Role of COX-2/PGE2 Mediated Inflammation in Oral Squamous Cell Carcinoma


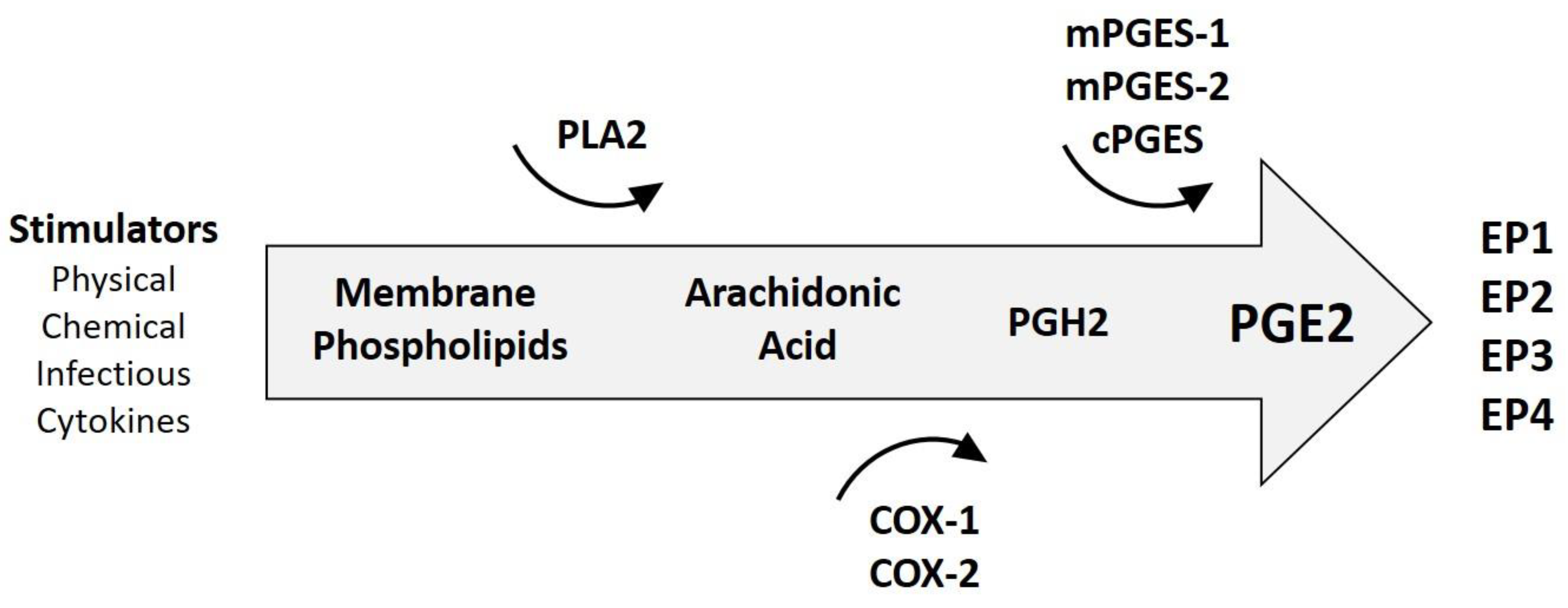
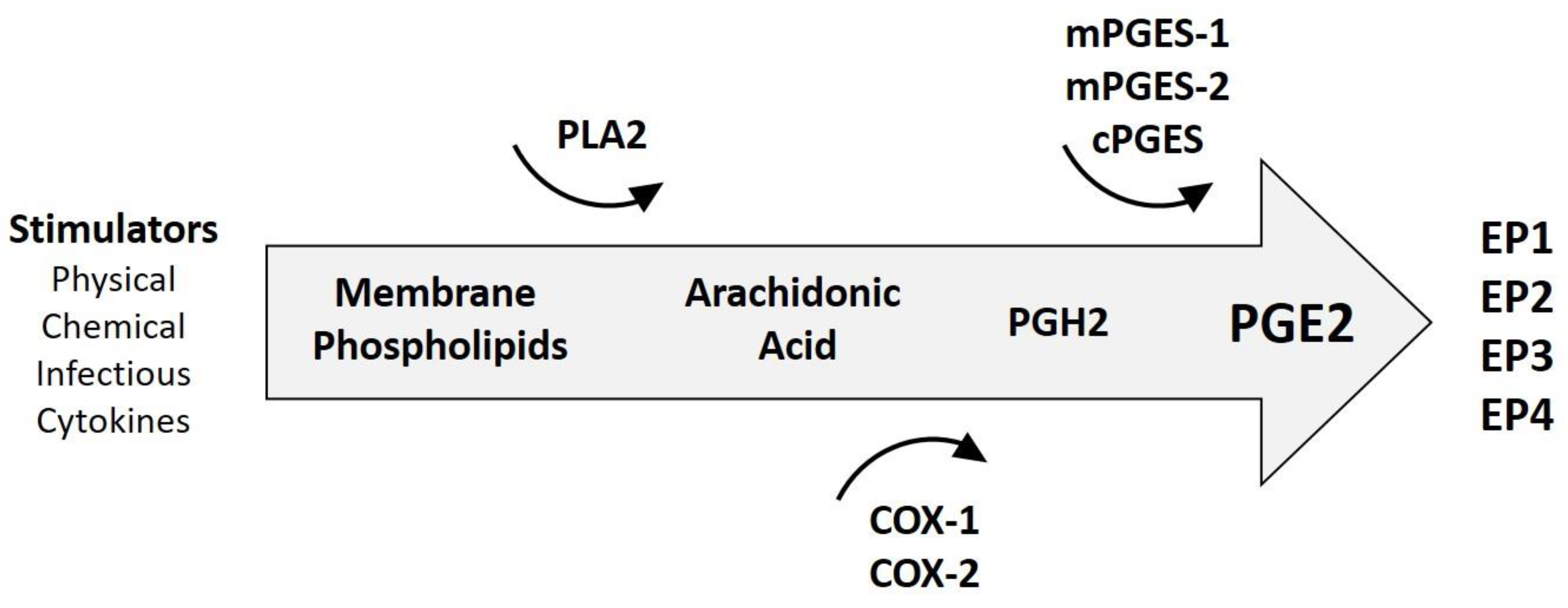{kind=link}
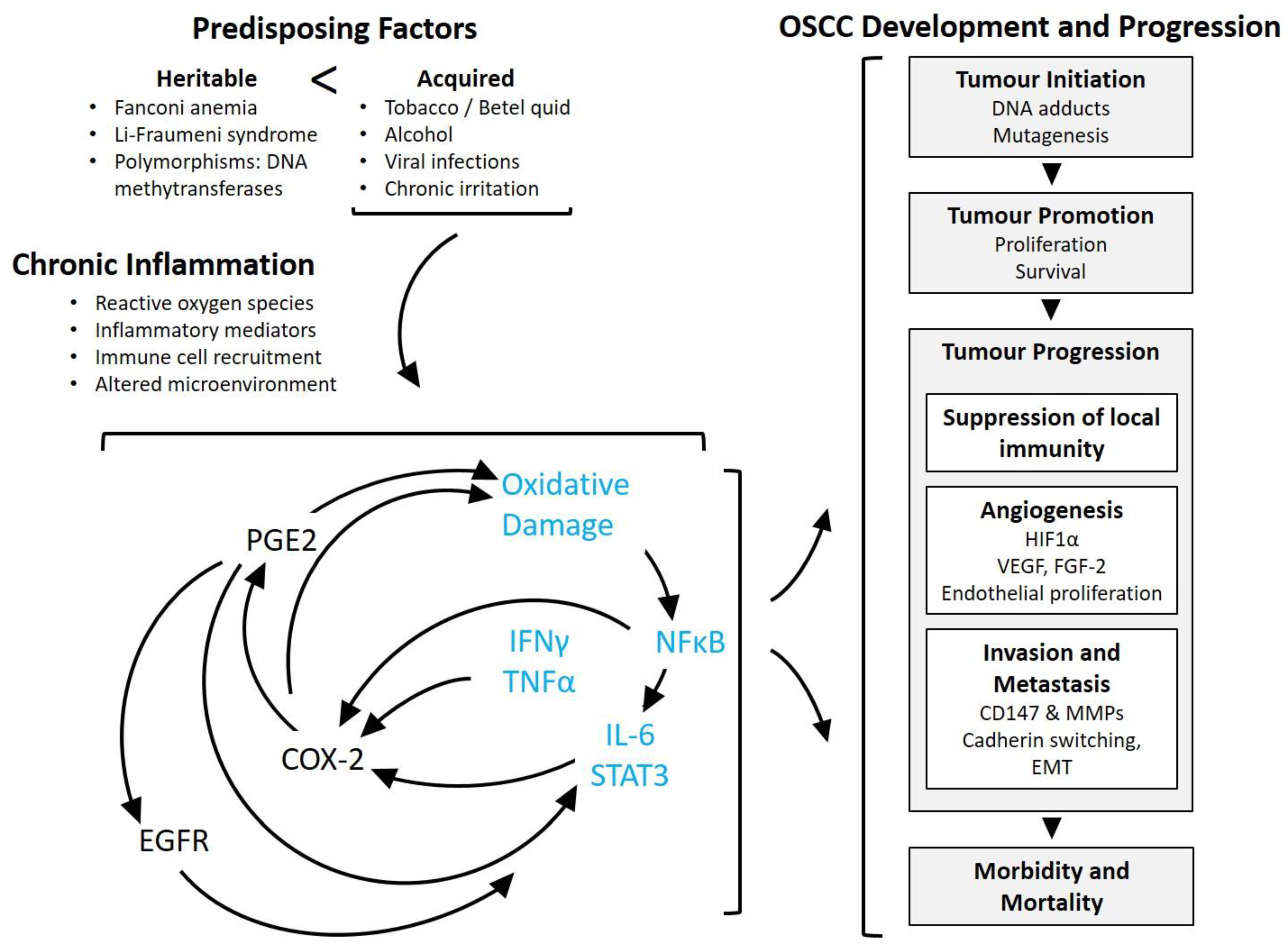
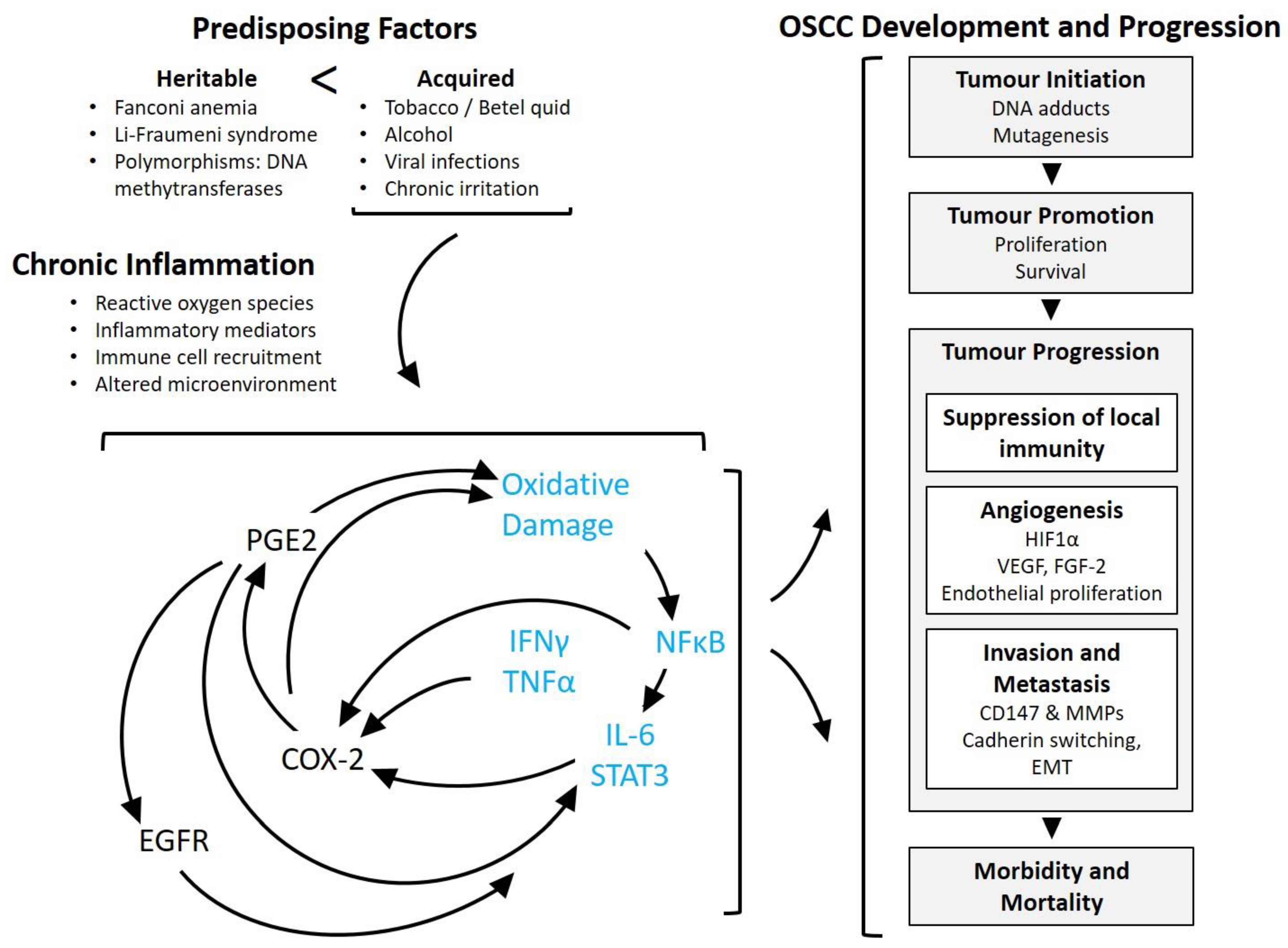{kind=link}
Abstract
:1. Introduction
2. General Epidemiology and Prognostic Factors for Oral Squamous Cell Carcinoma
2.1. Risk Factors of OSCC
2.2. Prognostic Indicators of OSCC
3. Inflammation-Related Mechanisms in Cancer Pathogenesis, with Emphasis on Oral Squamous Cell Carcinoma
3.1. Generalities of Inflammation in Cancer
3.2. Significance of Inflammation in Oral Squamous Cell Carcinoma
3.3. Inflammation, Tumour Initiation and Promotion
3.4. Inflammation and Anticancer Immunity
3.5. Inflammation and Angiogenesis
3.6. Inflammation, Invasion and Metastasis
3.6.1. Epithelial to Mesenchymal Transition
3.6.2. Tumour Invasion
3.6.3. The Role of CD147 in Inflammation and Invasion
4. Anti-Inflammatory Drugs in Cancer Prevention and Treatment
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sun, B.; Karin, M. Inflammation and liver tumorigenesis. Front. Med. 2013, 7, 242–254. [Google Scholar] [CrossRef] [PubMed]
- Manninen, P.; Karvonen, A.L.; Laukkarinen, J.; Aitola, P.; Huhtala, H.; Collin, P. Colorectal cancer and cholangiocarcinoma in patients with primary sclerosing cholangitis and inflammatory bowel disease. Scand. J. Gastroenterol. 2015, 50, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Thun, M.J.; Henley, S.J.; Gansler, T. Inflammation and Cancer: An Epidemiological Perspective. Novartis Foundation Symp. 2004, 256, 22–28, 49–52, 266–269. [Google Scholar]
- Maret, D.; Peters, O.A.; Vigarios, E.; Epstein, J.B.; van der Sluis, L. Dental screening of medical patients for oral infections and inflammation: Consideration of risk and benefit. Microbes Infect. 2017, 19, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Byatnal, A.A.; Byatnal, A.; Sen, S.; Guddattu, V.; Solomon, M.C. Cyclooxygenase-2—An Imperative Prognostic Biomarker in Oral Squamous Cell Carcinoma—An Immunohistochemical Study. Pathol. Oncol. Res. 2015, 21, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N. Tobacco use and oral cancer: A global perspective. J. Dent. Educ. 2001, 65, 328–339. [Google Scholar] [PubMed]
- Parkin, D.M.; Bray, F.; Ferlay, J.; Pisani, P. Global cancer statistics, 2002. CA Cancer J Clin 2005, 55, 74–108. [Google Scholar] [CrossRef] [PubMed]
- De Camargo Cancela, M.; Voti, L.; Guerra-Yi, M.; Chapuis, F.; Mazuir, M.; Curado, M.P. Oral cavity cancer in developed and in developing countries: Population-based incidence. Head Neck 2010, 32, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Warnakulasuriya, S. Global epidemiology of oral and oropharyngeal cancer. Oral Oncol. 2009, 45, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2013. CA Cancer J. Clin. 2013, 63, 11–30. [Google Scholar] [CrossRef] [PubMed]
- Canadian Cancer Society’s Advisory Committee on Cancer Statistics. Canadian Cancer Statistics 2017; Canadian Cancer Society: Toronto, ON, Canada, 2017. [Google Scholar]
