lupski, J.R. Non-coding genetic variants in human disease. Hum. Mol. Genet. 2015, 24, R102–R110. [Google Scholar] [CrossRef] [PubMed]
- Stacey, S.N.; Manolescu, A.; Sulem, P.; Rafnar, T.; Gudmundsson, J.; Gudjonsson, S.A.; Masson, G.; Jakobsdottir, M.; Thorlacius, S.; Helgason, A.; et al. Common variants on chromosomes 2q35 and 16q12 confer susceptibility to estrogen receptor-positive breast cancer. Nat. Genet. 2007, 39, 865–869. [Google Scholar] [CrossRef] [PubMed]
- Horn, S.; Figl, A.; Rachakonda, P.S.; Fischer, C.; Sucker, A.; Gast, A.; Kadel, S.; Moll, I.; Nagore, E.; Hemminki, K.; et al. TERT promoter mutations in familial and sporadic melanoma. Science 2013, 339, 959–961. [Google Scholar] [CrossRef] [PubMed]
- Cong, Y.-S.; Wright, W.E.; Shay, J.W. Human telomerase and its regulation. MMBR 2002, 66, 407–425. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Li, S.; Stohr, B.A. The role of telomere biology in cancer. Annu. Rev. Pathol. 2013, 8, 49–78. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.L.; Dobbins, T.; Lindor, N.M.; Rapkins, R.W.; Hitchins, M.P. Identification of constitutional MLH1 epimutations and promoter variants in colorectal cancer patients from the Colon Cancer Family Registry. Genet. Med. 2013, 15, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Hitchins, M.P.; Wong, J.J.L.; Suthers, G.; Suter, C.M.; Martin, D.I.K.; Hawkins, N.J.; Ward, R.L. Inheritance of a cancer-associated MLH1 germ-line epimutation. N. Engl. J. Med. 2007, 356, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.-P.; Waite, K.A.; Pilarski, R.; Hampel, H.; Fernandez, M.J.; Bos, C.; Dasouki, M.; Feldman, G.L.; Greenberg, L.A.; Ivanovich, J.; et al. Germline PTEN promoter mutations and deletions in Cowden/Bannayan-Riley-Ruvalcaba syndrome result in aberrant PTEN protein and dysregulation of the phosphoinositol-3-kinase/Akt pathway. Am. J. Hum. Genet. 2003, 73, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Hitchins, M.P.; Rapkins, R.W.; Kwok, C.-T.; Srivastava, S.; Wong, J.J.L.; Khachigian, L.M.; Polly, P.; Goldblatt, J.; Ward, R.L. Dominantly inherited constitutional epigenetic silencing of MLH1 in a cancer-affected family is linked to a single nucleotide variant within the 5’UTR. Cancer Cell 2011, 20, 200–213. [Google Scholar] [CrossRef] [PubMed]
- Pomerantz, M.M.; Ahmadiyeh, N.; Jia, L.; Herman, P.; Verzi, M.P.; Doddapaneni, H.; Beckwith, C.A.; Chan, J.A.; Hills, A.; Davis, M.; et al. The 8q24 cancer risk variant rs6983267 shows long-range interaction with MYC in colorectal cancer. Nat. Genet. 2009, 41, 882–884. [Google Scholar] [CrossRef] [PubMed]
- Vaughn, J.P.; Davis, P.L.; Jarboe, M.D.; Huper, G.; Evans, A.C.; Wiseman, R.W.; Berchuck, A.; Iglehart, J.D.; Futreal, P.A.; Marks, J.R. BRCA1 expression is induced before DNA synthesis in both normal and tumor-derived breast cells. Cell Growth Differ. 1996, 7, 711–715. [Google Scholar] [PubMed]
- Misra, S.; Sharma, S.; Agarwal, A.; Khedkar, S.V.; Tripathi, M.K.; Mittal, M.K.; Chaudhuri, G. Cell cycle-dependent regulation of the bi-directional overlapping promoter of human BRCA2/ZAR2 genes in breast cancer cells. Mol. Cancer 2010, 9, 50. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.F.; Brown, M.A.; Chambers, J.A.; Griffiths, B.; Nicolai, H.; Solomon, E. Distinct transcription start sites generate two forms of BRCA1 mRNA. Hum. Mol. Genet. 1995, 4, 2259–2264. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.F.; Chambers, J.A.; Solomon, E. Complex regulation of the BRCA1 gene. J. Biol. Chem. 1997, 272, 20994–20997. [Google Scholar] [CrossRef] [PubMed]
