Abstract
Radiation and certain anticancer drugs damage DNA, resulting in apoptosis induction in cancer cells. Currently, the major limitations on the efficacy of such therapies are development of resistance and adverse side effects. Sensitization is an important strategy for increasing therapeutic efficacy while minimizing adverse effects. In this manuscript, we review possible sensitization strategies for radiation and anticancer drugs that cause DNA damage, focusing especially on modulation of damage repair pathways and the associated reactions.
1. Introduction
Radiation and anticancer drugs that damage DNA were developed many years ago, and are still widely used for cancer therapy [1,2]. These methods achieve their clinical efficacy by promoting the induction of apoptosis in response to DNA damage and cellular stress [3,4,5,6]. A major problem that arises when using such anticancer drugs is development of resistance, which causes treatment to fail [7,8,9,10]. Other major problems include side effects, in which toxicity in a non-targeted tissue limits the tolerable dosage, thereby decreasing the therapeutic efficacy and leading, ultimately, to recurrence [11,12]. The existence of these problems emphasizes the importance of using sensitizers to efficiently induce cancer cell death [11]. Sensitization strategies include combination therapies with multiple drugs, which can achieve synergistic induction of apoptosis in cancer cells.
One important strategy for sensitizing cancer cells to radiation or DNA-damaging drugs is modulation of DNA repair pathways. For example, susceptibility to the DNA-damaging agent cisplatin is higher in cells harboring mutations in BRCA1, BRCA2, and Rad51, which cause deficiencies in homologous recombination (HR) [13,14]. In fact, even in such HR-defective backgrounds, damaged cells exhibit normal checkpoint responses [14], allowing them to induce apoptosis when they sense DNA damage. Such an effect could be produced by simultaneous administration of multiple drugs, as when a poly (ADP-ribose) polymerase (PARP) inhibitor is used to sensitize cells to the DNA methylation agent temozolomide [15,16,17].
In this manuscript, we review recently developed methods for efficiently inducing cancer cell death, focusing on sensitization strategies for chemotherapy with camptothecin (CPT) and radiotherapy. Many of these strategies are based on the modulation of DNA repair pathways. In addition, we draw special attention to mechanistic insights.
2. PARP Inhibitor as a Potential Sensitizer to Top1 Inhibitor
2.1. Top1 Inhibitor Treatment in the Presence of PARP Inhibitor
Topoisomerase 1 (Top 1) is an enzyme that cuts one strand of the DNA duplex and religates the broken ends to relax DNA supercoiling stress, which often arises when DNA or RNA polymerases are operating [18,19] (Figure 1A). CPT is a naturally occurring Top1 inhibitor isolated from Camptotheca acuminate [20]. Derivatives of CPT, such as topotecan and irinotecan, are widely used for cancer chemotherapy [21]. Multiple studies show that PARP inhibitors are potential sensitizers for chemotherapy with CPT, based on the fundamental observation that induction of apoptosis by CPT in vitro is stronger when cells are simultaneously treated with PARP inhibitor [22,23]. To efficiently induce the desired therapeutic outcomes while minimizing side effects, it is important to carefully determine how sensitization is achieved. Recent work showed that, when a PARP inhibitor is administered simultaneously with CPT or its derivatives, multiple reaction steps are modulated [24,25] (Figure 1B,C).
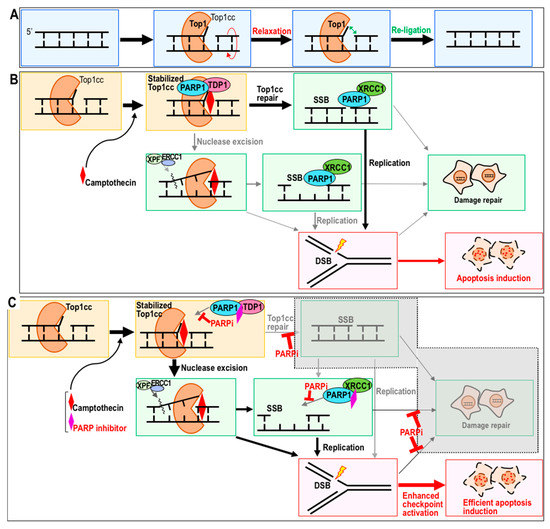
Figure 1.
Model of the topoisomerase 1 reaction and its inhibition by camptothecin (CPT) and poly (ADP-ribose) polymerase (PARP) inhibitor. (A,B) Top1 cuts a single strand of DNA to relax super-coiled DNA stress (A). CPT blocks the ligation step and, hence, induces toxicity during the subsequent S phase in association with replication stress (B). (C) PARP inhibitor sensitizes the cell to CPT by blocking multiple steps of the repair pathway. In this cellular background, apoptosis is induced more efficiently.
CPT (or its derivatives) binds to the Top1–DNA cleavage complex (Top1cc) and inhibits the religation step (Figure 1B). Top1cc is trapped and stabilized by CPT, as well as by endogenous DNA lesions, including a basic sites, mismatches, oxidized bases, and nicks [26,27]. Therefore, Top1cc, like the intermediates of its repair process, is a cause of DNA double-strand break (DSB) formation, mainly during replication stress arising during the subsequent S phase (Figure 1B). Removal of Top1cc can be mediated by either PARP–TDP1 (tyrosyl-DNA phosphodiesterase 1) complexes or the XPF (xeroderma pigmentosum complementation group F)-ERCC1 (Excision repair cross-complementing group 1) endonuclease [28,29] (Figure 1B); consequently, when CPT is administered in the presence of PARP inhibitor, one of the major pathways is blocked.
