Immunoliposomes with Simvastatin as a Potential Therapeutic in Treatment of Breast Cancer Cells Overexpressing HER2—An In Vitro Study

Abstract
1. Introduction
2. Results
2.1. Design of Simvastatin Liposomes: Cholesterol as a Key Factor Limiting the Capacity and Toxicity of Liposomes
2.2. Characterisation of Non-Targeted Liposomes and Immunoliposomes Containing Simvastatin
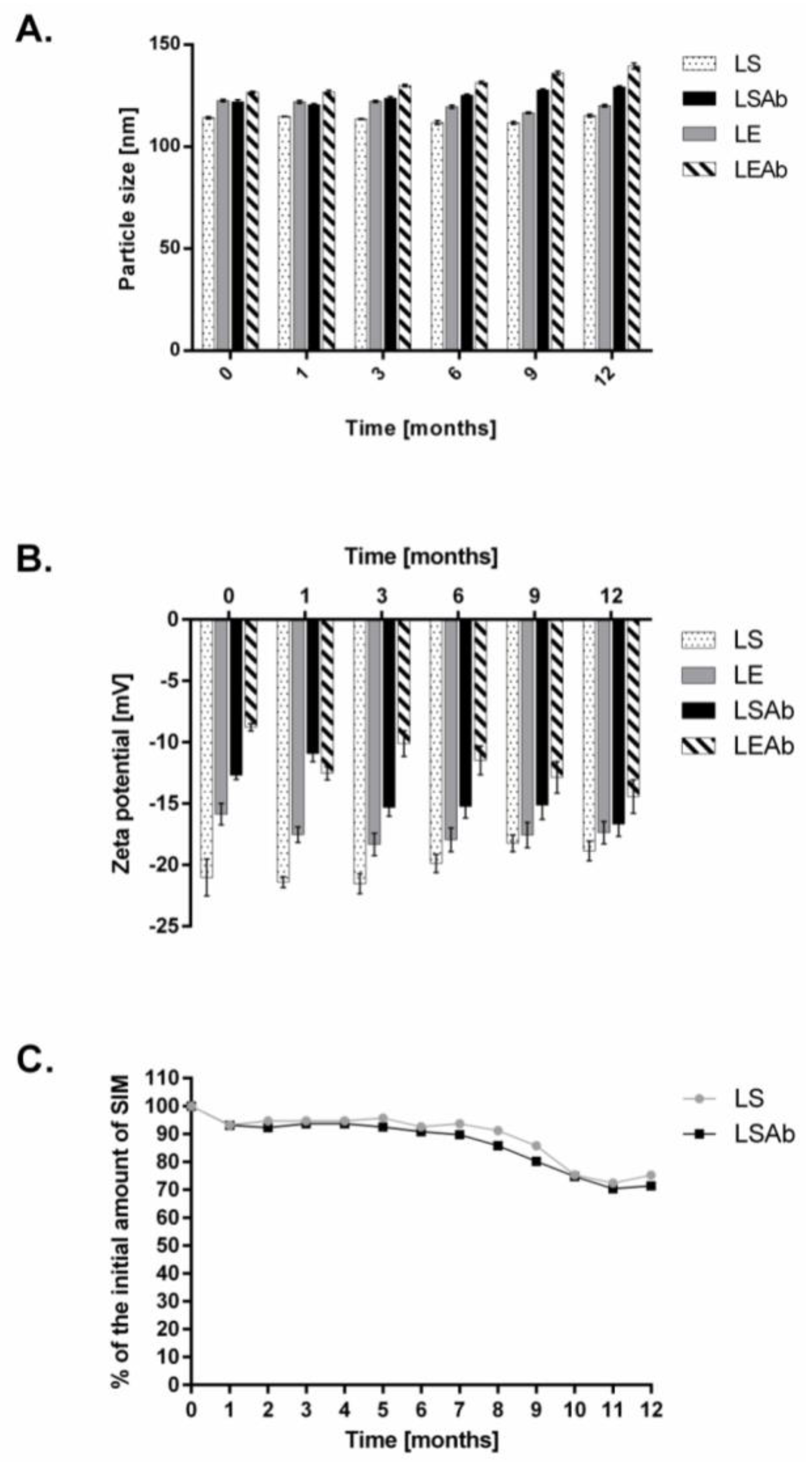
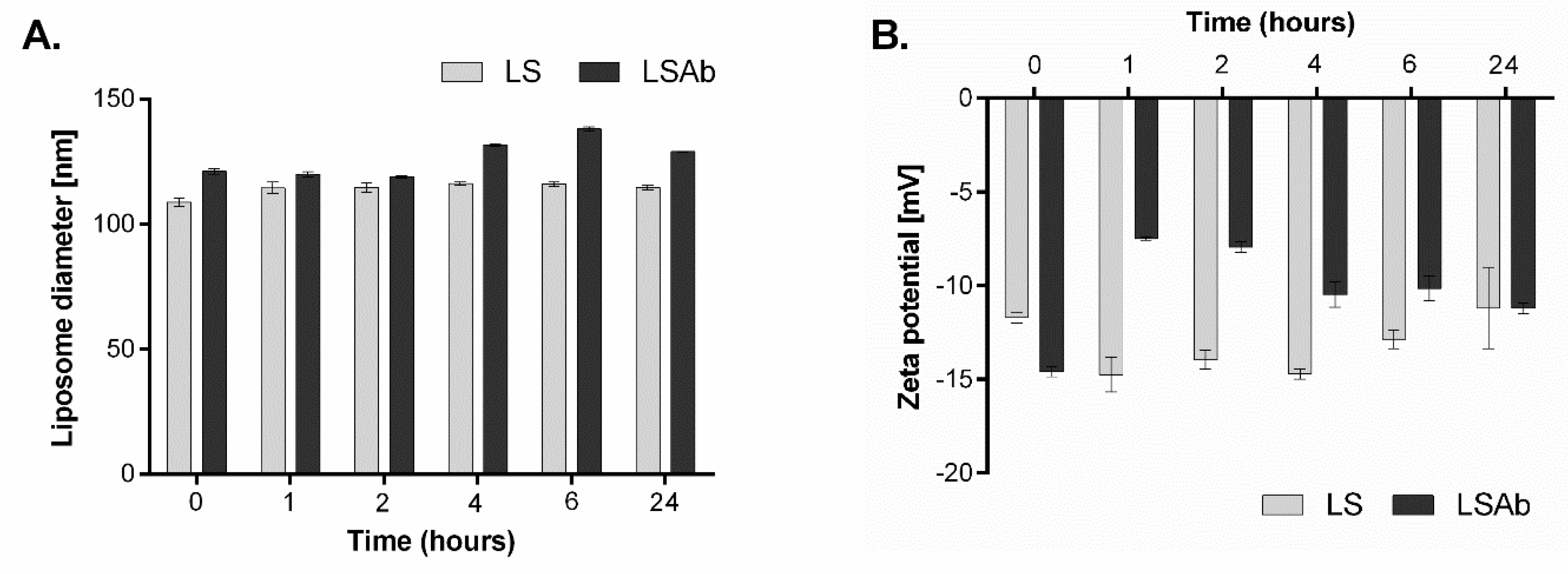
2.3. Stability of Designed Liposomes and Immunoliposomes
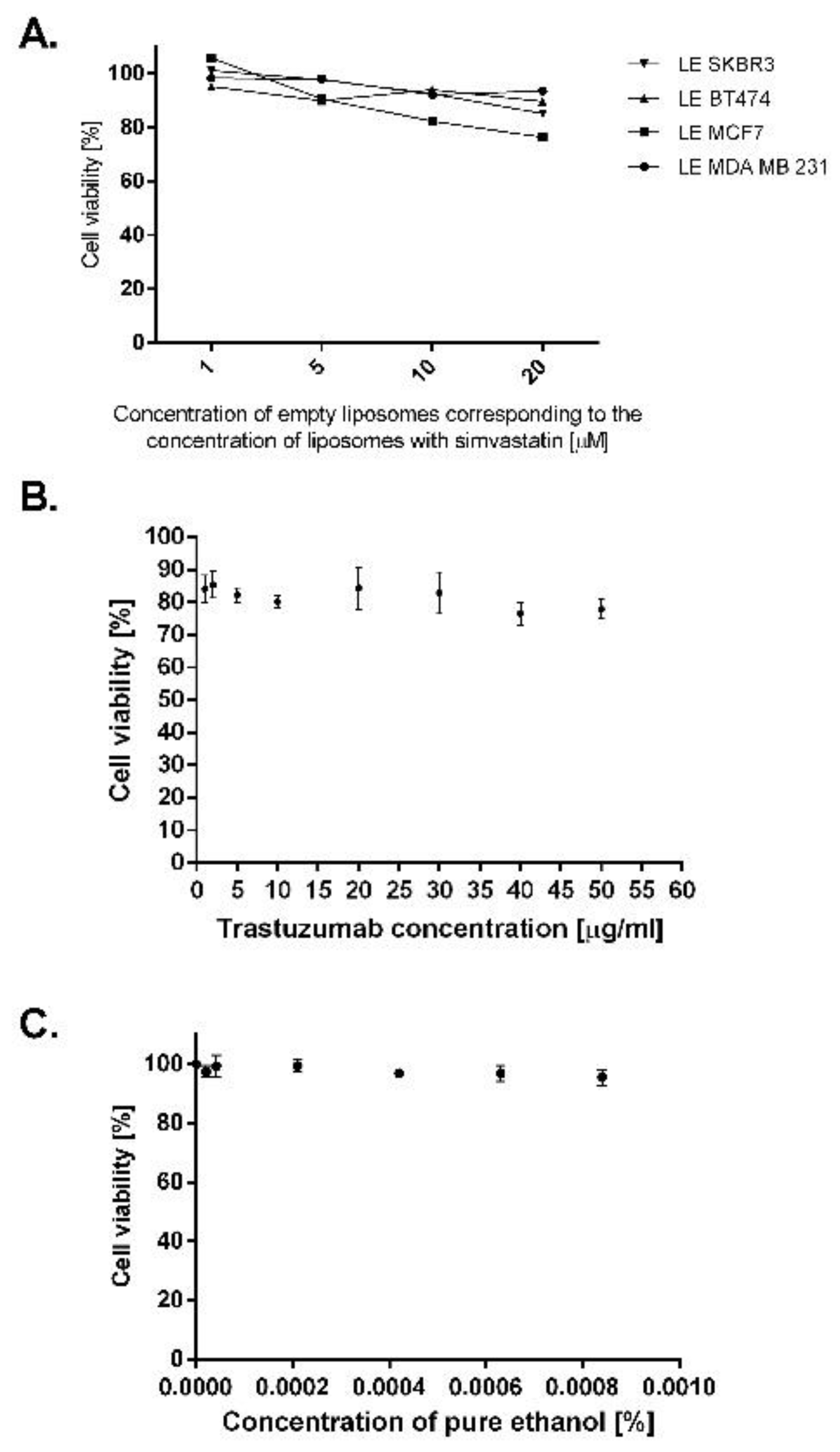
2.4. Breast Cancer Cell Lines Overexpressing EGFR Are Sensitive to Treatment with Liposomal and Immunoliposomal Forms of Simvastatin
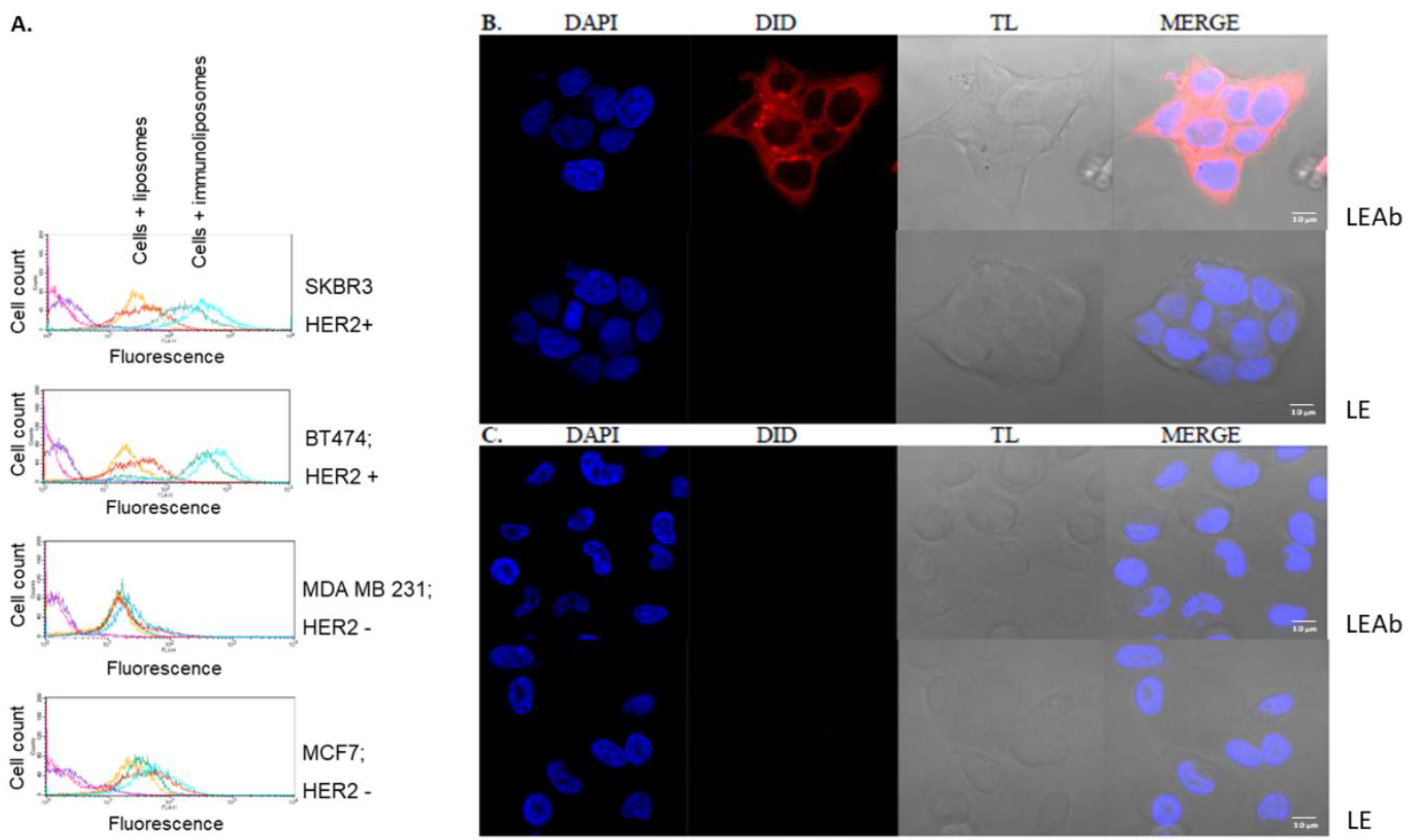
2.5. Simvastatin Immunoliposomes Are Selective towards Breast Cancer Cells Overexpressing HER2
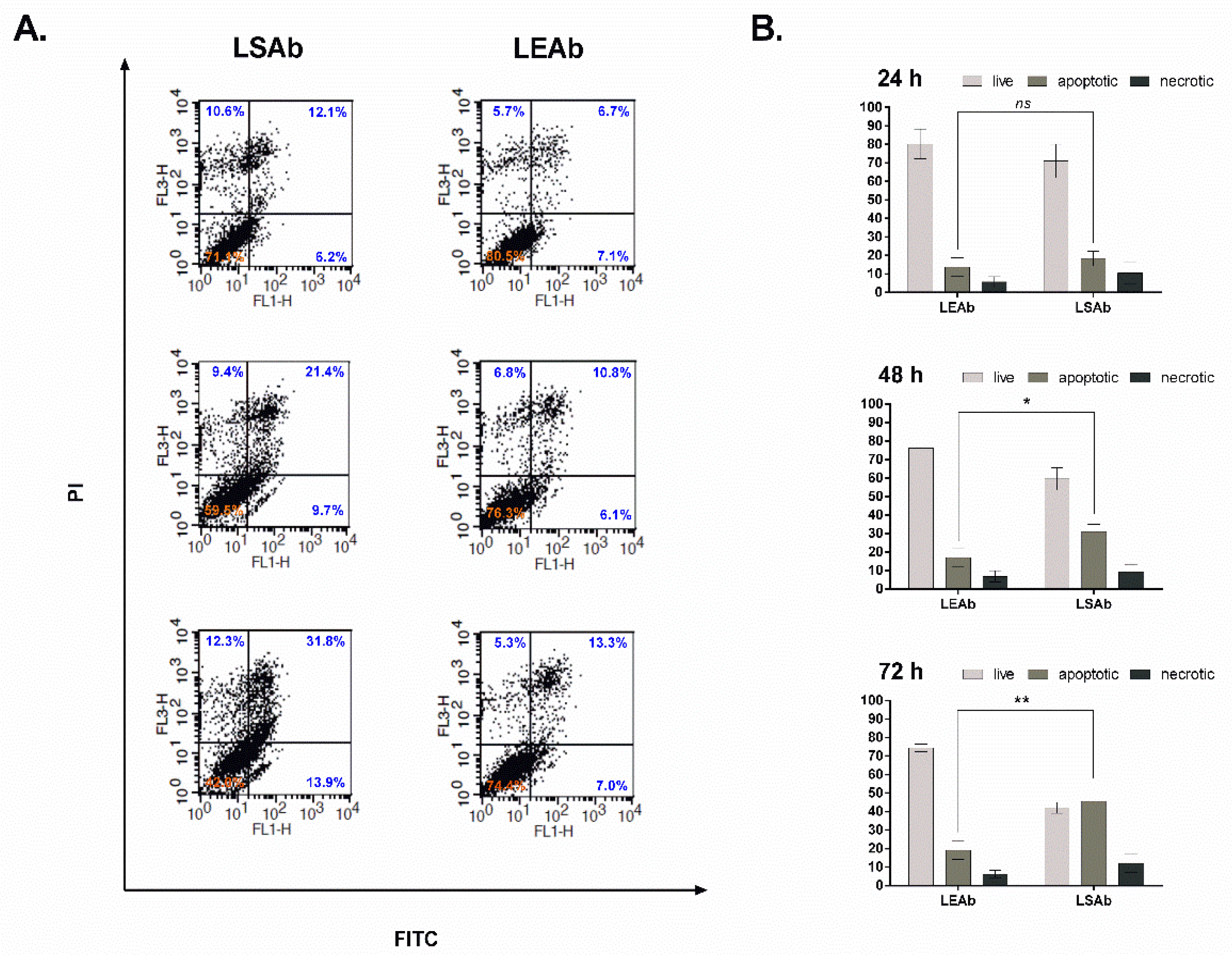
2.6. Immunoliposomal Simvastatin Induces Apoptosis in SKBR3 Cells
2.7. Immunoliposomal Form of Simvastatin Inhibits Signalling Pathways Involving Akt and Erk
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Preparation of Non-Targeted Liposomes with Simvastatin
4.3. Antibody Conjugation to Liposomes
4.4. Particle Size and Zeta Potential Analyses
4.5. Determination of Drug Encapsulation Efficiency and Drug Loading
4.6. Stability of Simvastatin Immunoliposomes
4.7. Cell Culture
4.8. Selectivity of Immunoliposomes towards Breast Cancer Cells Overexpressing HER2
4.9. Cellular Viability Assay
4.10. Determination of Apoptosis
4.11. Signalling of Pathways Involving Akt and Erk
4.12. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Subik, K.; Lee, J.F.; Baxter, L.; Strzepek, T.; Costello, D.; Crowley, P.; Xing, L.; Hung, M.C.; Bonfiglio, T.; Hicks, D.G.; et al. The Expression Patterns of ER, PR, HER2, CK5/6, EGFR, Ki-67 and AR by Immunohistochemical Analysis in Breast Cancer Cell Lines. Breast Cancer 2010, 4, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Goard, C.A.; Chan-Seng-Yue, M.; Mullen, P.J.; Quiroga, A.D.; Wasylishen, A.R.; Clendening, J.W.; Sendorek, D.H.; Haider, S.; Lehner, R.; Boutros, P.C.; et al. Identifying molecular features that distinguish fluvastatin-sensitive breast tumor cells. Breast Cancer Res. Treat. 2014, 143, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; Mcguire, W.L. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987, 235, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Carter, P.; Presta, L.; Gorman, C.M.; Ridgway, J.B.; Henner, D.; Wong, W.L.; Rowland, A.M.; Kotts, C.; Carver, M.E.; Shepard, H.M. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc. Natl. Acad. Sci. USA 1992, 89, 4285–4289. [Google Scholar] [CrossRef] [PubMed]