- Sobczak, K.; Krzyzosiak, W.J. Structural determinants of BRCA1 translational regulation. J. Biol. Chem. 2002, 277, 17349–17358. [Google Scholar] [CrossRef] [PubMed]
- Suen, T.-C.; Tang, M.-S.; Goss, P.E. Model of transcriptional regulation of the BRCA1-NBR2 bi-directional transcriptional unit. Biochim. Biophys. Acta 2005, 1728, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Curtis, C.; Shah, S.P.; Chin, S.-F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Atlas, E.; Stramwasser, M.; Whiskin, K.; Mueller, C.R. GA-binding protein alpha/beta is a critical regulator of the BRCA1 promoter. Oncogene 2000, 19, 1933–1940. [Google Scholar] [CrossRef] [PubMed]
- Atlas, E.; Stramwasser, M.; Mueller, C.R. A CREB site in the BRCA1 proximal promoter acts as a constitutive transcriptional element. Oncogene 2001, 20, 7110–7114. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Schneider-Broussard, R.; Kumar, A.P.; MacLeod, M.C.; Johnson, D.G. Regulation of BRCA1 expression by the Rb-E2F pathway. J. Biol. Chem. 2000, 275, 4532–4536. [Google Scholar] [CrossRef] [PubMed]
- Norris, J.; Fan, D.; Aleman, C.; Marks, J.R.; Futreal, P.A.; Wiseman, R.W.; Iglehart, J.D.; Deininger, P.L.; McDonnell, D.P. Identification of a new subclass of Alu DNA repeats which can function as estrogen receptor-dependent transcriptional enhancers. J. Biol. Chem. 1995, 270, 22777–22782. [Google Scholar] [CrossRef] [PubMed]
- Suen, T.C.; Goss, P.E. Identification of a novel transcriptional repressor element located in the first intron of the human BRCA1 gene. Oncogene 2001, 20, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Herman, J.G.; Baylin, S.B. Gene silencing in cancer in association with promoter hypermethylation. N. Engl. J. Med. 2003, 349, 2042–2054. [Google Scholar] [CrossRef] [PubMed]
- Gazzoli, I.; Loda, M.; Garber, J.; Syngal, S.; Kolodner, R.D. A hereditary nonpolyposis colorectal carcinoma case associated with hypermethylation of the MLH1 gene in normal tissue and loss of heterozygosity of the unmethylated allele in the resulting microsatellite instability-high tumor. Cancer Res. 2002, 62, 3925–3928. [Google Scholar] [PubMed]
- Miyakura, Y.; Sugano, K.; Akasu, T.; Yoshida, T.; Maekawa, M.; Saitoh, S.; Sasaki, H.; Nomizu, T.; Konishi, F.; Fujita, S.; et al. Extensive but hemiallelic methylation of the hMLH1 promoter region in early-onset sporadic colon cancers with microsatellite instability. Clin. Gastroenterol. Hepatol. 2004, 2, 147–156. [Google Scholar] [CrossRef]
- Suter, C.M.; Martin, D.I.K.; Ward, R.L. Germline epimutation of MLH1 in individuals with multiple cancers. Nat. Genet. 2004, 36, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Dobrovic, A.; Simpfendorfer, D. Methylation of the BRCA1 gene in sporadic breast cancer. Cancer Res. 1997, 57, 3347–3350. [Google Scholar] [PubMed]
- Magdinier, F.; Ribieras, S.; Lenoir, G.M.; Frappart, L.; Dante, R. Down-regulation of BRCA1 in human sporadic breast cancer; analysis of DNA methylation patterns of the putative promoter region. Oncogene 1998, 17, 3169–3176. [Google Scholar] [CrossRef] [PubMed]
