Epidemiology of Moderate Alcohol Consumption and Breast Cancer: Association or Causation?

Abstract
1. Introduction
1.1. Molecular Characteristics of Breast Cancer Subtypes
1.2. Risk Factors for Breast Cancer
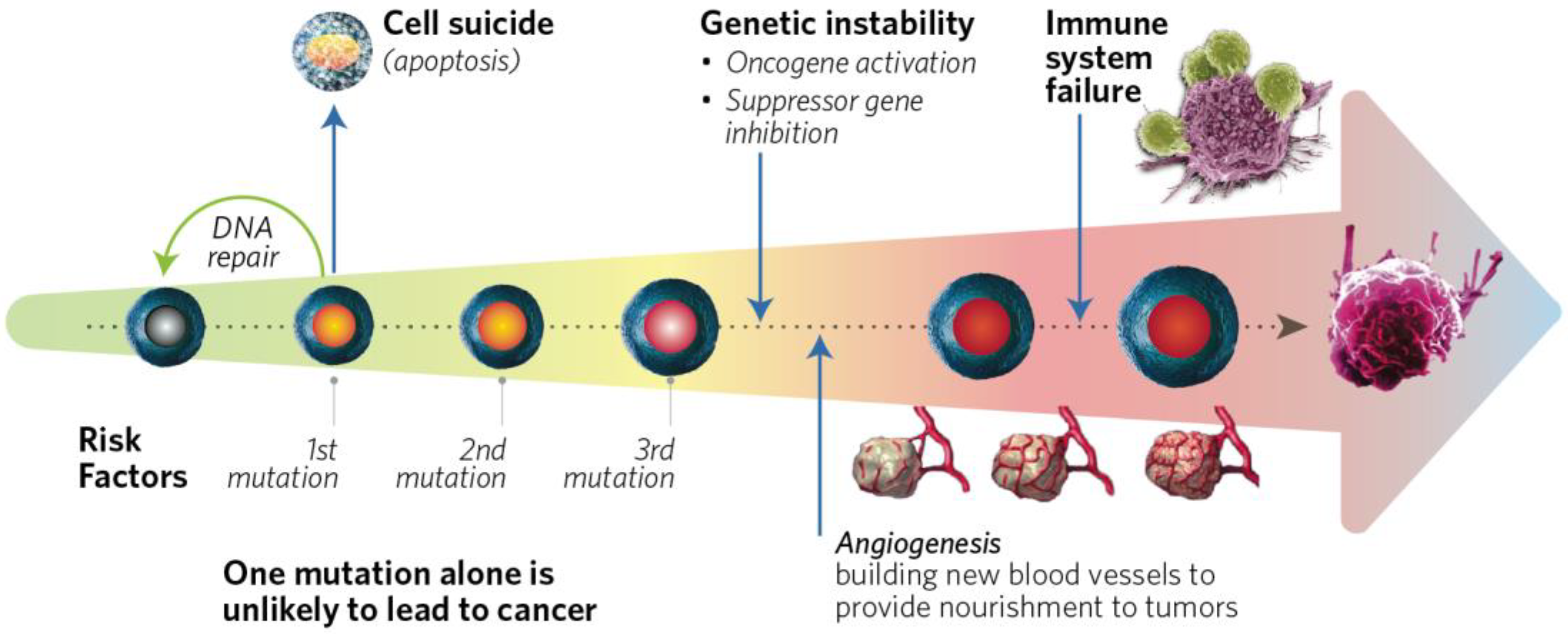
1.2.1. Genetic Factors
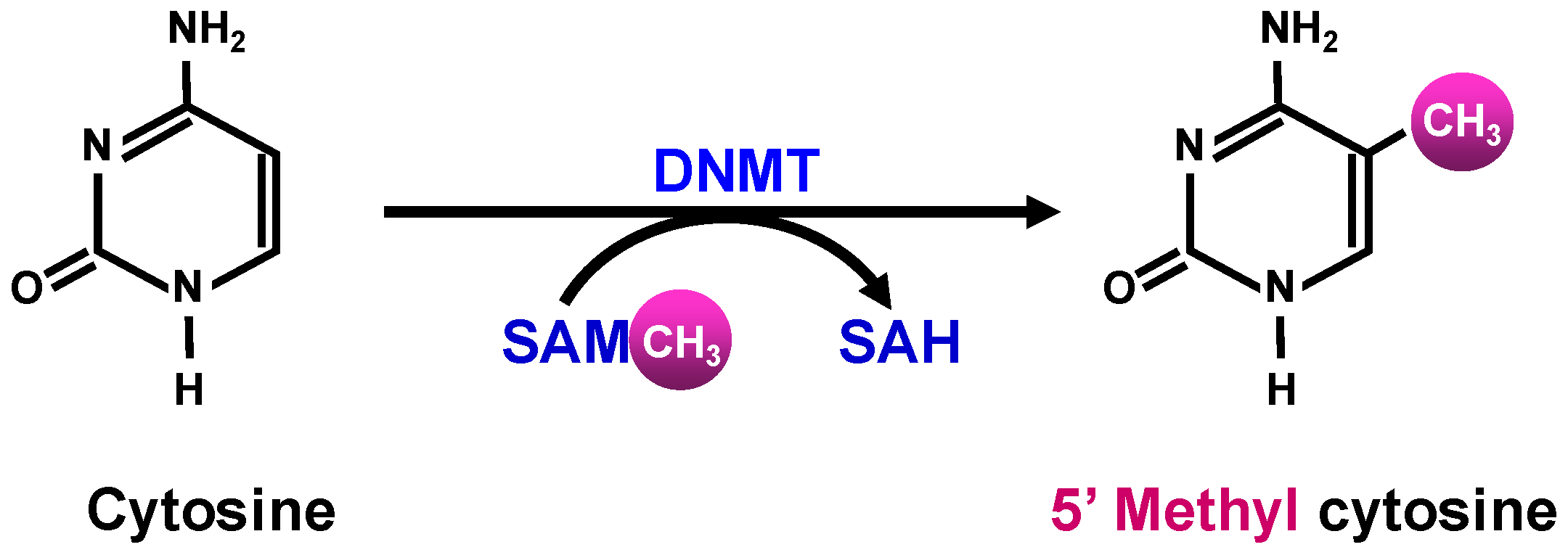
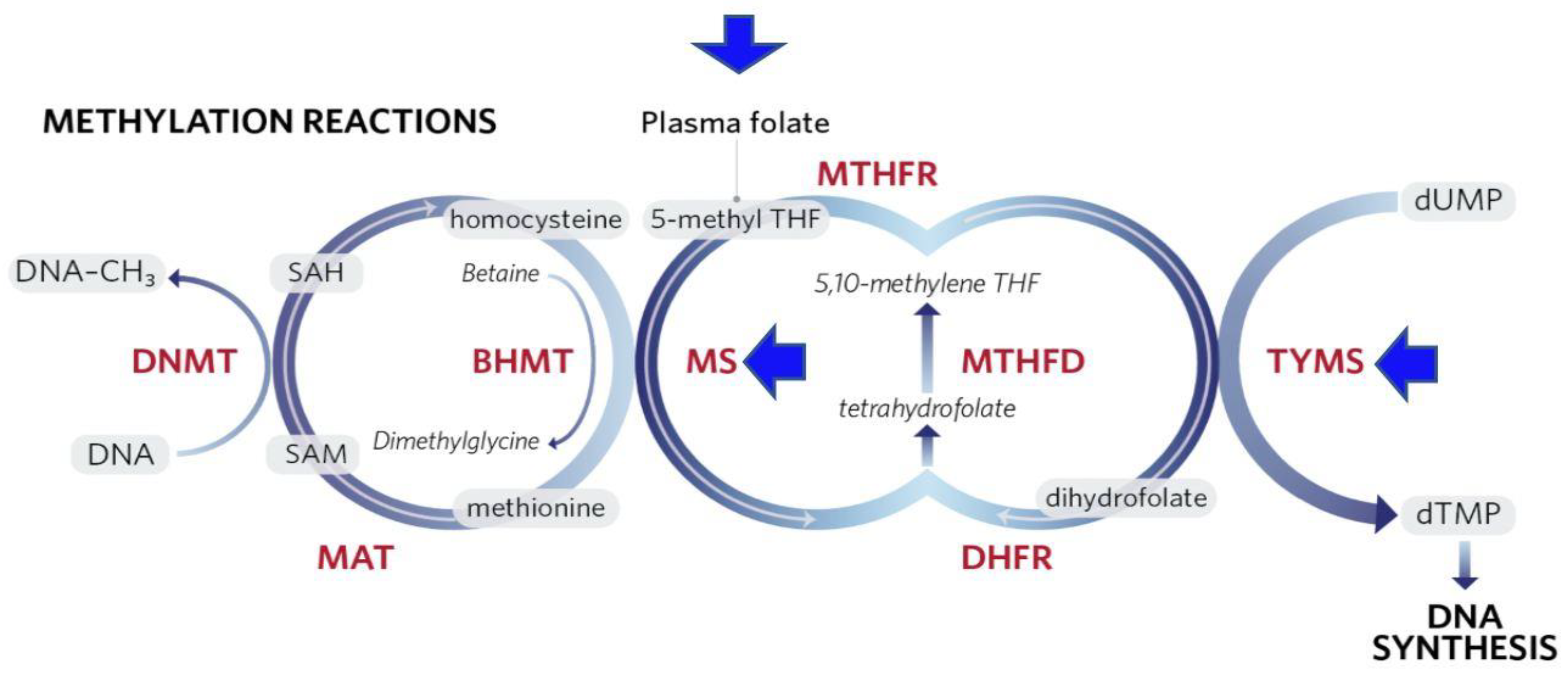
1.2.2. Epigenetic Modifications
2. Alcohol Consumption and Breast Cancer: Epidemiological Studies
3. Alcohol Consumption and Breast Cancer: Meta-Analyses
4. Association or Causation
4.1. Strengthening Causal Inference by Genetic Epidemiology through Mendelian Randomization
4.2. Marginal Structural Modeling and Agent-Based Modeling
4.3. Molecular Pathological Epidemiology
5. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Cancer Stat Facts: Female Breast Cancer. Available online: https://seer.cancer.gov/statfacts/html/breast.html (accessed on 22 January 2018).