- Slamon, D.J.; Leyland-Jones, B.; Shak, S.; Fuchs, H.; Paton, V.; Bajamonde, A.; Fleming, T.; Eiermann, W.; Wolter, J.; Pegram, M.; et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N. Engl. J. Med. 2001, 344, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Bangham, A.D.; Standish, M.M.; Miller, N. Cation permeability of phospholipid model membranes: Effect of narcotics. Nature 1965, 208, 1295–1297. [Google Scholar] [CrossRef] [PubMed]
- Bangham, A.D.; Standish, M.M.; Watkins, J.C. Diffusion of univalent ions across the lamellae of swollen phospholipids. J. Mol. Biol. 1965, 13, 238–252. [Google Scholar] [CrossRef]
- Allen, T.M.; Cullis, P.R. Liposomal drug delivery systems: From concept to clinical applications. Adv. Drug Deliv. Rev. 2013, 65, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Goren, D.; Horowitz, A.T.; Zalipsky, S.; Woodle, M.C.; Yarden, Y.; Gabizon, A. Targeting of stealth liposomes to erbB-2 (Her/2) receptor: In vitro and in vivo studies. Br. J. Cancer 1996, 74, 1749–1756. [Google Scholar] [CrossRef] [PubMed]
- Papahadjopoulos, D.; Allen, T.M.; Gabizon, A.; Mayhew, E.; Matthay, K.; Huang, S.K.; Lee, K.D.; Woodle, M.C.; Lasic, D.D.; Redemann, C.; et al. Sterically stabilized liposomes: Improvements in pharmacokinetics and antitumor therapeutic efficacy. Proc. Natl. Acad. Sci. USA 1991, 88, 11460–11464. [Google Scholar] [CrossRef] [PubMed]
- Toporkiewicz, M.; Meissner, J.; Matusewicz, L.; Czogalla, A.; Sikorski, A.F. Toward a magic or imaginary bullet? Ligands for drug targeting to cancer cells: Principles, hopes, and challenges. Int. J. Nanomed. 2015, 10, 1399–1414. [Google Scholar]
- Hu, M.; Cheung, B.M.; Tomlinson, B. Safety of statins: An update. Ther. Adv. Drug Saf. 2015, 3, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Schachter, M. Chemical, pharmacokinetic and pharmacodynamic properties of statins: An update. Fundam. Clin. Pharmacol. 