- Rice, J.C.; Massey-Brown, K.S.; Futscher, B.W. Aberrant methylation of the BRCA1 CpG island promoter is associated with decreased BRCA1 mRNA in sporadic breast cancer cells. Oncogene 1998, 17, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Vos, S.; van Diest, P.J.; Moelans, C.B. A systematic review on the frequency of BRCA promoter methylation in breast and ovarian carcinomas of BRCA germline mutation carriers: Mutually exclusive, or not? Crit. Rev. Oncol. Hematol. 2018, 127, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Wardrop, S.L.; Brown, M.A. kConFab Investigators Identification of two evolutionarily conserved and functional regulatory elements in intron 2 of the human BRCA1 gene. Genomics 2005, 86, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, E.S.; Caputo, S.M.; Castera, L.; Gendrot, M.; Briaux, A.; Breault, M.; Krieger, S.; Rogan, P.K.; Mucaki, E.J.; Burke, L.J.; et al. Assessment of the functional impact of germline BRCA1/2 variants located in non-coding regions in families with breast and/or ovarian cancer predisposition. Breast Cancer Res. Treat. 2017. [Google Scholar] [CrossRef] [PubMed]
- Pongsavee, M.; Yamkamon, V.; Dakeng, S.; O-charoenrat, P.; Smith, D.R.; Saunders, G.F.; Patmasiriwat, P. The BRCA1 3’-UTR: 5711+421T/T_5711+1286T/T genotype is a possible breast and ovarian cancer risk factor. Genet. Test Mol. Biomarkers. 2009, 13, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Garcia, A.I.; Buisson, M.; Damiola, F.; Tessereau, C.; Barjhoux, L.; Verny-Pierre, C.; Sornin, V.; Dondon, M.-G.; Eon-Marchais, S.; GENESIS Investigators; et al. Mutation screening of MIR146A/B and BRCA1/2 3’-UTRs in the GENESIS study. EJHG 2016. [Google Scholar] [CrossRef] [PubMed]
- Brewster, B.L.; Rossiello, F.; French, J.D.; Edwards, S.L.; Wong, M.; Wronski, A.; Whiley, P.; Waddell, N.; Chen, X.; Bove, B.; et al. Identification of fifteen novel germline variants in the BRCA1 3’UTR reveals a variant in a breast cancer case that introduces a functional miR-103 target site. Hum. Mutat. 2012, 33, 1665–1675. [Google Scholar] [CrossRef] [PubMed]
- Lheureux, S.; Lambert, B.; Krieger, S.; Legros, A.; Vaur, D.; Denoyelle, C.; Berthet, P.; Poulain, L.; Hardouin, A. Two novel variants in the 3’UTR of the BRCA1 gene in familial breast and/or ovarian cancer. Breast Cancer Res. Treat. 2011, 125, 885–891. [Google Scholar] [CrossRef] [PubMed]
- Davis, P.L.; Miron, A.; Andersen, L.M.; Iglehart, J.D.; Marks, J.R. Isolation and initial characterization of the BRCA2 promoter. Oncogene 1999, 18, 6000–6012. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Jiang, S.W.; Thangaraju, M.; Wu, G.; Couch, F.J. Induction of the BRCA2 promoter by nuclear factor-kappa B. J. Biol. Chem. 2000, 275, 35548–35556. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Bian, C.; Li, J.; Couch, F.J.; Wu, K.; Zhao, R.C. Poly(ADP-ribose) polymerase-1 down-regulates BRCA2 expression through the BRCA2 promoter. J. Biol. Chem. 2008, 283, 36249–36256. [Google Scholar] [CrossRef] [PubMed]
- Sharan, C.; Hamilton, N.M.; Parl, A.K.; Singh, P.K.; Chaudhuri, G. Identification and characterization of a transcriptional silencer upstream of the human BRCA2 gene. Biochem. Biophys. Res. Commun. 1999, 265, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Jiang, S.-W.; Couch, F.J. p53 mediates repression of the BRCA2 promoter and down-regulation of BRCA2 mRNA and protein levels in response to DNA damage. J. Biol. Chem. 2003, 278, 15652–15660. [Google Scholar] [CrossRef] [PubMed]
- Fraile-Bethencourt, E.; Valenzuela-Palomo, A.; Díez-Gómez, B.; Infante, M.; Durán, M.; Marcos, G.; Lastra, E.; Gómez-Barrero, S.; Velasco, E.A. Genetic dissection of the BRCA2 promoter and transcriptional impact of DNA variants. Breast Cancer Res. Treat. 2018. [Google Scholar] [CrossRef] [PubMed]
- Maia, A.-T.; Antoniou, A.C.; O’Reilly, M.; Samarajiwa, S.; Dunning, M.; Kartsonaki, C.; Chin, S.-F.; Curtis, C.N.; McGuffog, L.; Domchek, S.M.; et al. Effects of BRCA2 cis-regulation in normal breast and cancer risk amongst BRCA2 mutation carriers. Breast Cancer Res. 2012, 14, R63. [Google Scholar] [CrossRef] [PubMed]