- Canadian Cancer Society’s Advisory Committee on Cancer Statistics. Canadian Cancer Statistics 2016; Canadian Cancer Society: Toronto, ON, Canada, 2016; ISSN 0835-2976. [Google Scholar]
- Ribeiro, I.L.; de Medeiros, J.J.; Rodrigues, L.V.; Valenca, A.M.; Lima Neto Ede, A. Factors associated with lip and oral cavity cancer. Rev. Bras. Epidemiol. 2015, 18, 618–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Dakkak, I. Socioeconomic status and head and neck cancer. Evid. Based Dent. 2010, 11, 57–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.H.; Ko, A.M.; Warnakulasuriya, S.; Yin, B.L.; Sunarjo; Zain, R.B.; Ibrahim, S.O.; Liu, Z.W.; Li, W.H.; Zhang, S.S.; et al. Intercountry prevalences and practices of betel-quid use in south, southeast and eastern Asia regions and associated oral preneoplastic disorders: An international collaborative study by Asian betel-quid consortium of south and east. Asia. Int. J. Cancer 2011, 129, 1741–1751. [Google Scholar]
- Varoni, E.M.; Lodi, G.; Iriti, M. Ethanol versus Phytochemicals in Wine: Oral Cancer Risk in a Light Drinking Perspective. Int. J. Mol. Sci. 2015, 16, 17029–17047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro, G.D.; Castro, J.A. Alcohol drinking and mammary cancer: Pathogenesis and potential dietary preventive alternatives. World J. Clin. Oncol. 2014, 5, 713–729. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, A.K.; Engels, E.A.; Anderson, W.F.; Gillison, M.L. Incidence trends for human papillomavirus-related and -unrelated oral squamous cell carcinomas in the United States. J. Clin. Oncol. 2008, 26, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Sarode, G.S.; Batra, A.; Sarode, S.C.; Yerawadekar, S.; Patil, S. Oral Cancer-related Inherited Cancer Syndromes: A Comprehensive Review. J. Contemp. Dent. Pract. 2016, 17, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Supic, G.; Kozomara, R.; Zeljic, K.; Jovic, N.; Magic, Z. Prognostic value of the DNMTs mRNA expression and genetic polymorphisms on the clinical outcome in oral cancer patients. Clin. Oral Investig. 2017, 21, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Hardisson, D. Molecular pathogenesis of head and neck squamous cell carcinoma. Eur. Arch. Otorhinolaryngol. 2003, 260, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.B.; Greene, F.L.; Edge, S.B.; Compton, C.C.; Gershenwald, J.E.; Brookland, R.K.; Meyer, L.; Gress, D.M.; Byrd, D.R.; Winchester, D.P. The Eighth Edition AJCC Cancer Staging Manual: Continuing to build a bridge from a population-based to a more “personalized” approach to cancer staging. CA Cancer J. Clin. 2017, 67, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Thiagarajan, S.; Nair, S.; Nair, D.; Chaturvedi, P.; Kane, S.V.; Agarwal, J.P.; D’Cruz, A.K. Predictors of prognosis for squamous cell carcinoma of oral tongue. J. Surg. Oncol. 2014, 109, 639–644. [Google Scholar] [CrossRef] [PubMed]
- Dillon, J.K.; Brown, C.B.; McDonald, T.M.; Ludwig, D.C.; Clark, P.J.; Leroux, B.G.; Futran, N.D. How does the close surgical margin impact recurrence and survival when treating oral squamous cell carcinoma? J. Oral Maxillofac. Surg. 2015, 73, 1182–1188. [Google Scholar] [CrossRef] [PubMed]