In the presence of PARP inhibitor olaparib, Top1cc stably accumulates after CPT treatment [30] (Figure 1C). Moreover, the rate of DSB formation caused by CPT treatment is dramatically elevated in the presence of PARP inhibitor. Although replication stress-associated DSBs caused by CPT are primarily targeted by HR, induction of HR is suppressed in the presence of PARP inhibitor [25]. In addition, microhomology-mediated end joining (MMEJ) is also blocked by PARP inhibitor [31]. Therefore, non-homologous end joining (NHEJ) is the only repair pathway available to the cell under these conditions. The checkpoint response is much more effectively activated in cells treated with CPT and PARP inhibitor together, than in cells treated with CPT alone; consequently, dual treatment leads to more effective induction of apoptosis [32,33]. Thus, PARP inhibition causes multiple effects, probably because PARP1 and 2 mediate multiple repair pathways, including base excision repair, MMEJ, and HR [34,35,36].
2.2. Sensitization to CPT by PARP Inhibitor
Given that PARP inhibitors cause multiple effects in CPT-treated cells, it is important to determine which of these effects is critical for sensitization. Recent studies showed that the PARP inhibitor ABT-888 (veliparib) increases CPT-induced cytotoxicity by mediating DSB accumulation, without increasing the level of Top1cc [29]. This implies that CPT sensitization by PARP inhibitor is correlated with DSB accumulation, but not directly associated with stabilization or accumulation of Top1cc. In mechanistic terms, sensitization could be mediated by promotion of the associated checkpoint response, leading to more effective induction of apoptosis [37]. DSBs caused by CPT in the presence of PARP inhibitor are targeted by NHEJ factors. In particular, enlargement of γH2AX/p-ATM foci are often observed in association with heightened damage checkpoint signaling, resulting in more efficient induction of apoptosis (Figure 2). By contrast, these features are not effectively activated during HR, which is usually triggered when CPT is administered alone. Given that apoptosis is induced as a consequence of damage checkpoint signaling [38,39,40], it is reasonable to expect that apoptosis would be strongly induced when checkpoint signaling is strongly activated.
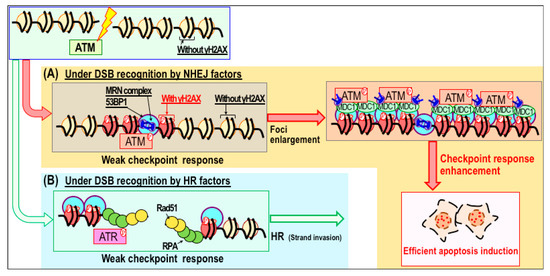
Figure 2.
Model of checkpoint response enhancement through modulation of DNA repair pathways. (A,B) In response to double-strand breaks (DSBs), γH2AX/53BP1 foci form immediately, and are subsequently enlarged in association when the damage checkpoint response is stimulated (A). Under these conditions, repair factors associated with non-homologous end joining (NHEJ) accumulate at DSB sites. Stimulation of the checkpoint response increases the efficiency of apoptosis induction. By contrast, DSBs recognized by homologous recombination (HR) factors are usually not associated with the enlargement of γH2AX foci or stimulation of the damage checkpoint response (B).
In support of this hypothesis, synthetic lethality is induced by pharmacologic inhibition of PARP1/2 in HR-defective cancer cells. It is well established that the PARP inhibitor olaparib (or veliparib) selectively kills BRCA1/2-mutated breast and ovarian cancers [22,23,41,42]. In the presence of PARP inhibitor, these cells spontaneously accumulate DSBs. Since those DSBs are not efficiently repaired by HR or MMEJ (due to the presence of the BRCA1/2 mutation and inhibition of PARP), NHEJ is the only pathway available to repair those DSBs [43]. However, despite having functional NHEJ, cells in this context undergo apoptosis rather than repair, analogous to the situation in cells treated with CPT in the presence of PARP inhibitor (Figure 2).
2.3. Potential Combination Therapy with CPT and a PARP Inhibitor as a Sensitizer
Although combination treatment with CPT (or its derivatives) and olaparib effectively induces cancer cell killing in vitro, a phase I study concluded that this combination is not suitable for clinical use, due to dose-limiting adverse effects, causing the maximum tolerated dose to be subtherapeutic [44]. The main dose-limiting adverse effects were neutropenia and thrombocytopenia, as previously reported for topotecan treatment [45], but these toxicities were observed at substantially lower doses of both drugs [44]. Thus, combination therapy with CPT (or its derivatives) and olaparib has, thus far, failed as a strategy for cancer chemotherapy. However, a series of studies using this combination demonstrated that modulation of repair pathways is conceptually useful as a strategy for efficient induction of apoptosis when cells are treated with DNA-damaging agents.
3. Radiation Therapy and Its Sensitizers
3.1. Radiosensitizers and Their Clinical Use
Clinical trials of radiosensitizers for various cancers have been reported; these trials were based on improvements in killing efficiency in vitro [46,47,48,49]. Examples of increased killing efficiency include radiosensitization of glioblastoma using temozolomide (DNA alkylating agent) [50], prostate cancer using gefitinib (EGFR inhibitor) [51], and non-small-cell lung cancer (NSCLC) using paclitaxel (mitotic inhibitor) [46,52,53]. However, combination therapy has some limitations; for example, elderly NSCLC patients cannot tolerate standard chemoradiation regimens [46]. Survival rates after combined use of radiotherapy plus gefitinib are better that those after radiation therapy only; the most common dose-limiting toxicity is a grade 3 to 4 increase in transaminase activity [51]. Nevertheless, it is important to gain mechanistic insight to improve the therapeutic efficiency of combined therapy in general.