- Plon, S.E.; Eccles, D.M.; Easton, D.; Foulkes, W.D.; Genuardi, M.; Greenblatt, M.S.; Hogervorst, F.B.L.; Hoogerbrugge, N.; Spurdle, A.B.; Tavtigian, S.V.; et al. Sequence variant classification and reporting: Recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum. Mutat. 2008, 29, 1282–1291. [Google Scholar] [CrossRef] [PubMed]
- Spurdle, A.B.; Healey, S.; Devereau, A.; Hogervorst, F.B.; Monteiro, A.N.; Nathanson, K.L.; Radice, P.; Stoppa-Lyonnet, D.; Tavtigian, S.; Wappenschmidt, B.; et al. ENIGMA—Evidence-based Network for the Interpretation of Germline Mutant Alleles: An international initiative to evaluate risk and clinical significance associated with sequence variation in BRCA1 and BRCA2 genes. Hum. Mutat. 2012, 33, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Troyanskaya, O.G. Predicting effects of noncoding variants with deep learning–based sequence model. Nat. Methods 2015, 12, 931–934. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-F.; Gulko, B.; Siepel, A. Fast, scalable prediction of deleterious noncoding variants from functional and population genomic data. Nat. Genet. 2017, 49, 618–624. [Google Scholar] [CrossRef] [PubMed]
- Mucaki, E.J.; Caminsky, N.G.; Perri, A.M.; Lu, R.; Laederach, A.; Halvorsen, M.; Knoll, J.H.M.; Rogan, P.K. A unified analytic framework for prioritization of non-coding variants of uncertain significance in heritable breast and ovarian cancer. BMC Med. Genomics 2016, 9, 19. [Google Scholar] [CrossRef] [PubMed]
- Halvorsen, M.; Martin, J.S.; Broadaway, S.; Laederach, A. Disease-associated mutations that alter the RNA structural ensemble. PLoS Genet. 2010, 6, e1001074. [Google Scholar] [CrossRef] [PubMed]
- Steen, K.-A.; Siegfried, N.A.; Weeks, K.M. Selective 2′-hydroxyl acylation analyzed by protection from exoribonuclease (RNase-detected SHAPE) for direct analysis of covalent adducts and of nucleotide flexibility in RNA. Nat. Protoc. 2011, 6, 1683–1694. [Google Scholar] [CrossRef] [PubMed]
- Spurdle, A.B.; Couch, F.J.; Hogervorst, F.B.L.; Radice, P.; Sinilnikova, O.M.; IARC Unclassified Genetic Variants Working Group. Prediction and assessment of splicing alterations: Implications for clinical testing. Hum. Mutat. 2008, 29, 1304–1313. [Google Scholar] [CrossRef] [PubMed]
- Anczuków, O.; Buisson, M.; Léoné, M.; Coutanson, C.; Lasset, C.; Calender, A.; Sinilnikova, O.M.; Mazoyer, S. BRCA2 deep intronic mutation causing activation of a cryptic exon: Opening toward a new preventive therapeutic strategy. Hum. Cancer Bio. 2012, 18, 4903–4909. [Google Scholar] [CrossRef] [PubMed]
- Dutil, J.; Godoy, L.; Rivera-Lugo, R.; Arroyo, N.; Albino, E.; Negrón, L.; Monteiro, A.N.; Matta, J.L.; Echenique, M. No Evidence for the Pathogenicity of the BRCA2 c.6937 + 594T>G Deep Intronic Variant: A Case-Control Analysis. Genet. Test. Mol. Biomark. 2018, 22, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Van Heetvelde, M.; Van Loocke, W.; Trypsteen, W.; Baert, A.; Vanderheyden, K.; Crombez, B.; Vandesompele, J.; De Leeneer, K.; Claes, K.B.M. Evaluation of relative quantification of alternatively spliced transcripts using droplet digital PCR. Biomol. Detect. Quantif. 2017, 13, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Wimmer, K.; Eckart, M.; Rehder, H.; Fonatsch, C. Illegitimate splicing of the NF1 gene in healthy individuals mimics mutation-induced splicing alterations in NF1 patients. Hum. Genet. 2000, 106, 311–313. [Google Scholar] [PubMed]