- Shetty, D.; Jayade, B.V.; Joshi, S.K.; Gopalkrishnan, K. Accuracy of palpation, ultrasonography, and computed tomography in the evaluation of metastatic cervical lymph nodes in head and neck cancer. Indian J. Dent. 2015, 6, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Angadi, P.V.; Savitha, J.K.; Rao, S.S.; Sivaranjini, Y. Oral field cancerization: Current evidence and future perspectives. Oral Maxillofac. Surg. 2012, 16, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Francis, D.O.; Weymuller, E.A.J.; Parvathaneni, U.; Merati, A.L.; Yueh, B. Dysphagia, stricture, and pneumonia in head and neck cancer patients: Does treatment modality matter? Ann. Otol. Rhinol. Laryngol. 2010, 119, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Platteaux, N.; Dirix, P.; Dejaeger, E.; Nuyts, S. Dysphagia in head and neck cancer patients treated with chemoradiotherapy. Dysphagia 2010, 25, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Munn, L.L. Cancer and inflammation. Wiley Interdiscip. Rev. Syst. Biol. Med. 2017, 9, e1370. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B.; Vijayalekshmi, R.V.; Sung, B. Targeting inflammatory pathways for prevention and therapy of cancer: Short-term friend, long-term foe. Clin. Cancer Res. 2009, 15, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Hasselbalch, H.C. Smoking as a contributing factor for development of polycythemia vera and related neoplasms. Leuk. Res. 2015, 39, 1137–1145. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Dubois, R.N. Eicosanoids and cancer. Nat. Rev. Cancer 2010, 10, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Smalley, W.E.; DuBois, R.N. Colorectal cancer and nonsteroidal anti-inflammatory drugs. Adv. Pharmacol. 1997, 39, 1–20. [Google Scholar] [PubMed]
- Arun, B.; Goss, P. The role of COX-2 inhibition in breast cancer treatment and prevention. Semin. Oncol. 2004, 31, 22–29. [Google Scholar] [CrossRef] [PubMed]
- St John, M.A. Inflammatory mediators drive metastasis and drug resistance in head and neck squamous cell carcinoma. Laryngoscope 2015, 125, S1–S11. [Google Scholar] [CrossRef] [PubMed]
- Kalinski, P. Regulation of Immune Responses by Prostaglandin E2. J. Immunol. 2012, 188, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, M.; Rosenberg, D.W. Multifaceted roles of PGE2 in inflammation and cancer. Semin. Immunopathol. 2013, 35, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Legler, D.F.; Bruckner, M.; Uetz-von Allmen, E.; Krause, P. Prostaglandin E2 at new glance: Novel insights in functional diversity offer therapeutic chances. Int. J. Biochem. Cell Biol. 2010, 42, 198–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallo, O.; Franchi, A.; Magnelli, L.; Sardi, I.; Vannacci, A.; Boddi, V.; Chiarugi, V.; Masini, E. Cyclooxygenase-2 pathway correlates with VEGF expression in head and neck cancer. Implications for tumor angiogenesis and metastasis. Neoplasia 2001, 3, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Erovic, B.M.; Pelzmann, M.; Turhani, D.; Pammer, J.; Niederberger, V.; Neuchrist, C.; Grasl, M.C.; Thurnher, D. Differential expression pattern of cyclooxygenase-1 and -2 in head and neck squamous cell carcinoma. Acta Otolaryngol. 2003, 123, 950–953. [Google Scholar] [CrossRef] [PubMed]