2005, 19, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Yanamandra, S.; Venkatesan, N.; Kadajji, V.G.; Wang, Z.; Issar, M.; Betageri, G.V. Proliposomes as a drug delivery system to decrease the hepatic first-pass metabolism: Case study using a model drug. Eur. J. Pharm. Sci. 2014, 64, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Afergan, E.; Ben David, M.; Epstein, H.; Koroukhov, N.; Gilhar, D.; Rohekar, K.; Danenberg, H.D. Golomb G Liposomal simvastatin attenuates neointimal hyperplasia in rats. AAPS J. 2010, 12, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Alupei, M.C.; Licarete, E.; Patras, L.; Banciu, M. Liposomal simvastatin inhibits tumor growth via targeting tumor-associated macrophages-mediated oxidative stress. Cancer Lett. 2015, 356, 946–952. [Google Scholar] [CrossRef] [PubMed]
- Coimbra, M.; Banciu, M.; Fens, M.H.; De Smet, L.; Cabaj, M.; Metselaar, J.M.; Storm, G.; Schiffelers, R.M. Liposomal pravastatin inhibits tumor growth by targeting cancer-related inflammation. J. Control Release 2010, 148, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Porfire, A.; Tomuta, I.; Muntean, D.; Luca, L.; Licarete, E.; Alupei, M.C.; Achim, M.; Vlase, L.; Banciu, M. Optimizing long-circulating liposomes for delivery of simvastatin to C26 colon carcinoma cells. J. Liposome Res. 2015, 25, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Tuerdi, N.; Xu, L.; Zhu, B.; Chen, C.; Cao, Y.; Wang, Y.; Zhang, Q.; Li, Z.; Qi, R. Preventive effects of simvastatin nanoliposome on isoproterenol-induced cardiac remodeling in mice. Nanomedicine 2016, 12, 1899–1907. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lee, H.; Huesca, M.; Young, A.; Allen, C. Liposome formulation of a novel hydrophobic aryl-imidazole compound for anti-cancer therapy. Cancer Chemother. Pharmacol. 2006, 58, 306–318. [Google Scholar] [CrossRef] [PubMed]
- Mahmud, M.; Piwoni, A.; Filipczak, N.; Janicka, M.; Gubernator, J. Long-Circulating Curcumin-Loaded Liposome Formulations with High Incorporation Efficiency, Stability and Anticancer Activity towards Pancreatic Adenocarcinoma Cell Lines In Vitro. PLoS ONE 2016, 11, e0167787. [Google Scholar] [CrossRef] [PubMed]
- Meissner, J.M.; Toporkiewicz, M.; Czogalla, A.; Matusewicz, L.; Kuliczkowski, K.; Sikorski, A.F. Novel antisense therapeutics delivery systems: In vitro and in vivo studies of liposomes targeted with anti-CD20 antibody. J. Control Release 2015, 220, 515–528. [Google Scholar] [CrossRef] [PubMed]