- Benson, J.R.; Jatol, I. The global breast cancer burden. Future Oncol. 2012, 8, 697–702. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. World Health Organization Classification of Tumors of the Breast, 4th ed.; IARC Press: Lyon, France, 2012. [Google Scholar]
- Vinay, K.; Abul, K.A.; Jon, C.A.; Nelson, F. Robbins and Cotran Pathologic Basis of Disease, 8th ed.; Elsevier: Lyon, France, 2010. [Google Scholar]
- Parker, J.S.; Mullins, M.; Cheang, M.C.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Suprvised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 2009, 27, 1160–1167. [Google Scholar] [CrossRef] [PubMed]
- Turkoz, F.P.; Solak, M.; Petekkaya, I.; Keskin, O.; Kertmen, N.; Sarici, F.; Arik, Z.; Babacan, T.; Ozisik, Y.; Altundag, K.; et al. Association between common risk factors and molecular subtypes in breast cancer patients. Breast 2013, 22, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Sørlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; Van De Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [PubMed]
- Millikan, R.C.; Newman, B.; Tse, C.K.; Moorman, P.G.; Conway, K.; Smith, L.V.; Labbok, M.H.; Geradts, J.; Bensen, J.T.; Jackson, S.; et al. Epidemiology of basal-like breast cancer. Breast Cancer Res. Treat. 2008, 109, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Kwan, M.L.; Kushi, L.H.; Weltzien, E.; Maring, B.; Kutner, S.E.; Fulton, R.S.; Lee, M.M.; Ambrosone, C.B.; Caan, B.J.; et al. Epidemiology of breast cancer subtypes in two prospective cohort studies of breast cancer survivors. Breast Cancer Res. 2009, 11, R31. [Google Scholar] [CrossRef] [PubMed]
- Polyak, K. Breast cancer: Origins and evolution. J. Clin. Invest. 2007, 117, 3155–3163. [Google Scholar] [CrossRef] [PubMed]
- Zakhari, S.; Hoek, J. Alcohol and Breast Cancer: Reconciling Epidemiological and Molecular Data. Adv. Exp. Med. Biol. 2015, 815, 7–39. [Google Scholar] [PubMed]
- Urbaniak, C.; Gloor, G.B.; Brackstone, M.; Scott, L.; Tangney, M.; Reid, G. The Microbiota of Breast Tissue and Its Association with Breast Cancer. Appl. Environ. Microbiol. 2016, 82, 5039–5048. [Google Scholar] [CrossRef] [PubMed]
- Barnes, B.B.E.; Steindorf, K.; Hein, R.; Flesch-Janys, D.; Chang-Claude, J. Population attributable risk of invasive postmenopausal breast cancer and breast cancer subtypes for modifiable and non-modifiable risk factors. Cancer Epidemiol. 2011, 35, 345–352. [Google Scholar] [CrossRef] [PubMed]
- Collaborative Group on Hormonal Factors in Breast Cancer. Familial breast cancer: Collaborative analysis of individual data from 52 epidemiological studies including 58,209 women with breast cancer and 101,986 women without the disease. Lancet 2001, 358, 1389–1399. [Google Scholar] [CrossRef]
- Apostolou, P.; Fostira, F. Hereditary Breast Cancer: The Era of New Susceptibility Genes. Biomed. Res. Int. 2013, 2013, 747318. [Google Scholar] [CrossRef] [PubMed]
- Skol, A.D.; Sasaki, M.M.; Onel, K. The genetics of breast cancer risk in the post-genome era: Thoughts on study design to move past BRCA and towards clinical relevance. Breast Cancer Res. 2016, 18, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Miki, Y.; Swensen, J.; Shattuck-Eidens, D.; Futreal, P.A.; Harshman, K.; Tavtigian, S.; Liu, Q.; Cochran, C.; Bennett, L.M.; Ding, W. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994, 266, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Wooster, R.; Bignell, G.; Lancaster, J.; Swift, S.; Seal, S.; Mangion, J.; Collins, N.; Gregory, S.; Gumbs, C.; Micklem, G.; et al. Identification of the breast cancer susceptibility gene BRCA2. Nature 1995, 378, 789–792. [Google Scholar] [CrossRef] [PubMed]
- Lasa, A.; Cajal, T.R.; Llort, G.; Suela, J.; Cigudosa, J.C.; Cornet, M.; Alonso, C.; Barnadas, A.; Baiget, M. Copy number variations are not modifiers of phenotypic expression in a pair of identical twins carrying a BRCA1 mutation. Breast Cancer Res. Treat. 2010, 123, 901–905. [Google Scholar] [CrossRef] [PubMed]
- Fostira, F.; Tsitlaidou, M.; Papadimitriou, C.; Pertesi, M.; Timotheadou, E.; Stavropoulou, A.V.; Glentis, S.; Bournakis, E.; Bobos, M.; Pectasides, D.; et al. Prevalence of BRCA1 mutations among 403 women with triple-negative breast cancer: Implications for genetic screening selection criteria: A Hellenic Cooperative Oncology Group Study. Breast Cancer Res. Treat. 2012, 134, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Hirshfield, K.M.; Ganesan, S. Triple-negative breast cancer: Molecular subtypes and targeted therapy. Curr. Opin. Obstet. Gynecol. 2014, 26, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Shuen, A.W.; Foulkers, W.D. Inherited mutations in breast cancer genes—Risk and response. J. Mammary Gland. Biol. Neoplasia 2011, 16, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Dennis, J.; Ghadirian, P.; Little, J.; Lubinski, J.; Gronwald, J.; Kim-Sing, C.; Foulkes, W.; Moller, P.; Lynch, H.T.; Neuhausen, S.L.; et al. Alcohol consumption and the risk of breast cancer among BRCA1 and BRCA2 mutation carriers. Breast 2010, 19, 479–483. [Google Scholar] [CrossRef] [PubMed]
- McGuire, V.; John, E.M.; Felberg, A.; Haile, R.W.; Boyd, N.F.; Thomas, D.C.; Jenkins, M.A.; Milne, R.L.; Daly, M.B.; Ward, J.; et al. No increased risk of breast cancer associated with alcohol consumption among carriers of BRCA1 and BRCA2 mutations ages <50 years. Cancer Epidemiol. Biomark. Prev. 2006, 15, 1565–1567. [Google Scholar] [CrossRef] [PubMed]
- Shiovitz, S.; Korde, L.A. Genetics of breast cancer: A topic in evolution. Ann. Oncol. 2015, 26, 1291–1299. [Google Scholar] [CrossRef] [PubMed]
- Erkko, H.; Xia, B.; Nikkilä, J.; Schleutker, J.; Syrjäkoski, K.; Mannermaa, A.; Kallioniemi, A.; Pylkäs, K.; Karppinen, S.M.; Rapakko, K.; et al. A recurrent mutation in PALB2 in Finnish cancer families. Nature 2007, 446, 316–319. [Google Scholar] [CrossRef] [PubMed]
- The CHEK2 breast cancer case-control consortium. CHECK2*1100delC and susceptibility to breast cancer: A collaborative analysis involving 10,860 breast cancer cases and 9065 controls from ten studies. Am. J. Hum. Genet. 2004, 74, 1175–1182. [Google Scholar] [CrossRef] [PubMed]
- Renwick, A.; Thompson, D.; Seal, S.; Kelly, P.; Chagtai, T.; Ahmed, M.; North, B.; Jayatilake, H.; Barfoot, R.; Spanova, K.; et al. ATM mutations that cause ataxia-telangiectasia are breast cancer susceptibility alleles. Nat. Genet. 2006, 38, 837–875. [Google Scholar] [CrossRef] [PubMed]