- Jia, R.; Chai, P.; Zhang, H.; Fan, X. Novel insights into chromosomal conformations in cancer. Mol. Cancer 2017, 16, 173. [Google Scholar] [CrossRef] [PubMed]
- Lawrenson, K.; Kar, S.; McCue, K.; Kuchenbaeker, K.; Michailidou, K.; Tyrer, J.; Beesley, J.; Ramus, S.J.; Li, Q.; Delgado, M.K.; et al. Functional mechanisms underlying pleiotropic risk alleles at the 19p13.1 breast-ovarian cancer susceptibility locus. Nat. Commun. 2016, 7, 12675. [Google Scholar] [CrossRef] [PubMed]
- Hinrichsen, I.; Brieger, A.; Trojan, J.; Zeuzem, S.; Nilbert, M.; Plotz, G. Expression defect size among unclassified MLH1 variants determines pathogenicity in Lynch syndrome diagnosis. Clin. Cancer Res. 2013, 19, 2432–2441. [Google Scholar] [CrossRef] [PubMed]
- Garner, M.M.; Revzin, A. A gel electrophoresis method for quantifying the binding of proteins to specific DNA regions: Application to components of the Escherichia coli lactose operon regulatory system. Nucleic. Acids Res. 1981, 9, 3047–3060. [Google Scholar] [CrossRef] [PubMed]
- Orlando, V.; Strutt, H.; Paro, R. Analysis of chromatin structure by in vivo formaldehyde cross-linking. Methods San Diego. Calif. 1997, 11, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Mavaddat, N.; Rebbeck, T.R.; Lakhani, S.R.; Easton, D.F.; Antoniou, A.C. Incorporating tumour pathology information into breast cancer risk prediction algorithms. Breast Cancer Res. 2010, 12, R28. [Google Scholar] [CrossRef] [PubMed]
- Spurdle, A.B.; Couch, F.J.; Parsons, M.T.; McGuffog, L.; Barrowdale, D.; Bolla, M.K.; Wang, Q.; Healey, S.; Schmutzler, R.; Wappenschmidt, B.; et al. kConFab Investigators Refined histopathological predictors of BRCA1 and BRCA2 mutation status: A large-scale analysis of breast cancer characteristics from the BCAC, CIMBA, and ENIGMA consortia. Breast Cancer Res. 2014, 16, 3419. [Google Scholar] [CrossRef] [PubMed]
- Burke, L.J.; Sevcik, J.; Gambino, G.; Tudini, E.; Mucaki, E.J.; Shirley, B.C.; Whiley, P.; Parsons, M.T.; De Leeneer, K.; Gutiérrez-Enríquez, S.; et al. BRCA1 and BRCA2 5′ non-coding region variants identified in breast cancer patients alter promoter activity and protein binding. Hum. Mutat. 2018. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, G.; Stramwasser, M.; Mueller, C.R. Characterization of a negative transcriptional element in the BRCA1 promoter. Breast Cancer Res. 2007, 9, R49. [Google Scholar] [CrossRef] [PubMed]
- Kao, J.; Salari, K.; Bocanegra, M.; Choi, Y.-L.; Girard, L.; Gandhi, J.; Kwei, K.A.; Hernandez-Boussard, T.; Wang, P.; Gazdar, A.F.; et al. Molecular profiling of breast cancer cell lines defines relevant tumor models and provides a resource for cancer gene discovery. PLoS ONE 2009, 4, e6146. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.G.R.; van Veen, E.M.; Byers, H.J.; Wallace, A.J.; Ellingford, J.M.; Beaman, G.; Santoyo-Lopez, J.; Aitman, T.J.; Eccles, D.M.; Lalloo, F.I.; et al. A Dominantly Inherited 5′ UTR Variant Causing Methylation-Associated Silencing of BRCA1 as a Cause of Breast and Ovarian Cancer. Am. J. Hum. Genet. 2018, 103, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lu, C.; Min, D.; Wang, Z.; Ma, X. A mutation in the 5′ untranslated region of the BRCA1 gene in sporadic breast cancer causes downregulation of translation efficiency. J. Int. Med. Res. 2007, 35, 564–573. [Google Scholar] [CrossRef] [PubMed]
- Signori, E.; Bagni, C.; Papa, S.; Primerano, B.; Rinaldi, M.; Amaldi, F.; Fazio, V.M. A somatic mutation in the 5’UTR of BRCA1 gene in sporadic breast cancer causes down-modulation of translation efficiency. Oncogene 2001, 20, 4596–4600. [Google Scholar] [CrossRef] [PubMed]