- Catania, A.; Barrajon-Catalan, E.; Nicolosi, S.; Cicirata, F.; Micol, V. Immunoliposome encapsulation increases cytotoxic activity and selectivity of curcumin and resveratrol against HER2 overexpressing human breast cancer cells. Breast Cancer Res. Treat 2013, 141, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Matusewicz, L.; Ligas, J.; Sikorski, A.F. Liposomal Forms of Statins; Unpublished Data; Department of Cytobiochemistry, Faculty of Biotechnology, University of Wroclaw: Wroclaw, Poland, 2016. [Google Scholar]
- Matusewicz, L.; Podkalicka, J.; Sikorski, A.F. Selectivity of Simvastatin Immunoliposomes; Unpublished Data; Department of Cytobiochemistry, Faculty of Biotechnology, University of Wroclaw: Wroclaw, Poland, 2015. [Google Scholar]
- Wang, T.; Seah, S.; Loh, X.; Chan, C.W.; Hartman, M.; Goh, B.C.; Lee, S.C. Simvastatin-induced breast cancer cell death and deactivation of PI3K/Akt and MAPK/ERK signalling are reversed by metabolic products of the mevalonate pathway. Oncotarget 2016, 7, 2532–2544. [Google Scholar] [CrossRef] [PubMed]
- Barrajon-Catalan, E.; Menendez-Gutierrez, M.P.; Falco, A.; Carrato, A.; Saceda, M.; Micol, V. Selective death of human breast cancer cells by lytic immunoliposomes: Correlation with their HER2 expression level. Cancer Lett. 2010, 290, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Matusewicz, L.; Meissner, J.; Toporkiewicz, M.; Sikorski, A.F. The effect of statins on cancer cells—Review. Tumour Biol. 2015, 36, 4889–4904. [Google Scholar] [CrossRef] [PubMed]
- Luput, L.; Licarete, E.; Drotar, D.M.; Nagy, A.L.; Sesarman, A.; Patras, L.; Rauca, V.F.; Porfire, A.; Muntean, D.; Achim, M.; et al. In Vivo Double Targeting of C26 Colon Carcinoma Cells and Microenvironmental Protumor Processes Using Liposomal Simvastatin. J. Cancer 2018, 9, 440–449. [Google Scholar] [CrossRef] [PubMed]
- Budman, D.R.; Tai, J.; Calabro, A. Fluvastatin enhancement of trastuzumab and classical cytotoxic agents in defined breast cancer cell lines in vitro. Breast Cancer Res. Treat 2007, 104, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Funakoshi, Y.; Iwao, Y.; Noguchi, S.; Itai, S. Effect of Alkyl Chain Length and Unsaturation of the Phospholipid on the Physicochemical Properties of Lipid Nanoparticles. Chem. Pharm. Bull. 2015, 63, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Grzybek, M.; Stebelska, K.; Wyrozumska, P.; Grieb, P.; Langner, M.; Jaszewski, A.; Jezierski, A.; Sikorski, A.F. ESR and monolayer study of the localization of coenzyme Q10 in artificial membranes. Gen. Physiol. Biophys. 2005, 24, 449–460. [Google Scholar] [PubMed]