3.2. Sensitization to Radiation Therapy through Modulation of Repair Pathways
Radiation exposure primarily causes DNA damage. Therefore, as with anticancer drugs that cause DNA damage, its therapeutic effects are mainly due to induction of apoptosis in response to DSBs. One strategy for sensitizing cancer cells to radiation therapy is modulation of DNA repair pathways. For example, the chemotherapeutic drug cisplatin, which crosslinks DNA strands, is used in conjunction with ionizing radiation (IR) to treat various types of cancer, including cervical carcinomas, and head and neck cancers [54,55]. Cells can be sensitized to cisplatin through inhibition of NHEJ [56], conceptually analogous to the aforementioned sensitization to DNA-damaging anticancer drugs through the modulation of repair pathways [32,33,43].
DNA-dependent protein kinase (DNA-PK) and PARP inhibitors also increase the cytotoxicity of radiation [17,57,58,59,60]. In the presence of PARP inhibitor, damage checkpoint activation in response to radiation exposure is significantly elevated [61], leading to efficient induction of cancer cell death through apoptosis. Radiosensitization by PARP inhibition is primarily due to suppression of PARP-mediated repair pathways, analogous to PARP inhibitor-mediated sensitization to CPT [32,33]. Thus, repair pathway modulation is a feasible strategy for sensitization to radiotherapy. This idea is further supported by the observation that inhibition of DNA-PK, which inhibits NHEJ, also increases the cytotoxicity of radiation [57,58].
IR causes multiple types of DNA damage, including DSBs and single-strand breaks (SSBs). IR also causes formation of reactive oxygen species (ROS) which, in turn, promote production of oxidized nucleotide adducts, such as 8-oxoguanine [62,63,64]. In addition, SSBs and ROS cause replication stress, which is itself associated with formation of DSBs [65]. Although apoptosis can be induced in response to DSBs [66], the DSBs caused directly by therapeutic radiation are usually repaired within a few hours [67]. In mechanistic terms, it remains unclear which damage, stresses, and adducts make the greatest contributions to cancer cell killing. A recent study revealed that persistent DSBs in irradiated cells form in association with replication stress during the S phase following the repair of radiation-induced DSBs [68], suggesting that these later DSBs are primarily responsible for cytotoxicity. In this case, the sensitization effect caused by PARP inhibitor might be identical to that observed during CPT sensitization.
3.3. Radiotherapy in Conjunction with Molecularly Targeted Agents and Immune Checkpoint Inhibitors
Some molecularly targeted agents efficiently sensitize specific types of cancer to radiation. For example, cetuximab, an anti-epidermal growth factor receptor (EGFR) antibody, is currently used as a sensitizer for radiation therapy in cases of head and neck squamous cell carcinoma. The sensitization effect is induced by inhibiting the radiation-induced upregulation of HIF-1α [69], suggesting that modulation of growth factor signaling represents another potential target for sensitization to radiation therapy.
Radiotherapy might also be complemented by immune checkpoint blockade [70,71]. Radiation, initially thought to be an immunosuppressive, was recently shown to be a promising candidate for such a combination [72]. Victor et al. demonstrated that radiation therapy, in conjunction with anti-cytotoxic-T lymphocyte-associated protein 4 (CTLA4) antibody, is more effective than either treatment alone [72]. Furthermore, addition of anti-prognostic of programmed cell death ligand 1 (PD-L1) further suppresses the adaptive immune resistance that arises in patients treated with radiation therapy and anti-CTLA4 antibody [73]. These observations indicate that the combination of radiation therapy with dual immune checkpoint blockade represents a promising strategy for radiotherapy sensitization. In the future, it may be possible to further develop sensitization strategies by combining multiple sensitizers.
3.4. Radiosensitivity by Autophagy Regulatory Drugs
Multiple studies show that radiation sensitivity is associated with the activation status of autophagy, which is normally involved in maintaining intracellular metabolic balance [74,75]. Although it remains unclear how autophagy is associated with radiation sensitivity, it is clear that autophagy is induced by radiation. Moreover, it is further activated by simultaneous treatment with DNA-PK and PARP inhibitors, cisplatin, and anti-EGFR antibody [60,75,76,77]. Importantly, autophagy activation is associated with the efficiency of cancer cell killing [78]. However, in some other contexts, radioresistance is promoted when autophagy is activated [79]. Thus, although autophagy activation is usually associated with radiosensitivity, it can also induce resistance. Currently, we do not have a way to simultaneously control the cancer-suppressing and -promoting effects of autophagy [80].
3.5. Proton Beam and Carbon-Ion Beam Therapy
Proton beam therapy (PBT) and carbon-ion beam radiotherapy are more recent modes of radiation therapy; both have fewer adverse effects and higher therapeutic efficacy than conventional radiation therapy [81]. Like radiation therapy, PBT also causes DNA damage, leading to induction of apoptosis in tumor cells [82]. The advantage of PBT is that the proton beam penetrates deeply into tissues, and can be controlled to efficiently target tumors while reducing exposure of the surrounding normal tissues. Carbon-ion beam therapy also has several advantages over radiotherapy; these include higher relative biological effectiveness, lack of an oxygen effect, and lower cell cycle-related radiosensitivity [83,84]. Importantly, carbon-ion beams have a marked killing effect even on cancer cells that are resistant to X- and γ-radiation [85]. The increased killing effect appears to be due to complex DSBs caused by the high linear energy transfer effect of carbon ions [86]. Such complex DSBs are much harder to repair than DSBs caused by X- or γ-radiation [87,88]. Such differences in repair efficiency might be due to differences in the repair mechanisms involved. Damage caused by proton- and carbon-ion radiation is repaired mainly by NHEJ; however, damage caused by the latter is also repaired by HR [89].