- Seal, S.; Thompson, D.; Renwick, A.; Elliott, A.; Kelly, P.; Barfoot, R.; Chagtai, T.; Jayatilake, H.; Ahmed, M.; Spanova, K.; et al. Truncating mutations in the Fanconi anemia gene BRIP1 are low-penetrance breast cancer susceptibility alleles. Nat. Genet. 2006, 38, 1239–1241. [Google Scholar] [CrossRef] [PubMed]
- Afghahi, A.; Telli, M.L.; Kurian, A.W. Genetics of triple-negative breast cancer: Implications for patient care. Curr. Probl. Cancer 2016, 40, 130–140. [Google Scholar] [CrossRef] [PubMed]
- Easton, D.F.; Pooley, K.A.; Dunning, A.M.; Pharoah, P.D.; Thompson, D.; Ballinger, D.G.; Struewing, J.P.; Morrison, J.; Field, H.; Luben, R.; et al. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 2007, 447, 1087–1092. [Google Scholar] [CrossRef] [PubMed]
- Pharoah, P.D.; Antoniou, A.; Bobrow, M.; Zimmern, R.L.; Easton, D.F.; Ponder, B.A. Polygenic susceptibility to breast cancer and implications for prevention. Nat. Genet. 2002, 31, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Haiman, C.A.; Chen, G.K.; Vachon, C.M.; Canzian, F.; Dunning, A.; Millikan, R.C.; Wang, X.; Ademuyiwa, F.; Ahmed, S.; Ambrosone, C.B.; et al. A common variant at the TERT-CLPTM1L locus is associated with estrogen receptor–negative breast cancer. Nat. Genet. 2011, 43, 1210–1215. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Galán, J.; Torres, B.; del Moral, R.; Muñoz-Gámez, J.A.; Martín-Oliva, D.; Villalobos, M.; Núñez, M.I.; Luna, J.D.D.; Oliver, F.J.; Almodóvar, J.M.R.D. Quantitative detection of methylated ESR1 and 14-3-3-sigma gene promoters in serum as candidate biomarkers for diagnosis of breast cancer and evaluation of treatment efficacy. Cancer Biol. Ther. 2008, 7, 958–965. [Google Scholar] [CrossRef] [PubMed]
- Cen, Y.L.; Qi, M.L.; Li, H.G.; Su, Y.; Chen, L.J.; Lin, Y.; Chen, W.Q.; Xie, X.M.; Tang, L.Y.; Ren, Z.F. Associations of polymorphisms in the genes of FGFR2, FGF1, and RBFOX2 with breast cancer risk by estrogen/ progesterone receptor status. Mol. Carcinog. 2013, 52, E52–E59. [Google Scholar] [CrossRef] [PubMed]
- Andersen, S.W.; Trentham-Dietz, A.; Gangnon, R.E.; Hampton, J.M.; Figueroa, J.D.; Skinner, H.G.; Engelman, C.D.; Klein, B.E.; Titus, L.J.; Egan, K.M.; et al. Reproductive windows, genetic loci and breast cancer risk. Ann. Epidemiol. 2014, 24, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Purrington, K.S.; Slager, S.; Eccles, D.; Yannoukakos, D.; Fasching, P.A.; Miron, P.; Carpenter, J.; Chang-Claude, J.; Martin, N.G.; Montgomery, G.W.; et al. Genome-wide association study identifies 25 known breast cancer susceptibility loci as risk factors for triple-negative breast cancer. Carcinogenesis 2014, 35, 1012–1019. [Google Scholar] [CrossRef] [PubMed]
- Michailidou, K.; Hall, P.; Gonzalez-Neira, A.; Ghoussaini, M.; Dennis, J.; Milne, R.L.; Schmidt, M.K.; Chang-Claude, J.; Bojesen, S.E.; Bolla, M.K.; et al. Large-scale genotyping identifies 41 new loci associated with breast cancer risk. Nat. Genet. 2013, 45, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Michailidou, K.; Beesley, J.; Lindstrom, S.; Canisius, S.; Dennis, J.; Lush, M.J.; Maranian, M.J.; Bolla, M.K.; Wang, Q.; Shah, M.; et al. Genome-wide association analysis of more than 120,000 individuals identifies 15 new susceptibility loci for breast cancer. Nat. Genet. 2015, 47, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Schoeps, A.; Rudolph, A.; Seibold, P.; Dunning, A.M.; Milne, R.L.; Bojesen, S.E.; Swerdlow, A.; Andrulis, I.; Brenner, H.; Behrens, S.; et al. Identification of New Genetic Susceptibility Loci for Breast Cancer Through Consideration of Gene-Environment Interactions. Genet. Epidemiol. 2014, 38, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Ruark, E.; Snape, K.; Humburg, P.; Loveday, C.; Bajrami, I.; Brough, R.; Rodrigues, D.N.; Renwick, A.; Seal, S.; Ramsay, E.; et al. Mosaic PPM1D mutations are associated with predisposition to breast and ovarian cancer. Nature 2013, 493, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Chen, J. Association of MHTFR Ala222Val (rs1801133) polymorphism and breast cancer susceptibility: An update meta-analysis based on 51 research studies. Diagn. Pathol. 2012, 7, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Burns, M.B.; Lackey, L.; Carpenter, M.A.; Rathore, A.; Land, A.M.; Leonard, B.; Refsland, E.W.; Kotandeniya, D.; Tretyakova, N.; Nikas, J.B.; et al. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 2013, 494, 366–370. [Google Scholar] [CrossRef] [PubMed]
- MacPherson, G.; Healey, C.S.; Teare, M.D.; Balasubramanian, S.P.; Reed, M.W.; Pharoah, P.D.; Ponder, B.A.; Meuth, M.; Bhattacharyya, N.P.; Cox, A. Association of a common variant of the CASP8 gene with reduced risk of breast cancer. J. Natl. Cancer Inst. 2004, 96, 1866–1869. [Google Scholar] [CrossRef] [PubMed]
- Frank, B.; Bermejo, J.L.; Hemminki, K.; Klaes, R.; Bugert, P.; Wappenschmidt, B.; Schmutzler, R.K.; Burwinkel, B. Re: Association of a common variant of the CASP8 gene with reduced risk of breast cancer. J. Natl. Cancer Inst. 2005, 97, 1012. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.; Dunning, A.M.; Garcia-Closas, M.; Balasubramanian, S.; Reed, M.W.; Pooley, K.A.; Scollen, S.; Baynes, C.; Ponder, B.A.; Chanock, S.; et al. A common coding variant in CASP8 is associated with breast cancer risk. Nat. Genet. 2007, 39, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.H.; Sullivan, L.B.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; DeBerardinis, R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2012, 481, 385–388. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.L.; Kornbluth, S. The tangled circuitry of metabolism and apoptosis. Mol. Cell 2013, 49, 399–410. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.K.; Ayyasamy, V.; Owens, K.M.; Koul, M.S.; Vujcic, M. Mutations in mitochondrial DNA polymerase-gamma promote breast tumorigenesis. J. Hum. Genet. 2009, 54, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Platek, M.; Mahasneh, A.; Ambrosone, C.B.; Zhao, H. Mitochondrial copy number and risk of breast cancer: A pilot study. Mitochondrion 2010, 10, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Alhomidi, M.A.; Vedicherla, B.; Movva, S.; Rao, P.K.; Ahuja, Y.R.; Hasan, Q. Mitochondrial D310 instability in Asian Indian breast cancer patients. Tumour. Biol. 2013, 34, 2427–2432. [Google Scholar] [CrossRef] [PubMed]