- Bozzuto, G.; Molinari, A. Liposomes as nanomedical devices. Int. J. Nanomed. 2015, 10, 975–999. [Google Scholar] [CrossRef] [PubMed]
- Palchetti, S.; Colapicchioni, V.; Digiacomo, L.; Caracciolo, G.; Pozzi, D.; Capriotti, A.L.; La Barbera, G.; Lagana, A. The protein corona of circulating PEGylated liposomes. Biochim. Biophys. Acta 2016, 1858, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Drummond, D.C.; Meyer, O.; Hong, K.; Kirpotin, D.B.; Papahadjopoulos, D. Optimizing liposomes for delivery of chemotherapeutic agents to solid tumors. Pharmacol. Rev. 1999, 51, 691–743. [Google Scholar] [PubMed]
- Shen, S.; Wu, Y.; Liu, Y.; Wu, D. High drug-loading nanomedicines: Progress, current status, and prospects. Int. J. Nanomed. 2017, 12, 4085–4109. [Google Scholar] [CrossRef] [PubMed]
- Llaverias, G.; Danilo, C.; Mercier, I.; Daumer, K.; Capozza, F.; Williams, T.M.; Sotgia, F.; Lisanti, M.P.; Frank, P.G. Role of cholesterol in the development and progression of breast cancer. Am. J. Pathol. 2011, 178, 402–412. [Google Scholar] [CrossRef] [PubMed]
- Ahern, T.P.; Lash, T.L.; Damkier, P.; Christiansen, P.M.; Cronin-Fenton, D.P. Statins and breast cancer prognosis: Evidence and opportunities. Lancet. Oncol. 2014, 15, e461–e468. [Google Scholar] [CrossRef]
- Murtola, T.J.; Visvanathan, K.; Artama, M.; Vainio, H.; Pukkala, E. Statin use and breast cancer survival: A nationwide cohort study from Finland. PLoS ONE 2014, 9, e110231. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.C.; Park, M.J.; Ye, S.K.; Kim, C.W.; Kim, Y.N. Elevated levels of cholesterol-rich lipid rafts in cancer cells are correlated with apoptosis sensitivity induced by cholesterol-depleting agents. Am. J. Pathol. 2006, 168, 1107–1118; quiz 1404-1105. [Google Scholar] [CrossRef] [PubMed]
- Hryniewicz-Jankowska, A.; Augoff, K.; Biernatowska, A.; Podkalicka, J.; Sikorski, A.F. Membrane rafts as a novel target in cancer therapy. Biochim. Biophys. Acta 2014, 1845, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Podkalicka, J.; Biernatowska, A.; Olszewska, P.; Tabaczar, S.; Sikorski, A.F. The microdomain-organizing protein MPP1 is required for insulin-stimulated activation of H.-Ras. Oncotarget 2018, 9, 18410–18421. [Google Scholar] [CrossRef] [PubMed]
- Sikorski, A.F.; Podkalicka, J.; Jones, W.; Biernatowska, A. Membrane rafts in the erythrocyte membrane: A novel role of MPP1p55. Adv. Exp. Med. Biol. 2015, 2015 842, 61–78. [Google Scholar]