Sensitization studies were also reported for hadron radiation. Killing effect of cancer cells by carbon-ion beam irradiation was enhanced by carboplatin (cisplatin analogue) [90], paclitaxel [90], PU-H71 (HSP90 inhibitor) [91], and genistein (isoflavone compound) [92]. Vorinostat (histone deacetylase inhibitor) can sensitize cancer cells to proton beam and carbon ion beam radiations as well as γ-radiation [93]. Currently, several clinical trials involving combinations of these two radiation modalities plus chemotherapeutic agents are ongoing.
4. Conclusions and Prospects
Radiotherapy and chemotherapy with drugs that damage DNA are commonly used to treat cancer. However, the therapeutic efficacy of such approaches is often limited by multiple adverse effects. Recent studies suggest various strategies for sensitization, including modulation of DNA damage repair pathways and immune checkpoint blockade. Since it is difficult to predict side effects, it is important to carefully investigate the mechanisms underlying each sensitization strategy, both individually and in combination.
Author Contributions
Y.M., M.H., H.F., A.S. and K.Y. wrote the manuscript.
Funding
This work was supported by MEXT KAKENHI, No. 20770136.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Von Sonntag, C. Free DNA Damage Its Repair a Chemistry Perspect; Springer: Berlin, Germany, 2006; pp. 7–46. [Google Scholar]
- Adhikary, A.; Becker, D.; Sevilla, M.D. Electron Spin Resonance of Radicals in Irradiated DNA; Springer: Berlin, Germany, 2014; pp. 299–352. [Google Scholar]
- Yu, Q.; Rose, J.H.L.; Zhang, H.; Pommier, Y. Antisense inhibition of Chk2/hCds1 expression attenuates DNA damage-induced S and G2 checkpoints and enhances apoptotic activity in HEK-293 cells. FEBS Lett. 2001, 505, 7–12. [Google Scholar] [CrossRef]
- Hiu, W.C.; Jin, D.Y.; Ling, M.T.; Yong, C.W.; Wang, Q.; Sai, W.T.; Wang, X. Mitotic arrest deficient 2 expression induces chemosensitization to a DNA-damaging agent, cisplatin, in nasopharyngeal carcinoma cells. Cancer Res. 2005, 65, 1450–1458. [Google Scholar]
- Attardi, L.D.; de Vries, A.; Jacks, T. Activation of the p53-dependent G1 checkpoint response in mouse embryo fibroblasts depends on the specific DNA damage inducer. Oncogene 2004, 23, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Willis, J.; DeStephanis, D.; Patel, Y.; Gowda, V.; Yan, S. Study of the DNA damage checkpoint using Xenopus egg extracts. J. Vis. Exp. 2012, 69, e4449. [Google Scholar] [CrossRef] [PubMed]
- Sawicka, M.; Kalinowska, M.; Skierski, J.; Lewandowski, W. A review of selected anti-tumour therapeutic agents and reasons for multidrug resistance occurrence. J. Pharm. Pharmacol. 2004, 56, 1067–1081. [Google Scholar] [CrossRef] [PubMed]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug resistance in cancer: An overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [PubMed]
- Zahreddine, H.; Borden, K.L.B. Mechanisms and insights into drug resistance in cancer. Front. Pharmacol. 2013, 4, e28. [Google Scholar] [CrossRef] [PubMed]
- Luqmani, Y.A. Mechanisms of drug resistance in cancer chemotherapy. Med. Princ. Pract. 2005, 14, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Cheung-Ong, K.; Giaever, G.; Nislow, C. DNA-damaging agents in cancer chemotherapy: Serendipity and chemical biology. Chem. Biol. 2013, 20, 648–659. [Google Scholar] [CrossRef] [PubMed]
- Paoletti, X.; Le Tourneau, C.; Verweij, J.; Siu, L.L.; Seymour, L.; Postel-Vinay, S.; Collette, L.; Rizzo, E.; Ivy, P.; Olmos, D.; et al. Defining dose-limiting toxicity for phase 1 trials of molecularly targeted agents: Results of a DLT-TARGETT international survey. Eur. J. Cancer 2014, 50, 2050–2056. [Google Scholar] [CrossRef] [PubMed]
- Bartz, S.R.; Zhang, Z.; Burchard, J.; Imakura, M.; Martin, M.; Palmieri, A.; Needham, R.; Guo, J.; Gordon, M.; Chung, N.; et al. Small Interfering RNA Screens Reveal Enhanced Cisplatin Cytotoxicity in Tumor Cells Having both BRCA Network and TP53 Disruptions. Mol. Cell. Biol. 2006, 26, 9377–9386. [Google Scholar] [CrossRef] [PubMed]
- Bouwman, P.; Aly, A.; Escandell, J.M.; Pieterse, M.; Bartkova, J.; Van Der Gulden, H.; Hiddingh, S.; Thanasoula, M.; Kulkarni, A.; Yang, Q.; et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol. 2010, 17, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Engert, F.; Schneider, C.; Weiβ, L.M.; Probst, M.; Fulda, S. PARP Inhibitors Sensitize Ewing Sarcoma Cells to Temozolomide-Induced Apoptosis via the Mitochondrial Pathway. Mol. Cancer Ther. 2015, 14, 2818–2830. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.J.; Travers, J.; Pshenichnaya, I.; Kogera, F.A.; Barthorpe, S.; Mironenko, T.; Richardson, L.; Benes, C.H.; Stratton, M.R.; McDermott, U.; et al. Combinations of PARP Inhibitors with Temozolomide Drive PARP1 Trapping and Apoptosis in Ewing’s Sarcoma. PLoS ONE 2015, 10, e0140988. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, A.J. The potential role and application of PARP inhibitors in cancer treatment. Br. Med. Bull. 2009, 89, 23–40. [Google Scholar] [CrossRef] [PubMed]