- Imanishi, H.; Hattori, K.; Wada, R.; Ishikawa, K.; Fukuda, S.; Takenaga, K.; Nakada, K.; Hayashi, J.I. Mitochondrial DNA mutations regulate metastasis of human breast cancer cells. PLoS ONE 2011, 6, e23401. [Google Scholar] [CrossRef] [PubMed]
- Yadav, N.; Chandra, D. Mitochondrial DNA mutations and breast tumorigenesis. Biochem. Biophys. Acta 2013, 1836, 336–344. [Google Scholar] [CrossRef] [PubMed]
- Czarnecka, A.M.; Krawczyk, T.; Plak, K.; Klemba, A.; Zdrozny, M.; Arnold, R.S.; Kofler, B.; Golik, P.; Szybinska, A.; Lubinski, J.; et al. Mitochondrial genotype and breast cancer predisposition. Oncol. Rep. 2010, 24, 1521–1534. [Google Scholar] [PubMed]
- Bai, R.K.; Leal, S.M.; Covarrubias, D.; Liu, A.; Wong, L.J.C. Mitochondrial genetic background modifies breast cancer risk. Cancer Res. 2007, 67, 4687–4694. [Google Scholar] [CrossRef] [PubMed]
- Anderson, W.F.; Pfeiffer, R.M.; Dores, G.M.; Sherman, M.E. Comparison of age-distribution patterns of different histopathologic types of breast cancer. Cancer Epidemiol. Biomark. Prev. 2006, 15, 1899–1905. [Google Scholar] [CrossRef] [PubMed]
- Garaycoechea, J.I.; Crossan, G.P.; Langevin, F.; Mulderrig, L.; Louzada, S.; Yang, F.; Guilbaud, G.; Park, N.; Roerink, S.; Nik-Zainal, S.; et al. Alcohol and endogenous aldehydes damage chromosomes and mutate stem cells. Nature 2018, 171, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Mueller, S. Alcohol and cancer: An overview with special emphasis on the role of acetaldehyde and cytochrome P450 2E1. Adv. Exp. Med. Biol. 2015, 815, 59–70. [Google Scholar] [PubMed]
- Hoek, J.B.; Cahill, A.; Pastorino, J.G. Alcohol and mitochondria: A dysfunctional relationship. Gastroenterology 2002, 122, 2049–2063. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Lemasters, J.J. A Unifying Hypothesis Linking Hepatic Adaptations for Ethanol Metabolism to the Proinflammatory and Profibrotic Events of Alcoholic Liver Disease. Alcohol. Clin. Exp. Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Brooks, P.J.; Enoch, M.A.; Goldman, D.; Li, T.K.; Yokoyama, A. The Alcohol Flushing Response: An Unrecognized Risk Factor for Esophageal Cancer from Alcohol Consumption. PLoS Med. 2009, 6, e1000050. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Zakhari, S. Alcohol metabolism and epigenetics changes. Alcohol Res. Curr. Rev. 2013, 35, 6–16. [Google Scholar]
- Ndlovu, M.N.; Denis, H.; Fuke, F. Exposing the DNA methylome iceberg. Trends Biochem. Sci. 2011, 36, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Lo, P.K.; Sukumar, S. Epigenomics and breast cancer. Pharmacogenomics 2008, 9, 1879–1902. [Google Scholar] [CrossRef] [PubMed]
- Klarmann, G.; Decker, A.; Farrar, W.L. Epigenetic gene silencing in the Wnt pathway in breast cancer. Epigenetics 2008, 3, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Nowsheen, S.; Aziz, K.; Tran, P.T.; Gorgoulis, V.G.; Yang, E.S.; Georgakilas, A.G. Epigenetic inactivation of DNA repair in breast cancer. Cancer Lett. 2014, 342, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Lapidus, R.G.; Nass, S.J.; Butash, K.A.; Parl, F.F.; Weitzman, S.A.; Graff, J.G.; Herman, J.G.; Davidson, N.E. Mapping of ER gene CpG island methylation-specific polymerase chain reaction. Cancer Res. 1998, 58, 2515–2519. [Google Scholar] [PubMed]
- Ferguson, A.T.; Lapidus, R.G.; Baylin, S.B.; Davidson, N.E. Demethylation of the estrogen receptor gene in estrogen receptor-negative breast cancer cells can reactivate estrogen receptor gene expression. Cancer Res. 1995, 55, 2279–2283. [Google Scholar] [PubMed]
- Locke, W.J.; Clark, S.J. Epigenome remodelling in breast cancer: Insights from an early in vitro model of carcinogenesis. Breast Cancer Res. 2012, 14, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, M. Cancer-linked DNA hypomethylation and its relationship to hpermethylation. Curr. Top. Microbiol. Immunol. 2006, 310, 251–274. [Google Scholar] [PubMed]
- Soares, J.; Pinto, A.E.; Cunha, C.V.; André, S.; Barao, I.; Sousa, J.M.; Cravo, M. Global DNA hypomethylation in breast carcinoma: Correlation with prognostic factors and tumor progression. Cancer 1999, 85, 112–118. [Google Scholar] [CrossRef]
- Fan, M.; Yan, P.S.; Hartman-Frey, C.; Chen, L.; Paik, H.; Oyer, S.L.; Salisbury, J.D.; Cheng, A.S.; Li, L.; Abbosh, P.H.; et al. Diverse gene expression and DNA methylation profiles correlate with differential adaptation of breast cancer cells to the antiestrogen tamoxifen and fulvestrant. Cancer Res. 2006, 66, 11954–11966. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C.; Huang, Z.Z.; Yang, H.; Mato, J.M.; Avila, M.A.; Tsukamoto, H. Changes in methionine adenosyltransferase and S-adenosylmethionine homeostasis in the alcoholic rat liver. Am. J. Physiol. Gatrointest. Liver Physiol. 2000, 279, G178–G185. [Google Scholar] [CrossRef] [PubMed]
- Christensen, B.C.; Kelsey, K.T.; Zheng, S.; Houseman, E.A.; Marsit, C.J.; Wrensch, M.R.; Wiemels, J.L.; Nelson, H.H.; Karagas, M.R.; Kushi, L.H.; et al. Breast cancer DNA methylation profiles are associated with tumor size and alcohol and folate intake. PLoS Genet. 2010, 6, e1001043. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Davidson, N.E.; Hunter, S.; Yang, X.; Payne-Wilks, K.; Roland, C.L.; Phillips, D.; Bentley, C.; Dai, M.; Williams, S.M. Methyl-group dietary intake and risk of breast cancer among African-American women: A case-control study by methylation status of the estrogen receptor alpha genes. Cancer Causes Control 2003, 14, 827–836. [Google Scholar] [CrossRef] [PubMed]
- Tao, M.H.; Marian, C.; Shields, P.G.; Nie, J.; McCann, S.E.; Millen, A.; Ambrosone, C.; Hutson, A.; Edge, S.B.; Krishnan, S.S.; et al. Alcohol consumption in relation to aberrant DNA methylation in breast tumors. Alcohol 2011, 45, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Kato, I.; Dnistrian, A.M.; Schwartz, M.; Toniolo, P.; Koenig, K.; Shore, R.E.; Akhmedkhanov, A.; Zeleniuch-Jacquotte, A.; Riboli, E. Serum folate, homocysteine and colorectal cancer risk in women: A nested case-study. Br. J. Cancer 1999, 79, 1917–1922. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.I. Folate and DNA methylation: A mechanistic link between folate deficiency and colorectal cancer? Cancer Epidemiol. Biomark. Prev. 2004, 13, 511–519. [Google Scholar]