- Campbell, M.J.; Esserman, L.J.; Zhou, Y.; Shoemaker, M.; Lobo, M.; Borman, E.; Baehner, F.; Kumar, A.S.; Adduci, K.; Marx, C.; et al. Breast cancer growth prevention by statins. Cancer Res. 2006, 66, 8707–8714. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Kim, Y.S.; Han, W.; Ryu, H.S.; Chang, J.M.; Cho, N.; Moon, W.K. Tumor growth rate of invasive breast cancers during wait times for surgery assessed by ultrasonography. Medicine 2016, 95, e4874. [Google Scholar] [CrossRef] [PubMed]
- Kimbung, S.; Johansson, I.; Danielsson, A.; Veerla, S.; Egyhazi Brage, S.; Frostvik Stolt, M.; Skoog, L.; Carlsson, L.; Einbeigi, Z.; Lidbrink, E.; et al. Transcriptional Profiling of Breast Cancer Metastases Identifies Liver Metastasis-Selective Genes Associated with Adverse Outcome in Luminal A Primary Breast Cancer. Clin. Cancer Res. 2016, 22, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Nelson, E.R.; Wardell, S.E.; Jasper, J.S.; Park, S.; Suchindran, S.; Howe, M.K.; Carver, N.J.; Pillai, R.V.; Sullivan, P.M.; Sondhi, V.; et al. 27-Hydroxycholesterol links hypercholesterolemia and breast cancer pathophysiology. Science 2013, 342, 1094–1098. [Google Scholar] [CrossRef] [PubMed]
- Kochuparambil, S.T.; Al-Husein, B.; Goc, A.; Soliman, S.; Somanath, P.R. Anticancer efficacy of simvastatin on prostate cancer cells and tumor xenografts is associated with inhibition of Akt and reduced prostate-specific antigen expression. J. Pharmacol. Exp. Ther. 2011, 336, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Mullen, P.J.; Zahno, A.; Lindinger, P.; Maseneni, S.; Felser, A.; Krahenbuhl, S.; Brecht, K. Susceptibility to simvastatin-induced toxicity is partly determined by mitochondrial respiration and phosphorylation state of Akt. Biochim. Biophys. Acta 2011, 1813, 2079–2087. [Google Scholar] [CrossRef] [PubMed]
- Irwin, M.E.; Bohin, N.; Boerner, J.L. Src family kinases mediate epidermal growth factor receptor signaling from lipid rafts in breast cancer cells. Cancer Biol. Ther. 2011, 12, 718–726. [Google Scholar] [CrossRef] [PubMed]
- Irwin, M.E.; Mueller, K.L.; Bohin, N.; Ge, Y.; Boerner, J.L. Lipid raft localization of EGFR alters the response of cancer cells to the EGFR tyrosine kinase inhibitor gefitinib. J. Cell Physiol. 2011, 226, 2316–2328. [Google Scholar] [CrossRef] [PubMed]