- Saunus, J.M.; French, J.D.; Edwards, S.L.; Beveridge, D.J.; Hatchell, E.C.; Wagner, S.A.; Stein, S.R.; Davidson, A.; Simpson, K.J.; Francis, G.D.; et al. Posttranscriptional regulation of the breast cancer susceptibility gene BRCA1 by the RNA binding protein HuR. Cancer Res. 2008, 68, 9469–9478. [Google Scholar] [CrossRef] [PubMed]
- Mogilyansky, E.; Clark, P.; Quann, K.; Zhou, H.; Londin, E.; Jing, Y.; Rigoutsos, I. Post-transcriptional Regulation of BRCA2 through Interactions with miR-19a and miR-19b. Front. Genet. 2016, 7, 143. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the Interpretation of Sequence Variants: A Joint Consensus Recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Antoniou, A.C.; Cunningham, A.P.; Peto, J.; Evans, D.G.; Lalloo, F.; Narod, S.A.; Risch, H.A.; Eyfjord, J.E.; Hopper, J.L.; Southey, M.C.; et al. The BOADICEA model of genetic susceptibility to breast and ovarian cancers: updates and extensions. Br. J. Cancer 2008, 98, 1457–1466. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.A.; Iversen, E.S.; Gudbjartsson, D.F.; Hiller, E.H.; Garber, J.E.; Peshkin, B.N.; Lerman, C.; Watson, P.; Lynch, H.T. BRCAPRO validation, sensitivity of genetic testing of BRCA1/BRCA2, and prevalence of other breast cancer susceptibility genes. J. Clin. Oncol. 2002, 20, 2701–2712. [Google Scholar] [CrossRef] [PubMed]
- Tyrer, J.; Duffy, S.W.; Cuzick, J. A breast cancer prediction model incorporating familial and personal risk factors. Stat. Med. 2004, 23, 1111–1130. [Google Scholar] [CrossRef] [PubMed]


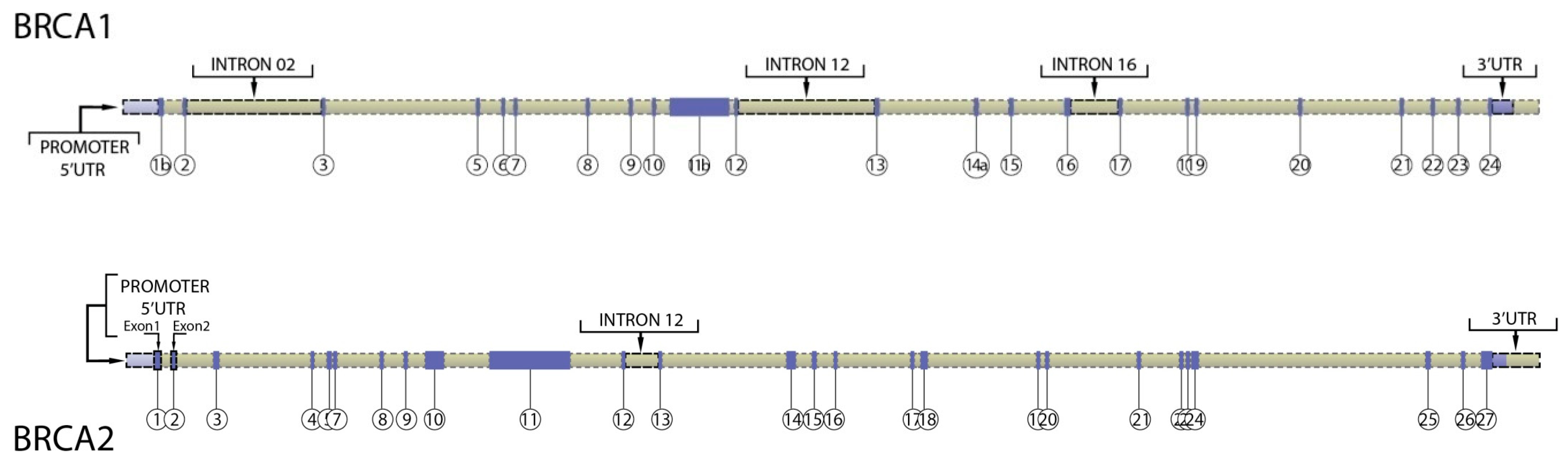{kind=link}
| Region of Interest | Hg19 Coordinates | Length | Comments |
|---|---|---|---|
| BRCA1 promoter | chr17: 41,277,500–41,278,500 | 1000 bases | Comprises 1 kb upstream on transcription start site |
| BRCA1 5′UTR (exon 1A) | chr17: 41,277,287–41,277,500 | 223 bases | Exon 1A |
| BRCA1 5′UTR (exon 1B) | chr17: 41,277,340–41,277,197 | 145 bases | Exon 1B |
| BRCA1 5′UTR + ATG (exon 2 to ATG) | chr17: 41,276,110–41,276,133 | 22 bases | 5′ end of Exon 2 |
| BRCA1 intron 2 | chr17: 41,271,250–41,272,100 | 850 bases | Includes validated enhancer and repressor elements that participate in gene looping and are conserved. Also contains sequences that UCSC/ENCODE indicates this region contains transcription factor binding sites, DnaseHS sites |
| BRCA1 intron 12 (region 1) | chr17: 41,237,500–41,237,850 | 350 bases | UCSC/ENCODE indicates this region contains transcription factor binding sites, DnaseHS sites and is conserved. |
| BRCA1 intron 12 (region 2) | chr17: 41,236,600–41,236,960 | 360 bases | UCSC/ENCODE indicates this region contains transcription factor binding sites, DnaseHS sites and is conserved. |
| BRCA1 intron 16 | Chr17: 41,220,900–41,221,250 | 350 bases | UCSC/ENCODE indicates this region contains transcription factor binding sites, DnaseHS sites |
| BRCA1 3′UTR (exon 24) | chr17: 41,196,311–41,197,698 | 1387 bases | From and including stop codon |
| BRCA2 promoter | chr13: 32,888,616–32,889,616 | 1000 bases | Comprises 1 kb upstream on transcription start site |
| BRCA2 5′UTR (exon 1) | chr13: 32,889,616–32,889,805 | 189 bases | Exon 1 (Refseq) |
| BRCA2 5′UTR (exon 2 to ATG) | chr13: 32,890,558–32,890,600 | 42 bases | Includes translation start codon |
| BRCA2 3′UTR | chr13: 32,972,904–32,973,809 | 905 bases | From and including stop codon |
| A | |||||