- Nazki, F.H.; Sameer, A.S.; Ganaie, B.A. Folate: Metabolism, genes, polymorphisms and the associated diseases. Gene 2014, 533, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Ma, E.; Iwasaki, M.; Junko, I.; Hamada, G.S.; Nishimoto, I.N.; Carvalho, S.M.T.; Motola, J.; Laginha, F.M.; Tsugane, S. Dietary intake of folate, vitamin B6, and vitamin B12, genetic polymorphism of related enzymes, and risk of breast cancer; a case-study in Brazilian women. BMC Cancer 2009, 9, 122. [Google Scholar] [CrossRef] [PubMed]
- Blount, B.C.; Mack, M.M.; Wehr, C.M.; MacGregor, J.T.; Hiatt, R.A.; Wang, G.; Wickramasinghe, S.N.; Everson, R.B.; Ames, B.N. Folate deficiency causes uracil misincorporation into human DNA and chromosome breakage: Implications for cancer and neuronal damage. Proc. Natl. Acad. Sci. USA 1997, 94, 3290–3295. [Google Scholar] [CrossRef]
- Platek, M.E.; Shields, P.G.; Marian, C.; McCann, S.E.; Bonner, M.R.; Nie, J.; Ambrosone, C.B.; Millen, A.E.; Ochs-Balcom, H.M.; Quick, S.K.; et al. Alcohol consumption and genetic variation in methylenetetrahydro-folate reductase and 5-methyltetrahydrofolate-homocysteine methyltransferase in relation to breast cancer risk. Cancer Epidemiol. Biomark. Prev. 2009, 18, 2453–2459. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Cui, L.H.; Ma, A.G.; Li, N.; Piao, J.M. Lack of effects of dietary folate intake on risk of breast cancer: An updated meta-analysis of prospective studies. Asian Pac. J. Cancer Prev. 2014, 15, 2323–2328. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.C.; Giovannucci, E.; Wolk, A. Folate and risk of breast cancer: A meta-analysis. J. Natl. Cancer Inst. 2007, 99, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Wade, P.A. Transcriptional control at regulatory checkpoints by histone deacetylases: Molecular connections between cancer and chromatin. Hum. Mol. Genet. 2001, 10, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Tryndyak, V.P.; Kovalchuk, O.; Pogribny, I.P. Loss of DNA methylation and histone H4 lysine 20 trimethylation in human breast cancer cells is associated with aberrant expression of DNA methyltransferases 1, Suv4-20h2 histone methyltransferases and methyl-binding proteins. Cancer Biol. Ther. 2006, 5, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.Y.; Bang, Y.J.; Robertson, K.D. Histone deacetylase inhibitors for cancer therapy. Epigenetics 2006, 1, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Vadigepalli, R.; Hoek, J.B. Introduction to the Virtual Issue Alcohol and Epigenetic Regulation: Do the Products of Alcohol Metabolism Drive Epigenetic Control of Gene Expression in Alcohol-Related Disorders? ACER 2018, 42, 845. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.S.; Culhane, A.C.; Chan, M.W.; Venkataramu, C.R.; Ehrich, M.; Nasir, A.; Rodriguez, B.A.; Liu, J.; Yan, P.S.; Quackenbush, J.; et al. Epithelial progeny of estrogen-exposed breast progenitor cells display a cancer-like methylome. Cancer Res. 2008, 68, 1786–1796. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.Y.; Deatherage, D.E.; Rodriguez, B.A.; Liyanarachchi, S.; Weng, Y.I.; Zuo, T.; Liu, J.; Cheng, A.S.; Huang, T.H. Xenoestrogen-induced epigenetic repression of microRNA-9-3 in breast epithelial cells. Cancer Res. 2009, 69, 5936–5945. [Google Scholar] [CrossRef] [PubMed]
- Hervouet, E.; Cartron, P.F.; Jouvenot, M.; Delage-Mourroux, R. Epigenetic regulation of estrogen signaling in breast cancer. Epigenetics 2013, 8, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.N.; Hata, A. MicroRNA in cancer – the involvement of aberrant microRNA biogenesis regulatory pathways. Genes Cancer 2010, 1, 1100–1114. [Google Scholar] [CrossRef] [PubMed]
- Barski, A.; Jothi, R.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Zhao, K. Chromatin poises miRNA- and protein-coding genes for expression. Genome Res. 2009, 19, 1742–1751. [Google Scholar] [CrossRef] [PubMed]
- Guil, S.; Esteller, M. DNA methylomes, histone codes and miRNAs: Tying it all together. Int. J. Biochem. Cell Biol. 2009, 41, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Ibarra, I.; Erlich, Y.; Muthuswamy, S.K.; Sachidanandam, R.; Hannon, G.J. A role for microRNAs in maintenance of mouse mammary epithelial progenitor cells. Genes Dev. 2007, 21, 3238–3243. [Google Scholar] [CrossRef] [PubMed]
- Shimono, Y.; Zabala, M.; Cho, R.W.; Lobo, N.; Dalerba, P.; Qian, D.; Diehn, M.; Liu, H.; Panula, S.P.; Chiao, E.; et al. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell 2009, 138, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Haneda, S.; Imakawa, K.; Sakai, S.; Nagaoka, K. A microRNA, miR-101a, controls mammary gland development by regulating cyclooxygenase-2 expression. Differentiation 2009, 77, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Greene, S.B.; Gunaratne, P.H.; Hammond, S.M.; Rosen, J.M. A putative role for microRNA-205 in progenitors of mammary epithelial cells. J. Cell Sci. 2010, 123, 606–618. [Google Scholar] [CrossRef] [PubMed]
- Place, R.F.; Li, L.C.; Pookot, D.; Noonan, E.J.; Dahiya, R. MicroRNA-373 induces expression of genes with complementary promoter sequence. Proc. Natl. Acad. Sci. USA 2008, 105, 1608–1613. [Google Scholar] [CrossRef] [PubMed]
- Iorio, M.V.; Ferracin, M.; Liu, C.G.; Veronese, A.; Spizzo, R.; Sabbioni, S.; Magri, E.; Pedriali, M.; Fabbri, M.; Campiglio, M.; et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005, 65, 7065–7070. [Google Scholar] [CrossRef] [PubMed]
- Simonini, P.D.S.R.; Breiling, A.; Gupta, N.; Malekpour, M.; Youns, M.; Omranipour, R.; Malekpour, F.; Volinia, S.; Croce, C.M.; Najmabadi, H.; et al. Epigenetically deregulated microRNA-375 is involved in a positive feedback loop with estrogen receptor alpha in breast cancer cells. Cancer Res. 2010, 70, 9175–9184. [Google Scholar] [CrossRef] [PubMed]
- Parrella, P. Epigenetic signature of breast cancer: Clinical perspective. Breast Care 2010, 5, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Boyle, P.; Boffetta, P. Alcohol Consumption and Breast Cancer Risk. 2009. Available online: http://breast-cancer-research.com/supplements/11/S3/S3 (accessed on 5 February 2018).