- Henjes, F.; Bender, C.; Von Der Heyde, S.; Braun, L.; Mannsperger, H.A.; Schmidt, C.; Wiemann, S.; Hasmann, M.; Aulmann, S.; Beissbarth, T.; et al. Strong EGFR signaling in cell line models of ERBB2-amplified breast cancer attenuates response towards ERBB2-targeting drugs. Oncogenesis 2012, 1, e16. [Google Scholar] [CrossRef] [PubMed]
- Ritter, C.A.; Perez-Torres, M.; Rinehart, C.; Guix, M.; Dugger, T.; Engelman, J.A.; Arteaga, C.L. Human breast cancer cells selected for resistance to trastuzumab in vivo overexpress epidermal growth factor receptor and ErbB ligands and remain dependent on the ErbB receptor network. Clin. Cancer Res. 2007, 13, 4909–4919. [Google Scholar] [CrossRef] [PubMed]
- Tseng, P.H.; Wang, Y.C.; Weng, S.C.; Weng, J.R.; Chen, C.S.; Brueggemeier, R.W.; Shapiro, C.L.; Chen, C.Y.; Dunn, S.E.; Pollak, M. Overcoming trastuzumab resistance in HER2-overexpressing breast cancer cells by using a novel celecoxib-derived phosphoinositide-dependent kinase-1 inhibitor. Mol. Pharmacol. 2006, 70, 1534–1541. [Google Scholar] [CrossRef] [PubMed]
- Arayne, M.S.; Sultana, N.; Siddiqui, F.A.; Zuberi, M.H.; Mirza, A.Z. Spectrophotometric methods for the simultaneous analysis of meclezine hydrochloride and pyridoxine hydrochloride in bulk drug and pharmaceutical formulations. Pak. J. Pharm. Sci. 2007, 20, 149–156. [Google Scholar] [PubMed]
- Rouser, G.; Fkeischer, S.; Yamamoto, A. Two dimensional then layer chromatographic separation of polar lipids and determination of phospholipids by phosphorus analysis of spots. Lipids 1970, 5, 494–496. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation No. | Formulation with mol % | Chol:SIM Molar Ratio | Stability (days) | EE (%) |
|---|---|---|---|---|
| 1 | HSPC:DSPE-PEG:chol:SIM 8.0:0.5:1.0:1.0 | 1:1 | 30 | 50.3 |
| 2 | HSPC:DSPE-PEG:SIM 8.5:0.6:1.5 | 0:1 | 45 | 40.8 |
| 3 | HSPC:DSPE-PEG:chol:SIM 8.5:0.6:0.5:1.0 | 0.5:1 | 40 | 51.3 |
| 4 | HSPC:DSPC:DSPE-PEG:chol:SIM 6.9:1.6:0.5:0.5:1.0 | 0.5:1 | 240 * | 67.8 |
| Lipid Composition of Empty Liposomes No. 1 | Lipid Composition of Empty Liposomes No. 2 | ||
|---|---|---|---|
| Component | Content (mol %) | Component | Content (mol %) |
| HSPC | 75.15 | HSPC | 65.65 |
| DSPC | 15 | DSPC | 15 |
| DSPE-Peg | 5.35 | DSPE-Peg | 5.35 |
| Cholesterol | 4.5 | Cholesterol | 14 |
| Formulation | Size (nm) | PDI | Zeta Potential (mV) | EE (%) | DL (%) |
|---|---|---|---|---|---|