| Gene | Region Screened | Population Screened (n) | Number of Samples Presenting a Variant | References | |
| BRCA2 | Promoter | 95 | 3 | [68] | |
| BRCA1 | Promoter (255 bp) | 3926 | 55 | [58] | |
| BRCA2 | Promoter (380 bp) | 3910 | 21 | [58] | |
| BRCA1 | Intron 2 (326 bp) | 3624 | 30 | [58] | |
| BRCA1 | Intron 12 (360 bp) | 2973 | 11 | [58] | |
| BRCA1 | 5′UTR | 49 | 2 | [93] | |
| BRCA1 | 5′UTR | 117* | 2 (somatic) | [94] | |
| BRCA1 | 5′UTR | 96 | 1(somatic) | [95] | |
| BRCA1 | 5′UTR (2400 bp) | 6475 | 81 | [90] | |
| BRCA2 | 5′UTR (2000 bp | 6603 | 60 | [90] | |
| BRCA1 | 3′UTR (1561 bp) | 1612 | 7 | [61] | |
| BRCA1 | 3′UTR (1376 bp) | 70 | 2 | [62] | |
| BRCA1 | 3′UTR (1382 bp) | 716 | 5 | [60] | |
| BRCA2 | 3′UTR (902 bp) | 716 | 1 | [60] | |
| B | |||||
| Gene | Variant | Localization | Functional Test | Effect | References |
| BRCA1 | |||||
| BRCA1 | c.-395C>T | Promoter | Luciferase assay | NS | [90] |
| BRCA1 | c.-380G>A | Promoter | Luciferase assay | NS | [58,90] |
| BRCA1 | c.-378C>A | Promoter | Luciferase assay | NS | [90] |
| BRCA1 | c.-362T>G | Promoter | Luciferase assay | up regulation | [58] |
| BRCA1 | c.-359G>T | Promoter | Luciferase assay | NS | [58] |
| BRCA1 | c.-315del | Promoter | Luciferase assay | down regulation | [90] |
| BRCA1 | c.-192T>C | Promoter | Luciferase assay | down regulation | [90] |
| BRCA1 | c.-220C>A | Promoter | Luciferase assay | NS | [90] |
| BRCA1 | c.-264T>G | Promoter | Luciferase assay | NS | [90] |
| BRCA1 | c.-273G>A | Promoter | Luciferase assay | NS | [90] |
| BRCA1 | c.-287C>T | Promoter | Luciferase assay | up regulation | [90] |
| BRCA1 | c.-177C>T | Promoter | Luciferase assay | NS | [58] |
| BRCA1 | c.-130del | Promoter | Luciferase assay | down regulation | [58] |
| BRCA1 | c.-125C>T | Promoter | Luciferase assay | down regulation | [58] |
| BRCA1 | c.-121G>C | Promoter | Luciferase assay | up regulation | [58] |
| BRCA1 | c.-107A>T | Exon 1 | Promoter methylation assays; RNA analysis by RT-PCR | down regulation | [93] |
| BRCA1 | c.-71G>A | Exon 1 | Luciferase assay | NS | [58] |
| BRCA1 | c.-24T>C | Exon 1 | Luciferase assay | NS | [58] |
| BRCA1 | c.-3G>C (+117G>C) | Exon2 | Luciferase assay; RNA translation assay | down regulation | [95] |
| BRCA1 | c.-2A>T (+118A>T) | Exon2 | Luciferase assay; RNA analysis by RT-PCR. Protein analysis by IHC | down regulation | [94] |
| BRCA1 | c.81-3985A>T | Intron 2 | Luciferase assay | up-regulation | [58] |
| BRCA1 | c.81-3980A>G | Intron 2 | Luciferase assay | down regulation | [58] |
| BRCA1 | c.4186-2022C>T | Intron 12 | Luciferase assay | down regulation | [58] |
| BRCA1 | c.*291C>T | 3′UTR | Luciferase assay | up-regulation | [61] |
| BRCA1 | c.*528G>C | 3′UTR | Luciferase assay | down regulation (MDAMB231) up-regulation (MCF7) | [61] |
| BRCA1 | c.*713C>T | 3′UTR | Luciferase assay | up-regulation | [60] |
| BRCA1 | c.*718A>G | 3′UTR | Luciferase assay | down regulation | [61] |
| BRCA1 | c.*750A>G | 3′UTR | Luciferase assay | NS up-regulation | [60,62] |
| BRCA1 | c.*780C>T | 3′UTR | Luciferase assay | up-regulation | [62] |
| BRCA1 | c.*800T>C | 3′UTR | Luciferase assay | NS | [61] |
| BRCA1 | c.*1012A>G | 3′UTR | Luciferase assay | NS | [62] |
| BRCA1 | c.*1139G>T | 3′UTR | Luciferase assay | up-regulation (MDAMB231) down regulation (MCF7) | [61] |
| BRCA1 | c.*1271T>C | 3′UTR | Luciferase assay | down regulation | [61] |
| BRCA1 | c.*1286C>A | 3′UTR | Luciferase assay | down regulation | [62] |
| BRCA1 | c.*1340_1342del | 3′UTR | Luciferase assay | up-regulation | [61] |
| BRCA2 | |||||