- Seitz, H.K.; Pelucchi, C.; Bagnard, V.; La Vecchia, C. Epidemiology and pathophysiology of alcohol and breast cancer: Update 2012. Alcohol Alcohol. 2012, 47, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Falk, R.T.; Maas, P.; Schairer, C.; Chatterjee, N.; Mabie, J.E.; Cunningham, C.; Buys, S.S.; Isaacs, C.; Ziegler, R.G. Alcohol and Risk of Breast Cancer in Postmenopausal Women: An Analysis of Etiological Heterogeneity by Multiple Tumor Characteristics. Am. J. Epidemiol. 2014, 180, 705–717. [Google Scholar] [CrossRef] [PubMed]
- Mullooly, M.; Khodr, Z.G.; Dallal, C.M.; Nyante, S.J.; Sherman, M.E.; Falk, R.; Liao, L.M.; Love, J.; Brinton, L.A.; Gierach, G.L. Epidemiologic Risk Factors for In Situ and Invasive Breast Cancers Among Postmenopausal Women in the National Institutes of Health-AARP Diet and Health Study. Am. J. Epidemiol. 2017, 186, 1329–1340. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Jung, S.; Eliassen, A.H.; Chen, W.Y.; Willett, W.C.; Cho, E. Alcohol Consumption and Breast Cancer Risk in Younger Women According to Family History of Breast Cancer and Folate Intake. Am. J. Epidemiol. 2017, 186, 524–531. [Google Scholar] [CrossRef] [PubMed]
- White, A.J.; DeRoo, L.A.; Weinberg, C.R.; Sandler, D.P. Lifetime Alcohol Intake, Binge Drinking Behaviors, and Breast Cancer Risk. Am. J. Epidemiol. 2017, 186, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Hirko, K.A.; Chen, W.Y.; Willett, W.C.; Rosner, B.A.; Hankinson, S.E.; Beck, A.H.; Tamimi, R.M.; Eliassen, A.H. Alcohol consumption and risk of breast cancer by molecular subtype: Prospective analysis of the nurses’ health study after 26 years of follow-up. Int. J. Cancer 2016, 138, 1094–1101. [Google Scholar] [CrossRef] [PubMed]
- Williams, L.A.; Olshan, A.F.; Tse, C.K.; Bell, M.E.; Troester, M.A. Alcohol intake and invasive breast cancer risk by molecular subtype and race in the Carolina Breast Cancer Study. Cancer Causes Control 2016, 27, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Dartois, L.; Fagherazzi, G.; Baglietto, L.; Boutron-Ruault, M.C.; Delaloge, S.; Mesrine, S.; Clavel-Chapelon, F. Proportion of premenopausal and postmenopausal breast cancers attributable to known risk factors: Estimates from the E3N-EPIC cohort. Int. J. Cancer 2016, 138, 2415–2427. [Google Scholar] [CrossRef] [PubMed]
- Romieu, I.; Scoccianti, C.; Chajes, V.; de Batlle, J.; Biessy, C.; Dossus, L.; Baglietto, L.; Clavel-Chapelon, F.; Overvad, K.; Olsen, A.; et al. Alcohol intake and breast cancer in the European prospective investigation into cancer and nutrition. Int. J. Cancer 2015, 137, 1921–1930. [Google Scholar] [CrossRef] [PubMed]
- Strumylaite, L.; Sharp, S.J.; Kregzdyte, R.; Poskiene, L.; Bogusevicius, A.; Pranys, D. The Association of Low-To-Moderate Alcohol Consumption with Breast Cancer Subtypes Defined by Hormone Receptor Status. PLoS ONE 2015, 10, e0144680. [Google Scholar] [CrossRef] [PubMed]
- Shin, A.; Sandin, S.; Lof, M.; Margolis, K.L.; Kim, K.; Couto, E.; Adami, H.O.; Weiderpass, E. Alcohol consumption, body mass index and breast cancer risk by hormone receptor status: Women’ Lifestyle and Health Study. BMC Cancer 2015, 15, 881. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, X.; Beck, A.H.; Collins, L.C.; Chen, W.Y.; Tamimi, R.M.; Hazra, A.; Brown, M.; Rosner, B.; Hankinson, S.E. Alcohol Consumption and Risk of Breast Cancer by Tumor Receptor Expression. Horm. Cancer 2015, 6, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Hvidtfeldt, U.A.; Tjønneland, A.; Keiding, N.; Lange, T.; Andersen, I.; Sørensen, T.I.; Prescott, E.; Hansen, Å.M.; Grønbæk, M.; Bojesen, S.E. Risk of Breast Cancer in Relation to Combined Effects of Hormone Therapy, Body Mass Index, and Alcohol Use, by Hormone-receptor Status. Epidemiology 2015, 26, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Islam, T.; Ito, H.; Sueta, A.; Hosono, S.; Hirose, K.; Watanabe, M.; Iwata, H.; Tajima, K.; Tanaka, H.; Matsuo, K. Alcohol and dietary folate intake and the risk of breast cancer: A case-control study in Japan. Eur. J. Cancer Prev. 2013, 4, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.Y.; Rosner, B.; Hankinson, S.E.; Colditz, G.A.; Willett, W.C. Moderate alcohol consumption during adult life, drinking patterns, and breast cancer risk. JAMA 2011, 30617, 1884–1890. [Google Scholar] [CrossRef] [PubMed]
- Kawai, M.; Minami, Y.; Kakizaki, M.; Kakugawa, Y.; Nishino, Y.; Fukao, A.; Tsuji, I.; Ohuchi, N. Alcohol consumption and breast cancer risk in Japanese women: The Miyagi Cohort study. Breast Cancer Res. Treat. 2011, 1283, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, R.; Iwasaki, M.; Inoue, M.; Sasazuki, S.; Sawada, N.; Yamaji, T.; Shimazu, T.; Tsugane, S. Alcohol consumption-associated breast cancer incidence and potential effect modifiers: The Japan Public Health Center-based Prospective Study. Int. J. Cancer 2010, 1273, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Bessaoud, F.; Daures, J.P. Patterns of alcohol (especially wine) consumption and breast cancer risk; a case-control study among a population in Southern France. Ann. Epidemiol. 2008, 6, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Dumitrescu, R.G.; Shields, P.G. The etiology of alcohol-induced breast cancer. Alcohol 2005, 353, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Mahabir, S.; Pfeiffer, R.; Xu, X.; Baer, D.J.; Taylor, P.R. Effects of low-to-moderate alcohol supplementation on urinary estrogen metabolites in postmenopausal women in a controlled feeding study. Cancer Med. 2017, 6, 2419–2423. [Google Scholar] [CrossRef] [PubMed]
- Sangrajrang, S.; Sato, Y.; Sakamoto, H.; Ohnami, S.; Khuhaprema, T.; Yoshida, T. Genetic polymorphisms in folate and alcohol metabolism and breast cancer risk: A case-control study in Thai women. Breast Cancer Res. Treat. 2010, 123, 885–893. [Google Scholar] [CrossRef] [PubMed]
- Kawase, T.; Matsuo, K.; Hiraki, A.; Suzuki, T.; Watanabe, M.; Iwata, H.; Tanaka, H.; Tajima, K. Interaction of the effects of alcohol drinking and polymorphisms in alcohol-metabolizing enzymes on the risk of female breast cancer in Japan. J. Epidemiol. 2009, 19, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.Y.; Abel, J.; Neuhaus, T.; Ko, Y.; Harth, V.; Hamajima, N.; Tajima, K.; Yoo, K.Y.; Park, S.K.; Noh, D.Y.; et al. Role of alcohol and genetic polymorphisms of CYP2E1 and ALDH2 in breast cancer development. Pharmacogenetics 2003, 13, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Playdon, M.C.; Ziegler, R.G.; Sampson, J.N.; Stolzenberg-Solomon, R.; Thompson, H.J.; Irwin, M.L.; Mayne, S.T.; Hoover, R.N.; Moore, S.C. Nutritional metabolomics and breast cancer risk in a prospective study. Am. J. Clin. Nutr. 2017, 106, 637–649. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, M.; Ke, Z.; Luo, J. Cellular and Molecular Mechanism Underlying Alcohol-induced Aggressiveness of Breast Cancer. Pharmacol. Res. 2017, 115, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.D.; Ebrahim, S. ‘Mendelian Randomization’: Can genetic epidemiology contribute to understanding environmental determinants of disease? Int. J. Epidemiol. 2003, 32, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Peto, R.; Doll, R.; Buckley, J.D.; Sporn, M.B. Can dietary beta-carotene materially reduce human cancer rates? Nature 1981, 290, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, R.G.; Colavito, E.A.; Hartge, P.; McAdams, M.J.; Schoenberg, J.B.; Mason, T.J.; Fraumeni, J.F., Jr. Importance of a-carotene, b-carotene, and other phytochemicals in the etiology of lung cancer. J. Natl. Cancer Inst. 1996, 88, 612–615. [Google Scholar] [CrossRef] [PubMed]