- Pontes, H.A.; Pontes, F.S.; Fonseca, F.P.; de Carvalho, P.L.; Pereira, E.M.; de Abreu, M.C.; de Freitas Silva, B.S.; dos Santos Pinto, D.J. Nuclear factor kappaB and cyclooxygenase-2 immunoexpression in oral dysplasia and oral squamous cell carcinoma. Ann. Diagn. Pathol. 2013, 17, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Shibata, M.; Kodani, I.; Osaki, M.; Araki, K.; Adachi, H.; Ryoke, K.; Ito, H. Cyclo-oxygenase-1 and -2 expression in human oral mucosa, dysplasias and squamous cell carcinomas and their pathological significance. Oral Oncol. 2005, 41, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.Y.; Wan, H.C.; Lai, Y.L.; Chou, I.C.; Chen, Y.T.; Hung, S.L. Areca nut extracts increased the expression of cyclooxygenase-2, prostaglandin E2 and interleukin-1alpha in human immune cells via oxidative stress. Arch. Oral Biol. 2013, 58, 1523–1531. [Google Scholar] [CrossRef] [PubMed]
- Seyedmajidi, M.; Shafaee, S.; Siadati, S.; Khorasani, M.; Bijani, A.; Ghasemi, N. Cyclo-oxygenase-2 expression in oral squamous cell carcinoma. J. Cancer Res. Ther. 2014, 10, 1024–1029. [Google Scholar] [CrossRef] [PubMed]
- Husvik, C.; Khuu, C.; Bryne, M.; Halstensen, T.S. PGE2 production in oral cancer cell lines is COX-2-dependent. J. Dent. Res. 2009, 88, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Abrahao, A.C.; Castilho, R.M.; Squarize, C.H.; Molinolo, A.A.; dos Santos-Pinto, D.J.; Gutkind, J.S. A role for COX2-derived PGE2 and PGE2-receptor subtypes in head and neck squamous carcinoma cell proliferation. Oral Oncol. 2010, 46, 880–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoshikawa, H.; Goto, R.; Mori, T.; Mitani, T.; Mori, N. Expression of prostaglandin E2 receptors in oral squamous cell carcinomas and growth inhibitory effects of an EP3 selective antagonist, ONO-AE3-240. Int. J. Oncol. 2009, 34, 847–852. [Google Scholar] [CrossRef] [PubMed]
- Amano, H.; Hayashi, I.; Endo, H.; Kitasato, H.; Yamashina, S.; Maruyama, T.; Kobayashi, M.; Satoh, K.; Narita, M.; Sugimoto, Y.; et al. Host prostaglandin E(2)-EP3 signaling regulates tumor-associated angiogenesis and tumor growth. J. Exp. Med. 2003, 197, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Shoji, Y.; Takahashi, M.; Kitamura, T.; Watanabe, K.; Kawamori, T.; Maruyama, T.; Sugimoto, Y.; Negishi, M.; Narumiya, S.; Sugimura, T.; et al. Downregulation of prostaglandin E receptor subtype EP3 during colon cancer development. Gut 2004, 53, 1151–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.F.; Chen, M.K.; Hsieh, Y.S.; Chung, T.T.; Hsieh, Y.H.; Lin, C.W.; Su, J.L.; Tsai, M.H.; Tang, C.H. Prostaglandin E2/EP1 signaling pathway enhances intercellular adhesion molecule 1 (ICAM-1) expression and cell motility in oral cancer cells. J. Biol. Chem. 2010, 285, 29808–29816. [Google Scholar] [CrossRef] [PubMed]