- Rose, K.M.; Szopa, J.; Han, F.S.; Cheng, Y.C.; Richter, A.; Scheer, U. Association of DNA Topoisomerase I and RNA Polymerase I: A Possible Role for Topoisomerase I in Ribosomal Gene Transcription; Springer: Berlin, Germany, 1988; pp. 411–416. [Google Scholar]
- Tuduri, S.; Crabbé, L.; Conti, C.; Tourrière, H.; Holtgreve-Grez, H.; Jauch, A.; Pantesco, V.; De Vos, J.; Thomas, A.; Theillet, C.; et al. Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nat. Cell Biol. 2009, 11, 1315–1324. [Google Scholar] [CrossRef] [PubMed]
- Wall, M.E.; Warn, M.C. Camptothecin and Taxol: Discovery to Clinic—Thirteenth Bruce F. Cain Memorial Award Lecture. Cancer Res. 1995, 55, 753–760. [Google Scholar] [PubMed]
- Venditto, V.J.; Simanek, E.E. Cancer therapies utilizing the camptothecins: A review of the in vivo literature. Mol. Pharm. 2010, 7, 307–349. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Das, B.B.; Huang, S.N.; Murai, J.; Rehman, I.; Amé, J.-C.C.; Sengupta, S.; Das, S.K.; Majumdar, P.; Zhang, H.; Biard, D.; et al. PARP1-TDP1 coupling for the repair of topoisomerase I-induced DNA damage. Nucleic Acids Res. 2014, 42, 4435–4449. [Google Scholar] [CrossRef] [PubMed]
- Sugimura, K.; Takebayashi, S.-I.; Taguchi, H.; Takeda, S.; Okumura, K. PARP-1 ensures regulation of replication fork progression by homologous recombination on damaged DNA. J. Cell Biol. 2008, 183, 1203–1212. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Barcelo, J.M.; Rao, V.A.; Sordet, O.; Jobson, A.G.; Thibaut, L.; Miao, Z.-H.; Seiler, J.A.; Zhang, H.; Marchand, C.; et al. Repair of topoisomerase I—Mediated DNA damage. Prog. Nucleic Acid Res. Mol. Biol. 2006, 81, 179–229. [Google Scholar] [PubMed]
- Pommier, Y. Drugging topoisomerases: Lessons and Challenges. ACS Chem. Biol. 2013, 8, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Lebedeva, N.A.; Anarbaev, R.O.; Sukhanova, M.; Vasil’eva, I.A.; Rechkunova, N.I.; Lavrik, O.I. Poly(ADP-ribose)polymerase 1 stimulates the AP-site cleavage activity of tyrosyl-DNA phosphodiesterase 1. Biosci. Rep. 2015, 35, e00230. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-W.; Regairaz, M.; Seiler, J.; Agama, K.K.; Doroshow, J.H.; Pommier, Y. Poly (ADP-ribose) polymerase and XPF–ERCC1 participate in distinct pathways for the repair of topoisomerase I-induced DNA damage in mammalian cells. Nucleic Acids Res. 2011, 39, 3607–3620. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Redon, C.; Rao, V.A.; Seiler, J.A.; Sordet, O.; Takemura, H.; Antony, S.; Meng, L.; Liao, Z.; Kohlhagen, G.; et al. Repair of and checkpoint response to topoisomerase I-mediated DNA damage. Mutat. Res. Mol. Mech. Mutagen. 2003, 532, 173–203. [Google Scholar] [CrossRef]
- Konecny, G.E.; Kristeleit, R.S. PARP inhibitors for BRCA1/2-mutated and sporadic ovarian cancer: Current practice and future directions. Br. J. Cancer 2016, 115, 1157–1173. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.G.; Flatten, K.S.; Schneider, P.A.; Dai, N.T.; McDonald, J.S.; Poirier, G.G.; Kaufmann, S.H. Enhanced killing of cancer cells by poly(ADP-ribose) polymerase inhibitors and topoisomerase I inhibitors reflects poisoning of both enzymes. J. Biol. Chem. 2012, 287, 4198–4210. [Google Scholar] [CrossRef] [PubMed]
- Das, S.K.; Rehman, I.; Ghosh, A.; Sengupta, S.; Majumdar, P.; Jana, B.; Das, B.B. Poly(ADP-ribose) polymers regulate DNA topoisomerase i (Top1) nuclear dynamics and camptothecin sensitivity in living cells. Nucleic Acids Res. 2016, 44, 8363–8375. [Google Scholar] [CrossRef] [PubMed]
- Ström, C.E.; Johansson, F.; Uhlén, M.; Szigyarto, C.A.K.; Erixon, K.; Helleday, T. Poly (ADP-ribose) polymerase (PARP) is not involved in base excision repair but PARP inhibition traps a single-strand intermediate. Nucleic Acids Res. 2011, 39, 3166–3175. [Google Scholar] [CrossRef] [PubMed]
- Audebert, M.; Salles, B.; Calsou, P. Involvement of poly(ADP-ribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. J. Biol. Chem. 2004, 279, 55117–55126. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Petermann, E.; Schultz, N.; Jemth, A.S.; Loseva, O.; Issaeva, N.; Johansson, F.; Fernandez, S.; McGlynn, P.; Helleday, T. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 2009, 28, 2601–2615. [Google Scholar] [CrossRef] [PubMed]
- Atsumi, Y.; Inase, A.; Osawa, T.; Sugihara, E.; Sakasai, R.; Fujimori, H.; Teraoka, H.; Saya, H.; Kanno, M.; Tashiro, F.; et al. The Arf/p53 protein module, which induces apoptosis, down-regulates histone H2AX to allow normal cells to survive in the presence of anti-cancer drugs. J. Biol. Chem. 2013, 288, 13269–13277. [Google Scholar] [CrossRef] [PubMed]