- Gallicchio, L.; Boyd, K.; Matanoski, G.; Tao, X.; Chen, L.; Lam, T.K.; Shiels, M.; Hammond, E.; Robinson, K.A.; Caulfield, L.E.; et al. Carotenoids and the risk of developing lung cancer: A systematic review. Am. J. Clin. Nutr. 2008, 88, 372–383. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.M.; Cook, N.R.; Gaziano, J.M.; Gordon, D.; Ridker, P.M.; Manson, J.E.; Hennekens, C.H.; Buring, J.E. Vitamin E in the primary prevention of cardiovascular disease and cancer: The Women’s Health Study: A randomized controlled trial. JAMA 2005, 294, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Tatsioni, A.; Bonitsis, N.G.; Ioannidis, J.P. Persistence of contradicted claims in the literature. JAMA 2007, 298, 2517–2526. [Google Scholar] [CrossRef] [PubMed]
- Phillips, A.N.; Smith, G.D. How independent are ‘independent’ effects? Relative risk estimation when correlated exposures are measured imprecisely. J. Clin. Epidemiol. 1991, 44, 1223–1231. [Google Scholar] [CrossRef]
- Phillips, A.N.; Smith, G.D. The design of prospective epidemiological studies: More subjects or better measurements? J. Clin. Epidemiol. 1993, 46, 1203–1211. [Google Scholar] [CrossRef]
- Longnecker, M.P. Alcoholic beverage consumption in relation to risk of breast cancer: Meta-analysis and review. Cancer Causes Control 1994, 51, 73–82. [Google Scholar] [CrossRef]
- Corrao, G.; Bagnardi, V.; Zambon, A.; La Vecchia, C. A meta-analysis of alcohol consumption and the risk of 15 diseases. Prev. Med. 2004, 385, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Key, J.; Hodgson, S.; Omar, R.Z.; Jensen, T.K.; Thompson, S.G.; Boobis, A.R.; Davies, D.S.; Elliott, P. Meta-analysis of studies of alcohol and breast cancer with consideration of the methodological issues. Cancer Causes Control 2006, 6, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, R.; Orsini, N.; Mignone, L.; Saji, S.; Wolk, A. Alcohol intake and risk of breast cancer defined by estrogen and progesterone receptor status-a meta-analysis of epidemiological studies. Int. J. Cancer 2008, 1228, 1832–1841. [Google Scholar] [CrossRef] [PubMed]
- Bagnardi, V.; Rota, M.; Botteri, E.; Tramacere, I.; Islami, F.; Fedirko, V.; Scotti, L.; Jenab, M.; Turati, F.; Pasquali, E.; et al. Light alcohol drinking and cancer: A meta-analysis. Ann. Oncol. 2013, 24, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Bagnardi, V.; Rota, M.; Botteri, E.; Tramacere, I.; Islami, F.; Fedirko, V.; Scotti, L.; Jenab, M.; Turati, F.; Pasquali, E.; et al. Alcohol consumption and site-specific cancer risk: A comprehensive dose-response meta-analysis. Br. J. Cancer 2015, 112, 580–593. [Google Scholar] [CrossRef] [PubMed]
- Jayasekara, H.; MacInnis, R.J.; Room, R.; English, D.R. Long-Term Alcohol Consumption and Breast, Upper Aero-Digestive Tract and Colorectal Cancer Risk: A Systematic Review and Meta-Analysis. Alcohol Alcohol. 2016, 51, 315–330. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Myung, S.K.; Lee, J.H. Light Alcohol Drinking and Risk of Cancer: A Meta-analysis of Cohort Studies. Cancer Res. Treat. 2018, 50, 474–487. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Wang, M.; Anderson, K.; Baglietto, L.; Bergkvist, L.; Bernstein, L.; van den Brandt, P.A.; Brinton, L.; Buring, J.E.; et al. Alcohol consumption and breast cancer risk by estrogen receptor status: In a pooled analysis of 20 studies. Int. J. Epidemiol. 2016, 45, 916–928. [Google Scholar] [CrossRef] [PubMed]
- Schoenfeld, J.; Ioannidis, P.A. Is everything we eat associated with cancer? A Systematic cookbook review. Am. J. Clin. Nutr. 2013, 97, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Song, F.; Parekh, S.; Hooper, L.; Loke, Y.K.; Ryder, J.; Sutton, A.J.; Hing, C.; Kwok, C.S.; Pang, C.; et al. Dissemination and publication of research findings: An updated review of related biases. Health Technol. Assess. 2010, 14, 1–193. [Google Scholar] [CrossRef] [PubMed]
- Egger, M.; Schneider, M.; Smith, G.D. Spurious precision? Meta-analysis of observational studies. BMJ 1988, 316, 140–144. [Google Scholar] [CrossRef]
- Ioannidis, J.P.A. The challenge of reforming nutritional epidemiologic research. JAMA 2018, 320, 969–970. [Google Scholar] [CrossRef]
- Theodoratou, E.; Timofeeva, M.; Li, X.; Meng, X.; Ioannidis, J.P.A. Nature, Nurture, and Cancer Risks: Genetic and Nutritional Contributions to Cancer. Annu. Rev. Nutr. 2017, 37, 293–320. [Google Scholar] [CrossRef] [PubMed]
- Dietary Guidelines for Americans 2015–2020. Available online: https://health.gov/dietaryguidelines/2015/resources/2015-2020_Dietary_Guidelines.pdf (accessed on 18 February 2018).
- Klatsky, A.L.; Udaltsova, N.; Li, Y.; Baer, D.; Tran, H.N.; Friedman, G.D. Moderate alcohol intake and cancer: The role of underreporting. Cancer Causes Control 2014, 25, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Schatzkin, A.; Kipnis, V. Could exposure assessment problems give us wrong answers to nutrition and cancer questions? J. Natl. Cancer Inst. 2004, 96, 1564–1565. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, D.A.; Smith, G.D.; Kundu, D.; Bruckdorfer, K.R.; Ebrahim, S. Those confounded vitamins: What can we learn from the differences between observational versus randomised trial evidence? Lancet 2004, 363, 1724–1727. [Google Scholar] [CrossRef]
- Goodman, S.N.; Samet, J.A. Causal inference in cancer epidemiology. In Cancer Epidemiology and Prevention; Thun, M.J., Linet, M.S., Cerhan, J.R., Haiman, C.A., Schottenfeld, D., Eds.; Oxford University Press: Oxford, UK, 2017; pp. 97–104. [Google Scholar]
- Hill, A.B. The Environment and Disease: Association or Causation? Proc. R. Soc. Med. 1965, 58, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Collins, F.S.; Tabak, L.A. Policy: NIH plans to enhance reproducibility. Nature 2014, 505, 612–613. [Google Scholar] [CrossRef] [PubMed]
- Begley, C.G.; Ioannidis, J.P. Reproducibility in science: Improving the standard for basic and preclinical research. Circ. Res. 2015, 116, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.D.; Lawlor, D.A.; Harbord, R.; Timpson, N.; Day, I.; Ebrahim, S. Clustered Environments and Randomized Genes: A Fundamental Distinction between Conventional and Genetic Epidemiology. PLoS Med. 2007, 4, e352. [Google Scholar] [CrossRef] [PubMed]
- Haycock, P.C.; Burgess, S.; Wade, K.H.; Bowden, J.; Relton, C.; Smith, G.D. Best (but oft-forgotten) practices: The design, analysis, and interpretation of Mendelian randomization studies. Am. J. Clin. Nutr. 2016, 103, 965–978. [Google Scholar] [CrossRef] [PubMed]
- Marshall, B.D.; Galea, S. Formalizing the role of agent-based modeling in causal inference and epidemiology. Am. J. Epidemiol. 2015, 181, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Vanderweele, T.J.; Vansteelandt, S.; Robins, J.M. Marginal Structural Models for Sufficient Cause Interactions. Am. J. Epidemiol. 2010, 171, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Hernán, M.A. Invited Commentary: Agent-Based Models for Causal Inference—Reweighting Data and Theory in Epidemiology. Am. J. Epidemiol. 2015, 181, 103–105. [Google Scholar] [CrossRef] [PubMed]
- Collins, F.S.; Varmus, H. A new initiative on precision medicine. N. Engl. J. Med. 2015, 372, 793–795. [Google Scholar] [CrossRef] [PubMed]