- Li, W.Z.; Huo, Q.J.; Wang, X.Y.; Xu, F. Inhibitive effect of celecoxib on the adhesion and invasion of human tongue squamous carcinoma cells to extracellular matrix via down regulation of MMP-2 expression. Prostaglandins Other Lipid Mediat. 2010, 93, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Pannone, G.; Sanguedolce, F.; De Maria, S.; Farina, E.; Lo Muzio, L.; Serpico, R.; Emanuelli, M.; Rubini, C.; De Rosa, G.; Staibano, S.; et al. Cyclooxygenase isozymes in oral squamous cell carcinoma: A real-time RT-PCR study with clinic pathological correlations. Int. J. Immunopathol. Pharmacol. 2007, 20, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Morita, Y.; Hata, K.; Nakanishi, M.; Nishisho, T.; Yura, Y.; Yoneda, T. Cyclooxygenase-2 promotes tumor lymphangiogenesis and lymph node metastasis in oral squamous cell carcinoma. Int. J. Oncol. 2012, 41, 885–892. [Google Scholar] [CrossRef] [PubMed]
- Morita, Y.; Morita, N.; Hata, K.; Nakanishi, M.; Kimoto, N.; Omata, T.; Nakamura, Y.; Yoneda, T. Cyclooxygenase-2 expression is associated with vascular endothelial growth factor-c and lymph node metastasis in human oral tongue cancer. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2014, 117, 502–510. [Google Scholar] [CrossRef] [PubMed]
- Pannone, G.; Bufo, P.; Caiaffa, M.F.; Serpico, R.; Lanza, A.; Lo Muzio, L.; Rubini, C.; Staibano, S.; Petruzzi, M.; De Benedictis, M.; et al. Cyclooxygenase-2 expression in oral squamous cell carcinoma. Int. J. Immunopathol. Pharmacol. 2004, 17, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Qian, M.; Qian, D.; Jing, H.; Li, Y.; Ma, C.; Zhou, Y. Combined cetuximab and celecoxib treatment exhibits a synergistic anticancer effect on human oral squamous cell carcinoma in vitro and in vivo. Oncol. Rep. 2014, 32, 1681–1688. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Hao, Y.; Ji, H.; Fang, Y.; Guo, Y.; Sha, W.; Zhou, Y.; Pang, X.; Southerland, W.M.; Califano, J.A.; et al. Combination effects of salvianolic acid B with low-dose celecoxib on inhibition of head and neck squamous cell carcinoma growth in vitro and in vivo. Cancer Prev. Res. 2010, 3, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Speed, N.; Blair, I.A. Cyclooxygenase- and lipoxygenase-mediated DNA damage. Cancer Metastasis Rev. 2011, 30, 437–447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wartenberg, M.; Wirtz, N.; Grob, A.; Niedermeier, W.; Hescheler, J.; Peters, S.C.; Sauer, H. Direct current electrical fields induce apoptosis in oral mucosa cancer cells by NADPH oxidase-derived reactive oxygen species. Bioelectromagnetics 2008, 29, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Guttikonda, V.R.; Patil, R.; Kumar, G. DNA damage in peripheral blood leukocytes in tobacco users. J. Oral. Maxillofac. Pathol. 2014, 18, S16–S20. [Google Scholar] [CrossRef] [PubMed]