| BRCA2 | c.-492C>T | Promoter | Luciferase assay | NS | [68] |
| BRCA2 | c.-467T>G | Promoter | Luciferase assay | up-regulation | [68] |
| BRCA2 | c.-407G>A | Promoter | Luciferase assay | NS | [90] |
| BRCA2 | c.-408T>A | Promoter | Luciferase assay | NS | [90] |
| BRCA2 | c.-296C>T | Promoter | Luciferase assay | down regulation | [58,90] |
| BRCA2 | c.-280_272dup | Promoter | Luciferase assay | up-regulation | [58] |
| BRCA2 | c.-280del | Promoter | Luciferase assay | NS | [90] |
| BRCA2 | c.-273G>T | Promoter | Luciferase assay | NS | [58] |
| BRCA2 | c.-262G>A | Promoter | Luciferase assay | up-regulation | [68] |
| BRCA2 | c.-248G>A | Promoter | Luciferase assay | NS | [68] |
| BRCA2 | c.-220G>T | Exon 1 | Luciferase assay | NS | [58] |
| BRCA2 | c.-218G>A | Exon 1 | Luciferase assay | NS | [58] |
| BRCA2 | c.-213G>T | Exon 1 | Luciferase assay | NS | [58] |
| BRCA2 | c.-200C>T | Exon 1 | Luciferase assay | NS | [90] |
| BRCA2 | c.-197A>C | Exon 1 | Luciferase assay | down regulation | [90] |
| BRCA2 | c.-188C>T (+46C>T) | Exon 1 | Luciferaseassay | NS | [68] |
| BRCA2 | c.-175C>T | Exon 1 | Luciferase assay | NS | [90] |
| BRCA2 | c.-175C>T (+59C>T) | Exon 1 | Luciferase assay | down regulation | [68] |
| BRCA2 | c.-174G>A | Exon 1 | Luciferase assay | up-regulation | [68] |
| BRCA2 | c.-162G>A (+72G>A) | Exon 1 | Luciferase assay | up-regulation | [68] |
| BRCA2 | c.-159T>A | Exon 1 | Luciferase assay | up-regulation | [68] |
| BRCA2 | c.-133T>G | Exon 1 | Luciferase assay | NS | [90] |
| BRCA2 | c.-123G>A | Exon 1 | Luciferase assay | up-regulation | [58] |
| BRCA2 | c.-120G>A | Exon 1 | Luciferase assay | up-regulation | [68] |
| BRCA2 | c.-119A>G | Exon 1 | Luciferase assay | down regulation | [68] |
| BRCA2 | c.-94T>C | Exon 1 | Luciferase assay | up-regulation | [68] |
| BRCA2 | c.-87T>G | Exon 1 | Luciferase assay | NS | [90] |
| BRCA2 | c.-82G>C | Exon 1 | Luciferase assay | NS | [90] |
| BRCA2 | c.-77C>T | Exon 1 | Luciferase assay | down regulation | [68] |
| BRCA2 | c.-63C>T | Exon 1 | Luciferase assay | NS | [68] |
| BRCA2 | c.-52A>G | Exon 1 | Luciferase assay | NS | [58] |
| BRCA2 | c.-52A>G (+182A>G) | Exon 1 | Luciferase assay | NS | [68] |
| BRCA2 | c.*172 G>A | 3′UTR | Luciferase assay | up-regulation (HBL-100) down regulation (MCF7) | [60] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santana dos Santos, E.; Lallemand, F.; Burke, L.; Stoppa-Lyonnet, D.; Brown, M.; Caputo, S.M.; Rouleau, E. Non-Coding Variants in BRCA1 and BRCA2 Genes: Potential Impact on Breast and Ovarian Cancer Predisposition. Cancers 2018, 10, 453. https://doi.org/10.3390/cancers10110453
Santana dos Santos E, Lallemand F, Burke L, Stoppa-Lyonnet D, Brown M, Caputo SM, Rouleau E. Non-Coding Variants in BRCA1 and BRCA2 Genes: Potential Impact on Breast and Ovarian Cancer Predisposition. Cancers. 2018; 10(11):453. https://doi.org/10.3390/cancers10110453
Chicago/Turabian StyleSantana dos Santos, Elizabeth, François Lallemand, Leslie Burke, Dominique Stoppa-Lyonnet, Melissa Brown, Sandrine M. Caputo, and Etienne Rouleau. 2018. "Non-Coding Variants in BRCA1 and BRCA2 Genes: Potential Impact on Breast and Ovarian Cancer Predisposition" Cancers 10, no. 11: 453. https://doi.org/10.3390/cancers10110453
APA StyleSantana dos Santos, E., Lallemand, F., Burke, L., Stoppa-Lyonnet, D., Brown, M., Caputo, S. M., & Rouleau, E. (2018). Non-Coding Variants in BRCA1 and BRCA2 Genes: Potential Impact on Breast and Ovarian Cancer Predisposition. Cancers, 10(11), 453. https://doi.org/10.3390/cancers10110453