- Nishi, A.; Milner, D.A., Jr.; Giovannucci, E.L.; Nishihara, R.; Tan, A.S.; Kawachi, I.; Ogino, S. Integration of Molecular Pathology, Epidemiology, and Social Science for Global Precision Medicine. Expert Rev. Mol. Diagn. 2016, 16, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Nishihara, R.; VanderWeele, T.J.; Wang, M.; Nishi, A.; Lochhead, P.; Qian, Z.R.; Zhang, X.; Wu, K.; Nan, H.; et al. The role of molecular pathological epidemiology in the study of neoplastic and non-neoplastic diseases in the era of precision medicine. Epidemiology 2016, 27, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Stampfer, M. Lifestyle Factors and Microsatellite Instability in Colorectal Cancer: The Evolving Field of Molecular Pathological Epidemiology. JNCI 2010, 102, 365–367. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Lochhead, P.; Chan, A.T.; Nishihara, R.; Cho, E.; Wolpin, B.M.; Meyerhardt, J.A.; Meissner, A.; Schernhammer, E.S.; Fuchs, C.S.; et al. Molecular pathological epidemiology of epigenetics: Emerging integrative science to analyze environment, host, and disease. Mod. Pathol. 2013, 26, 465–484. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Nowak, J.A.; Hamada, T.; Phipps, A.I.; Peters, U.; Milner, D.A., Jr.; Giovannucci, E.L.; Nishihara, R.; Giannakis, M.; Garrett, W.S.; et al. Integrative analysis of exogenous, endogenous, tumour and immune factors for precision medicine. Gut 2018, 67, 1168–1180. [Google Scholar] [CrossRef] [PubMed]
- Babic, A.; Shah, S.M.; Song, M.; Wu, K.; Meyerhardt, J.A.; Ogino, S.; Yuan, C.; Giovannucci, E.L.; Chan, A.T.; Stampfer, M.J.; et al. Soluble tumour necrosis factor receptor type II and survival in colorectal cancer. Br. J. Cancer 2016, 114, 995–1002. [Google Scholar] [CrossRef] [PubMed]
- Babic, A.; Bao, Y.; Qian, Z.R.; Yuan, C.; Giovannucci, E.L.; Aschard, H.; Kraft, P.; Amundadottir, L.T.; Stolzenberg-Solomon, R.Z.; Morales-Oyarvide, V.; et al. Pancreatic cancer risk associated with prediagnostic plasma levels of leptin and leptin receptor genetic polymorphisms. Cancer Res. 2016, 76, 7160–7167. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.; Clish, C.B.; Wu, C.; Mayers, J.R.; Kraft, P.; Townsend, M.K.; Zhang, M.; Tworoger, S.S.; Bao, Y.; Qian, Z.R.; et al. Circulating Metabolites and Survival Among Patients with Pancreatic Cancer. JNCI J. Natl. Cancer Inst. 2016, 108, djv409. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Nishihara, R.; Wu, K.; Qian, Z.R.; Kim, S.A.; Sukawa, Y.; Mima, K.; Inamura, K.; Masuda, A.; Yang, J.; et al. Marine omega-3 Polyunsaturated Fatty Acids and Risk of Colorectal Cancer According to Microsatellite Instability. J. Natl. Cancer Inst. 2015, 107, djv007. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Chan, A.T.; Fuchs, C.S.; Giovannucci, E. Molecular pathological epidemiology of colorectal neoplasia: An emerging transdisciplinary and interdisciplinary field. Gut 2011, 60, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Begg, C.B. A Strategy for Distinguishing Optimal Cancer Sub-Types. Int. J. Cancer 2011, 129, 931–937. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Number | Study Design | Age (yearrs) at Baseline | Data Collection | Unit of Measurement | Outcomes | Ref |
|---|---|---|---|---|---|---|
| 2017 | ||||||
| 1 | Longitudinal cohort NIH-AARP Diet and Health Study 190,325 postmenopausal women | 55–70 | Self-report | Avg. alcohol consumption (g/day) in 12 mo. before questionnaire completion |
| [108] |
| 2 | Prospective cohort Nurses’ Health Study II 93,835 US women | 27–44 | Semi-quantitative food questionnaire | Calculated total daily alcohol consumption |
| [109] |
| 3 | Sister Study 50,884 women | 35–74 | Self-report | Lifetime alcohol intake |
| [110] |
| 2016 | ||||||
| 4 | Prospective cohort Nurse’s Health Study 105,972 women | 30–55 | Semi-quantitative food frequency questionnaire | Cumulative average alcohol intake |
| [111] |
| 5 | Case-control Carolina Breast Cancer Study; 781 Afr. Am. women; 1014 White women | 25–50 | Alcohol intake (self-report) most proximal to diagnosis | Drinks per week |
| [112] |
| 2015 | ||||||
| 6 | Cohort study French E3N-EPIC 66,481 women | 40–65 | Self-report diet-history questionnaire | Cumulative average drinks/day |
| [113] |
| 7 | Prospective EPIC Study 334,850 women | 35–70 | Dietary and lifestyle questionnaires | Average lifetime alcohol intake |
| [114] |
| 8 | Case-control 585 cases | 28–90 | Self-administered questionnaire | Total number of alcoholic drinks per week |
| [115] |
| 9 | Prospective 45,233 women | 30–49 | Self-report | Current number of drinks/week converted to g/day |
| [116] |
| 10 | Prospective NHS | 30–55 | Self-report | Cumulative average intake per day |
| [117] |
| 11 | Prospective cohorts (2) Danish | 50+ | Self-report | Avg. drinks per week |
| [118] |
| 2013 and previous | ||||||
| 12 | Case Control, 2013 Japanese cohort 1754 pre- and postmenopausal women | 20–79 | Self-reported alcohol drinking | Avg. consumption g/day |
| [119] |
| 13 | Prospective observational, 2011 Nurses’ Health Study 105,986 women | Avg. 60 | Semiquantitative food frequency questionnaire | Avg. daily consumption in g/day |
| [120] |
| 14 | Prospective control, 2011 Japanese cohort 19,227 patients | 40–64 | Food frequency questionnaire | Avg. consumption g/day |
| [121] |
| 15 | Prospective, 2010 50,757 pre- and postmenopausal Japanese women | 40–69 | Self-reported questionnaires | Average consumption g/week |
| [122] |
| 16 | Case control, 2008 437 women | 25–85 | Structured questionnaire administered by two interviewers | Average consumption g/day |
| [123] |
| Study | # of Studies Included | Definitions of Drinking (g/day) | Relative Risk | Confidence Interval (95%) | Comments | Ref. |
|---|---|---|---|---|---|---|
| A | 38 10 follow-up 28 case-control | 13 g alcohol (~1 drink)/day | 1.10 | 1.08–1.17 | “Modest size of the association and variation in results across studies leaves the causal role of alcohol in question” | [139] |
| B | 29 24 case-control 5 cohort | 25 g/d (~1.8 drinks) 50 g/d (~3.6 drinks) 100 g/d (~7.1 drinks) | 1.25 1.55 2.41 | 1.20–1.29 1.44–1.67 2.07–2.80 | All doses are higher than moderate drinking | [140] |
| C | 85 77 retrospective 8 prospective studies | Drinkers vs. non-drinkers Dose response | 1.11 | 1.06–1.17 increased risk by 12% for 10 g/day | No quantification of amount of alcohol consumed | [141] |
| D | 16 4 prospective cohort 12 case-control | Dose response for all ER+, ER−, PR+ & PR-tumors. Increased risk 10 g ethanol/day | 12% ER+ 07% ER− 11% ER+/PR+ 15% ER+/PR− ER−/PR− | 8%–15% 0%–14% 7%–14% 2%–30% No significant association |
| [142] |
| E | 110 39 cohort 71 case-control | 1.05 | 1.02–1.08 |
| [143] | |
| F | 118 43 cohort 75 case-control | Light (≤12.5 g or ~1 drink) Moderate (≤50 g or ~3.6 drinks) Heavy (>50 g or >3.6 drinks) | 1.04 1.23 1.61 | 1.01–1.07 1.19–1.28 1.33–1.94 | According to US dietary guidelines moderate drinking is no more than one drink/day | [144] |
| G | 16 13 case-control 3 cohort | Highest vs. lowest category of alcohol intake | 1.28 | 1.07–1.52 |
| [145] |
| H | 34 cohort studies | ≤0.5 drink/day ≤1.0 drink/day | 1.04 1.09 | 1.01–1.07 1.06–1.12 | A small number of cohort studies in Asian populations were included | [146] |
| I | 20 prospective cohort 1,089,273 women | ≥30 g/day 5 to <15 g/day | 1.35 ER+ 1.28 ER− BC even among women with high folate intake 1.12 ER+ 1.19 ER− | 1.23–1.48 1.10–1.49 1.07–1.18 1.08–1.31 |
| [147] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zakhari, S.; Hoek, J.B. Epidemiology of Moderate Alcohol Consumption and Breast Cancer: Association or Causation? Cancers 2018, 10, 349. https://doi.org/10.3390/cancers10100349
Zakhari S, Hoek JB. Epidemiology of Moderate Alcohol Consumption and Breast Cancer: Association or Causation? Cancers. 2018; 10(10):349. https://doi.org/10.3390/cancers10100349
Chicago/Turabian StyleZakhari, Samir, and Jan B. Hoek. 2018. "Epidemiology of Moderate Alcohol Consumption and Breast Cancer: Association or Causation?" Cancers 10, no. 10: 349. https://doi.org/10.3390/cancers10100349
APA StyleZakhari, S., & Hoek, J. B. (2018). Epidemiology of Moderate Alcohol Consumption and Breast Cancer: Association or Causation? Cancers, 10(10), 349. https://doi.org/10.3390/cancers10100349