- Choudhari, S.K.; Chaudhary, M.; Gadbail, A.R.; Sharma, A.; Tekade, S. Oxidative and antioxidative mechanisms in oral cancer and precancer: A review. Oral Oncol. 2014, 50, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Thanan, R.; Murata, M.; Ma, N.; Hammam, O.; Wishahi, M.; El Leithy, T.; Hiraku, Y.; Oikawa, S.; Kawanishi, S. Nuclear localization of COX-2 in relation to the expression of stemness markers in urinary bladder cancer. Mediators. Inflamm. 2012, 2012, e165879. [Google Scholar] [CrossRef] [PubMed]
- Li, H.J.; Reinhardt, F.; Herschman, H.R.; Weinberg, R.A. Cancer-stimulated mesenchymal stem cells create a carcinoma stem cell niche via prostaglandin E2 signaling. Cancer Discov. 2012, 2, 840–855. [Google Scholar] [CrossRef] [PubMed]
- Rudnick, J.A.; Arendt, L.M.; Klebba, I.; Hinds, J.W.; Iyer, V.; Gupta, P.B.; Naber, S.P.; Kuperwasser, C. Functional heterogeneity of breast fibroblasts is defined by a prostaglandin secretory phenotype that promotes expansion of cancer-stem like cells. PLoS ONE 2011, 6, e24605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wang, J.; Wheat, J.; Chen, X.; Jin, S.; Sadrzadeh, H.; Fathi, A.T.; Peterson, R.T.; Kung, A.L.; Sweetser, D.A.; et al. AML1-ETO mediates hematopoietic self-renewal and leukemogenesis through a COX/beta-catenin signaling pathway. Blood 2013, 121, 4906–4916. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Mao, R.; Yang, J. NF-kappaB and STAT3 signaling pathways collaboratively link inflammation to cancer. Protein Cell 2013, 4, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.H.; Kirschenbaum, A.; Yao, S.; Levine, A.C. Cross-talk between the interleukin-6 and prostaglandin E(2) signaling systems results in enhancement of osteoclastogenesis through effects on the osteoprotegerin/receptor activator of nuclear factor-κB (RANK) ligand/RANK system. Endocrinology 2005, 146, 1991–1998. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Karin, M. Dangerous liaisons: STAT3 and NF-kappaB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 2010, 21, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhang, J.; Arfuso, F.; Chinnathambi, A.; Zayed, M.E.; Alharbi, S.A.; Kumar, A.P.; Ahn, K.S.; Sethi, G. NF-kappaB in cancer therapy. Arch. Toxicol. 2015, 89, 711–731. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, A.; Feige, U.; Fontana, A.; Muller, K.; Dinarello, C.A. Interleukin-1 enhances pain reflexes. Mediation through increased prostaglandin E2 levels. Agents Actions 1988, 25, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Qu, L.; Yan, S. Cyclooxygenase-2 promotes tumor growth and suppresses tumor immunity. Cancer Cell Int. 2015, 15, e106. [Google Scholar] [CrossRef] [PubMed]
- Gately, S.; Li, W.W. Multiple roles of COX-2 in tumor angiogenesis: A target for antiangiogenic therapy. Semin. Oncol. 2004, 31, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Gonda, T.A.; Tu, S.; Wang, T.C. Chronic inflammation, the tumor microenvironment and carcinogenesis. Cell Cycle 2009, 8, 2005–2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harizi, H.; Juzan, M.; Pitard, V.; Moreau, J.F.; Gualde, N. Cyclooxygenase-2-issued prostaglandin e(2) enhances the production of endogenous IL-10, which down-regulates dendritic cell functions. J. Immunol. 2002, 168, 2255–2263. [Google Scholar] [CrossRef] [PubMed]