- Gartner, A.; Milstein, S.; Ahmed, S.; Hodgkin, J.; Hengartner, M.O. A Conserved Checkpoint Pathway Mediates DNA Damage–Induced Apoptosis and Cell Cycle Arrest in C. elegans. Mol. Cell 2000, 5, 435–443. [Google Scholar] [CrossRef]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death by apoptosis. Trends Mol. Med. 2006, 12, 440–450. [Google Scholar] [CrossRef] [PubMed]
- Maréchal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, e12716. [Google Scholar] [CrossRef] [PubMed]
- Chan, N.; Pires, I.M.; Bencokova, Z.; Coackley, C.; Luoto, K.R.; Bhogal, N.; Lakshman, M.; Gottipati, P.; Oliver, F.J.; Helleday, T.; et al. Contextual synthetic lethality of cancer cell kill based on the tumor microenvironment. Cancer Res. 2010, 70, 8045–8054. [Google Scholar] [CrossRef] [PubMed]
- Murai, J.; Huang, S.Y.N.; Das, B.B.; Renaud, A.; Zhang, Y.; Do roshow, J.H.; Ji, J.; Takeda, S.; Pommier, Y. Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res. 2012, 72, 5588–5599. [Google Scholar] [CrossRef] [PubMed]
- Hirai, T.; Shirai, H.; Fujimori, H.; Okayasu, R.; Sasai, K.; Masutani, M. Radiosensitization effect of poly(ADP-ribose) polymerase inhibition in cells exposed to low and high liner energy transfer radiation. Cancer Sci. 2012, 103, 1045–1050. [Google Scholar] [CrossRef] [PubMed]
- Samol, J.; Ranson, M.; Scott, E.; Macpherson, E.; Carmichael, J.; Thomas, A.; Cassidy, J. Safety and tolerability of the poly(ADP-ribose) polymerase (PARP) inhibitor, olaparib (AZD2281) in combination with topotecan for the treatment of patients with advanced solid tumors: A phase I study. Investig. New Drugs 2012, 30, 1493–1500. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, D.; O’Reilly, S. Clinical Guidelines for Managing Topotecan-Related Hematologic Toxicity. Oncologist 1998, 3, 4–10. [Google Scholar] [PubMed]
- Chen, Y.; Pandya, K.; Keng, P.C.; Johnstone, D.; Li, J.; Lee, Y.-J.; Smudzin, T.; Okunieff, P. Phase I/II clinical study of pulsed paclitaxel radiosensitization for thoracic malignancy: A therapeutic approach on the basis of preclinical research of human cancer cell lines. Clin. Cancer Res. 2003, 9, 969–975. [Google Scholar] [PubMed]
- Chinnaiyan, P.; Huang, S.; Vallabhaneni, G.; Armstrong, E.; Varambally, S.; Tomlins, S.A.; Chinnaiyan, A.M.; Harari, P.M. Mechanisms of enhanced radiation response following epidermal growth factor receptor signaling inhibition by erlotinib (Tarceva). Cancer Res. 2005, 65, 3328–3335. [Google Scholar] [CrossRef] [PubMed]
- van Nifterik, K.A.; van den Berg, J.; Stalpers, L.J.A.; Lafleur, M.V.M.; Leenstra, S.; Slotman, B.J.; Hulsebos, T.J.M.; Sminia, P. Differential Radiosensitizing Potential of Temozolomide in MGMT Promoter Methylated Glioblastoma Multiforme Cell Lines. Int. J. Radiat. Oncol. 2007, 69, 1246–1253. [Google Scholar] [CrossRef] [PubMed]
- Bobola, M.S.; Kolstoe, D.D.; Blank, A.; Silber, J.R. Minimally cytotoxic doses of temozolomide produce radiosensitization in human glioblastoma cells regardless of MGMT expression. Mol. Cancer Ther. 2010, 9, 1208–1218. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Joensuu, G.; Joensuu, T.; Nokisalmi, P.; Reddy, C.; Isola, J.; Ruutu, M.; Kouri, M.; Kupelian, P.A.; Collan, J.; Pesonen, S.; et al. A Phase I/II Trial of Gefitinib Given Concurrently with Radiotherapy in Patients with Nonmetastatic Prostate Cancer. Int. J. Radiat. Oncol. 2010, 78, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Choy, H.; Safran, H.; Akerley, W.; Graziano, S.L.; Bogart, J.A.; Cole, B.F. Phase II trial of weekly paclitaxel and concurrent radiation therapy for locally advanced non-small cell lung cancer. Clin. Cancer Res. 1998, 4, 1931–1936. [Google Scholar] [PubMed]
- Lau, D.; Ryu, J.; Gandara, D.; Morgan, R.; Doroshow, J.; Wilder, R.; Leigh, B. Concurrent twice-weekly paclitaxel and thoracic irradiation for stage III non-small cell lung cancer. Semin. Radiat. Oncol. 1999, 9, 117–120. [Google Scholar] [PubMed]
- Keys, H.M.; Bundy, B.N.; Stehman, F.B.; Muderspach, L.I.; Chafe, W.E.; Suggs, C.L.; Walker, J.L.; Gersell, D. Cisplatin, Radiation, and Adjuvant Hysterectomy Compared with Radiation and Adjuvant Hysterectomy for Bulky Stage IB Cervical Carcinoma. N. Engl. J. Med. 1999, 340, 1154–1161. [Google Scholar] [CrossRef] [PubMed]
- Rose, P.G.; Bundy, B.N.; Watkins, E.B.; Thigpen, J.T.; Deppe, G.; Maiman, M.A.; Clarke-Pearson, D.L.; Insalaco, S. Concurrent Cisplatin-Based Radiotherapy and Chemotherapy for Locally Advanced Cervical Cancer. N. Engl. J. Med. 1999, 340, 1144–1153. [Google Scholar] [CrossRef] [PubMed]