| Non-targeted empty liposomes | 122.9 ± 1.2 | 0.046 ± 0.010 | −16.7 ± 1.2 | - | - |
| Non-targeted liposomes containing simvastatin | 117.3 ± 1.6 | 0.037 ± 0.011 | −17.6 ± 3.7 | 63.3 ± 12.4 | 6.8 ± 1.1 |
| Empty immunoliposomes | 127.6 ± 2.9 | 0.039 ± 0.014 | −9.9 ± 2.7 | - | - |
| Immunoliposomes containing simvastatin | 123.1 ± 3.1 | 0.054 ± 0.015 | −11.1 ± 3.5 | 64.6 ± 9.7 | 6.8 ± 1.4 |
| Caption | MDA MB 231 | MCF7 | BT474 | SKBR3 |
|---|---|---|---|---|
| Simvastatin | 2.3 µM | 56.2 µM | 194 µM | 6.8 µM |
| Liposomal simvastatin | 8.6 µM | 44.6 µM | 76.9 µM | 7.1 µM |
| Immunoliposomal simvastatin | 6.9 µM | 49.8 µM | 25.2 µM | 6.5 µM |
| Feature | MDA MB 231 and SKBR3 | MCF7 and BT474 |
|---|---|---|
| External cholesterol requirement (see Figure 1) | low | high |
| Susceptibility to statin treatment [2,44], (see Table 4) | high | low |
| ER status [1] | negative | positive |
| Tumour growth rate [45] | high | low |
| Expression of “signature” genes, among others HMGCR [46]. | lower | Higher * |
| Transcription of genes involved in cholesterol synthesis after statin treatment | stays at the same level (lower levels of cholesterol required to survive) | increase (higher levels of cholesterol required to survive **) |
| Derived from | metastatic site | metastatic site/mammary gland |
| EGFR expression [1] | high | very low |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matusewicz, L.; Podkalicka, J.; Sikorski, A.F. Immunoliposomes with Simvastatin as a Potential Therapeutic in Treatment of Breast Cancer Cells Overexpressing HER2—An In Vitro Study. Cancers 2018, 10, 418. https://doi.org/10.3390/cancers10110418
Matusewicz L, Podkalicka J, Sikorski AF. Immunoliposomes with Simvastatin as a Potential Therapeutic in Treatment of Breast Cancer Cells Overexpressing HER2—An In Vitro Study. Cancers. 2018; 10(11):418. https://doi.org/10.3390/cancers10110418
Chicago/Turabian StyleMatusewicz, Lucyna, Joanna Podkalicka, and Aleksander F. Sikorski. 2018. "Immunoliposomes with Simvastatin as a Potential Therapeutic in Treatment of Breast Cancer Cells Overexpressing HER2—An In Vitro Study" Cancers 10, no. 11: 418. https://doi.org/10.3390/cancers10110418
APA StyleMatusewicz, L., Podkalicka, J., & Sikorski, A. F. (2018). Immunoliposomes with Simvastatin as a Potential Therapeutic in Treatment of Breast Cancer Cells Overexpressing HER2—An In Vitro Study. Cancers, 10(11), 418. https://doi.org/10.3390/cancers10110418