- Harizi, H.; Grosset, C.; Gualde, N. Prostaglandin E2 modulates dendritic cell function via EP2 and EP4 receptor subtypes. J. Leukoc. Biol. 2003, 73, 756–763. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A. Tumour-associated macrophages as a prototypic type II polarised phagocyte population: Role in tumour progression. Eur. J. Cancer 2004, 40, 1660–1667. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Allavena, P.; Mantovani, A. Cancer related inflammation: The macrophage connection. Cancer Lett. 2008, 267, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Na, Y.R.; Yoon, Y.N.; Son, D.I.; Seok, S.H. Cyclooxygenase-2 inhibition blocks M2 macrophage differentiation and suppresses metastasis in murine breast cancer model. PLoS ONE 2013, 8, e63451. [Google Scholar] [CrossRef] [PubMed]
- Ljunggren, H.G.; Malmberg, K.J. Prospects for the use of NK cells in immunotherapy of human cancer. Nat. Rev. Immunol. 2007, 7, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Bankhurst, A.D. The modulation of human natural killer cell activity by prostaglandins. J. Clin. Lab Immunol. 1982, 7, 85–91. [Google Scholar] [PubMed]
- Goto, T.; Herberman, R.B.; Maluish, A.; Strong, D.M. Cyclic AMP as a mediator of prostaglandin E-induced suppression of human natural killer cell activity. J. Immunol. 1983, 130, 1350–1355. [Google Scholar] [PubMed]
- Mailliard, R.B.; Alber, S.M.; Shen, H.; Watkins, S.C.; Kirkwood, J.M.; Herberman, R.B.; Kalinski, P. IL-18-induced CD83+CCR7+ NK helper cells. J. Exp. Med. 2005, 202, 941–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zelenay, S.; van der Veen, A.G.; Bottcher, J.P.; Snelgrove, K.J.; Rogers, N.; Acton, S.E.; Chakravarty, P.; Girotti, M.R.; Marais, R.; Quezada, S.A.; et al. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell 2015, 162, 1257–1270. [Google Scholar] [CrossRef] [PubMed]
- Mitsuhashi, M.; Liu, J.; Cao, S.; Shi, X.; Ma, X. Regulation of interleukin-12 gene expression and its anti-tumor activities by prostaglandin E2 derived from mammary carcinomas. J. Leukoc. Biol. 2004, 76, 322–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coronella-Wood, J.A.; Hersh, E.M. Naturally occurring B-cell responses to breast cancer. Cancer Immunol. Immunother. 2003, 52, 715–738. [Google Scholar] [CrossRef] [PubMed]
- Simkin, N.J.; Jelinek, D.F.; Lipsky, P.E. Inhibition of human B cell responsiveness by prostaglandin E2. J. Immunol. 1987, 138, 1074–1081. [Google Scholar] [PubMed]
- Mahic, M.; Yaqub, S.; Johansson, C.C.; Tasken, K.; Aandahl, E.M. FOXP3+CD4+CD25+ adaptive regulatory T cells express cyclooxygenase-2 and suppress effector T cells by a prostaglandin E2-dependent mechanism. J. Immunol. 2006, 177, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, C.; Strauss, L.; Zeidler, R.; Lang, S.; Whiteside, T.L. Expansion of human T regulatory type 1 cells in the microenvironment of cyclooxygenase 2 overexpressing head and neck squamous cell carcinoma. Cancer Res. 2007, 67, 8865–8873. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrand-Rosenberg, S.; Sinha, P. Myeloid-derived suppressor cells: Linking inflammation and cancer. J. Immunol. 