- Boeckman, H.J. Cisplatin Sensitizes Cancer Cells to Ionizing Radiation via Inhibition of Nonhomologous End Joining. Mol. Cancer Res. 2005, 3, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Ren, Y.; Zhang, T.; Wang, Z.; Ling, C.C.; Li, G.C.; He, F.; Wang, C.; Wen, B. Inactivation of DNA-PK by knockdown DNA-PKcs or NU7441 impairs non-homologous end-joining of radiation-induced double strand break repair. Oncol. Rep. 2018, 39, 912–920. [Google Scholar] [CrossRef] [PubMed]
- Timme, C.R.; Rath, B.H.; O’Neill, J.W.; Camphausen, K.; Tofilon, P.J. The DNA-PK Inhibitor VX-984 Enhances the Radiosensitivity of Glioblastoma Cells Grown In Vitro and as Orthotopic Xenografts. Mol. Cancer Ther. 2018, 17, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Verhagen, C.V.M.; de Haan, R.; Hageman, F.; Oostendorp, T.P.D.; Carli, A.L.E.; O’Connor, M.J.; Jonkers, J.; Verheij, M.; van den Brekel, M.W.; Vens, C. Extent of radiosensitization by the PARP inhibitor olaparib depends on its dose, the radiation dose and the integrity of the homologous recombination pathway of tumor cells. Radiother. Oncol. 2015, 116, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Alotaibi, M.; Sharma, K.; Saleh, T.; Povirk, L.F.; Hendrickson, E.A.; Gewirtz, D.A. Radiosensitization by PARP Inhibition in DNA Repair Proficient and Deficient Tumor Cells: Proliferative Recovery in Senescent Cells. Radiat. Res. 2016, 185, 229–245. [Google Scholar] [CrossRef] [PubMed]
- Aguilar-Quesada, R.; Muñoz-Gámez, J.; Martín-Oliva, D.; Peralta, A.; Valenzuela, M.T.; Matínez-Romero, R.; Quiles-Pérez, R.; Murcia, J.; de Murcia, G.; de Almodóvar, M.R.; et al. Interaction between ATM and PARP-1 in response to DNA damage and sensitization of ATM deficient cells through PARP inhibition. BMC Mol. Biol. 2007, 8, e29. [Google Scholar] [CrossRef] [PubMed]
- Harrison, L.; Hatahet, Z.; Wallace, S.S. In vitro repair of synthetic ionizing radiation-induced multiply damaged DNA sites. J. Mol. Biol. 1999, 290, 667–684. [Google Scholar] [CrossRef] [PubMed]
- Azzam, E.I.; Jay-Gerin, J.-P.; Pain, D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett. 2012, 327, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Reisz, J.A.; Bansal, N.; Qian, J.; Zhao, W.; Furdui, C.M. Effects of ionizing radiation on biological molecules—Mechanisms of damage and emerging methods of detection. Antioxid. Redox Signal. 2014, 21, 260–292. [Google Scholar] [CrossRef] [PubMed]
- Lomax, M.E.; Folkes, L.K.; O’Neill, P. Biological Consequences of Radiation-induced DNA Damage: Relevance to Radiotherapy. Clin. Oncol. 2013, 25, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Nowsheen, S.; Yang, E.S. The intersection between DNA damage response and cell death pathways. Exp. Oncol. 2012, 34, 243–254. [Google Scholar] [PubMed]
- Atsumi, Y.; Minakawa, Y.; Ono, M.; Dobashi, S.; Shinohe, K.; Shinohara, A.; Takeda, S.; Takagi, M.; Takamatsu, N.; Nakagama, H.; et al. ATM and SIRT6/SNF2H Mediate Transient H2AX Stabilization When DSBs Form by Blocking HUWE1 to Allow Efficient γH2AX Foci Formation. Cell Rep. 2015, 13, 2728–2740. [Google Scholar] [CrossRef] [PubMed]
- Minakawa, Y.; Atsumi, Y.; Shinohara, A.; Murakami, Y.; Yoshioka, K. Gamma-irradiated quiescent cells repair directly induced double-strand breaks but accumulate persistent double-strand breaks during subsequent DNA replication. Genes Cells 2016, 21, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Liang, K.; Lu, Y.; Fan, Z. The anti-EGFR antibody cetuximab sensitizes human head and neck squamous cell carcinoma cells to radiation in part through inhibiting radiation-induced upregulation of HIF-1alpha. Cancer Let. 2012, 322, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Verbrugge, I.; Hagekyriakou, J.; Sharp, L.L.; Galli, M.; West, A.; McLaughlin, N.M.; Duret, H.; Yagita, H.; Johnstone, R.W.; Smyth, M.J.; et al. Radiotherapy increases the permissiveness of established mammary tumors to rejection by immunomodulatory antibodies. Cancer Res. 2012, 72, 3163–3174. [Google Scholar] [CrossRef] [PubMed]
- Formenti, S.C.; Demaria, S. Combining Radiotherapy and Cancer Immunotherapy: A Paradigm Shift. JNCI J. Natl. Cancer Inst. 2013, 105, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Auh, S.L.; Wang, Y.; Burnette, B.; Wang, Y.; Meng, Y.; Sharma, R.; Chin, R.; Tu, T.; Weichselbaum, R.R.; et al. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells. Blood 2009, 114, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Twyman-Saint Victor, C.; Rech, A.J.; Maity, A.; Rengan, R.; Pauken, K.E.; Stelekati, E.; Benci, J.L.; Xu, B.; Dada, H.; Odorizzi, P.M.; et al. Radiation and dual checkpoint blockade activate non-redundant immune mechanisms in cancer. Nature 2015, 520, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Daido, S.; Kanzawa, T.; Kondo, S.; Kondo, Y. Radiation-induced autophagy is associated with LC3 and its inhibition sensitizes malignant glioma cells. Int. J. Oncol. 2005, 26, 1401–1410. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, S.; Comincini, S. Autophagy and ionizing radiation in tumors: The “survive or not survive” dilemma. J. Cell. Physiol. 2013, 228, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Daido, S.; Yamamoto, A.; Fujiwara, K.; Sawaya, R.; Kondo, S.; Kondo, Y. Inhibition of the DNA-dependent protein kinase catalytic subunit radiosensitizes malignant glioma cells by inducing autophagy. Cancer Res. 2005, 65, 4368–4375. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Ma, S.; Liu, M.; Hou, Y.; Liang, B.; Su, X.; Liu, X. Synergistic killing of lung cancer cells by cisplatin and radiation via autophagy and apoptosis. Oncol. Lett. 2014, 7, 1903–1910. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Wang, Y.; Pang, X.; Zhang, B. Inhibition of autophagy induced by TSA sensitizes colon cancer cell to radiation. Tumor Biol. 2014, 35, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Rehman, S.K.; Zhang, W.; Wen, A.; Yao, L.; Zhang, J. Autophagy is a therapeutic target in anticancer drug resistance. Biochim. Biophys. Acta 2010, 1806, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Xin, Y.; Jiang, F.; Yang, C.; Yan, Q.; Guo, W.; Huang, Q.; Zhang, L.; Jiang, G. Role of autophagy in regulating the radiosensitivity of tumor cells. J. Cancer Res. Clin. Oncol. 2017, 143, 2147–2157. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, H.; Ishikawa, H.; Okumura, T. Proton beam therapy in Japan: Current and future status. Jpn. J. Clin. Oncol. 2016, 46, 885–892. [Google Scholar] [CrossRef] [PubMed]
- Alan Mitteer, R.; Wang, Y.; Shah, J.; Gordon, S.; Fager, M.; Butter, P.-P.; Jun Kim, H.; Guardiola-Salmeron, C.; Carabe-Fernandez, A.; Fan, Y. Proton beam radiation induces DNA damage and cell apoptosis in glioma stem cells through reactive oxygen species. Sci. Rep. 2015, 5, e13961. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; David, G.H.; Piero, D.; Charles, F.B.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Sai, S.; Oonishi, K.; Yamada, S.; Kamada, T.; Okayasu, R. Effects of carbon ion beam on putative colon cancer stem cells and its correlation with radiocurability. Radiother. Oncol. 2011, 99, e393. [Google Scholar] [CrossRef]
- Mohamad, O.; Sishc, B.J.; Saha, J.; Pompos, A.; Rahimi, A.; Story, M.D.; Davis, A.J.; Kim, D.W.N. Carbon ion radiotherapy: A review of clinical experiences and preclinical research, with an emphasis on DNA damage/repair. Cancers 2017, 9, e66. [Google Scholar] [CrossRef] [PubMed]
- Ibañez, I.L.; Bracalente, C.; Molinari, B.L.; Palmieri, M.A.; Policastro, L.; Kreiner, A.J.; Burlón, A.A.; Valda, A.; Navalesi, D.; Davidson, J.; et al. Induction and Rejoining of DNA Double Strand Breaks Assessed by H2AX Phosphorylation in Melanoma Cells Irradiated with Proton and Lithium Beams. Int. J. Radiat. Oncol. Biol. Phys. 2009, 74, 1226–1235. [Google Scholar] [CrossRef] [PubMed]
- Oonishi, K.; Cui, X.; Hirakawa, H.; Fujimori, A.; Kamijo, T.; Yamada, S.; Yokosuka, O.; Kamada, T. Different effects of carbon ion beams and X-rays on clonogenic survival and DNA repair in human pancreatic cancer stem-like cells. Radiother. Oncol. 2012, 105, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Gerelchuluun, A.; Manabe, E.; Ishikawa, T.; Sun, L.; Itoh, K.; Sakae, T.; Suzuki, K.; Hirayama, R.; Asaithamby, A.; Chen, D.J.; et al. The Major DNA Repair Pathway after Both Proton and Carbon-Ion Radiation is NHEJ, but the HR Pathway is More Relevant in Carbon Ions. Radiat. Res. 2015, 183, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Kubo, N.; Noda, S.; Takahashi, A.; Yoshida, Y.; Oike, T.; Murata, K.; Musha, A.; Suzuki, Y.; Ohno, T.; Takahashi, T.; et al. Radiosensitizing effect of carboplatin and paclitaxel to carbon-ion beam irradiation in the non-small-cell lung cancer cell line H460. J. Radiat. Res. 2015, 56, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Li, H.K.; Matsumoto, Y.; Furusawa, Y.; Kamada, T. PU-H71, a novel Hsp90 inhibitor, as a potential cancer-specific sensitizer to carbon-ion beam therapy. J. Radiat. Res. 2016, 57, 572–575. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, P.; Hirayama, R.; Niu, Y.; Liu, X.; Chen, W.; Jin, X.; Zhang, P.; Ye, F.; Zhao, T.; et al. Genistein sensitizes glioblastoma cells to carbon ions via inhibiting DNA-PKcs phosphorylation and subsequently repressing NHEJ and delaying HR repair pathways. Radiother. Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Gerelchuluun, A.; Maeda, J.; Manabe, E.; Brents, C.A.; Sakae, T.; Fujimori, A.; Chen, D.J.; Tsuboi, K.; Kato, T.A. Histone Deacetylase Inhibitor Induced Radiation Sensitization Effects on Human Cancer Cells after Photon and Hadron Radiation Exposure. Int. J. Mol. Sci. 2018, 19, e496. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).