2009, 182, 4499–4506. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Jain, R.K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada, K.; Supriatno; Kawashima, Y.; Yoshida, H.; Sato, M. S-1 inhibits tumorigenicity and angiogenesis of human oral squamous cell carcinoma cells by suppressing expression of phosphorylated Akt, vascular endothelial growth factor and fibroblast growth factor-2. Int. J. Oncol. 2007, 30, 365–374. [Google Scholar] [PubMed]
- Lee, L.T.; Wong, Y.K.; Chan, M.Y.; Chang, K.W.; Chen, S.C.; Chang, C.T.; Wang, J. The correlation between HIF-1 alpha and VEGF in oral squamous cell carcinomas: Expression patterns and quantitative immunohistochemical analysis. J. Chin. Med. Assoc. 2018, 81, 370–375. [Google Scholar] [CrossRef] [PubMed]
- Gammon, L.; Mackenzie, I.C. Roles of hypoxia, stem cells and epithelial-mesenchymal transition in the spread and treatment resistance of head and neck cancer. J. Oral Pathol. Med. 2015, 45, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.J.; Isaacs, J.S.; Lee, S.; Trepel, J.; Neckers, L. IL-1beta-mediated up-regulation of HIF-1alpha via an NFkappaB/COX-2 pathway identifies HIF-1 as a critical link between inflammation and oncogenesis. FASEB J. 2003, 17, 2115–2117. [Google Scholar] [CrossRef] [PubMed]
- Erreni, M.; Mantovani, A.; Allavena, P. Tumor-associated Macrophages (TAM) and Inflammation in Colorectal Cancer. Cancer Microenviron. 2011, 4, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Bingle, L.; Lewis, C.E.; Corke, K.P.; Reed, M.W.; Brown, N.J. Macrophages promote angiogenesis in human breast tumour spheroids in vivo. Br. J. Cancer 2006, 94, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, T.; Robinson, S.C.; Schulz, M.; Trumper, L.; Balkwill, F.R.; Binder, C. Enhanced invasiveness of breast cancer cell lines upon co-cultivation with macrophages is due to TNF-alpha dependent up-regulation of matrix metalloproteases. Carcinogenesis 2004, 25, 1543–1549. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.Y.; Chen, Y.W.; Hsiao, J.R.; Liu, C.S.; Kuo, Y.Z.; Wang, Y.C.; Chang, K.C.; Tsai, S.T.; Chang, M.Z.; Lin, S.H.; et al. Elevated S100A9 expression in tumor stroma functions as an early recurrence marker for early-stage oral cancer patients through increased tumor cell invasion, angiogenesis, macrophage recruitment and interleukin-6 production. Oncotarget 2015, 6, 28401–28424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finger, E.C.; Giaccia, A.J. Hypoxia, inflammation, and the tumor microenvironment in metastatic disease. Cancer Metastasis Rev. 2010, 29, 285–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeisberg, M.; Neilson, E.G. Biomarkers for epithelial-mesenchymal transitions. J. Clin. Investig. 2009, 119, 1429–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derycke, L.D.; Bracke, M.E. N-cadherin in the spotlight of cell-cell adhesion, differentiation, embryogenesis, invasion and signalling. Int. J. Dev. Biol. 2004, 48, 463–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hazan, R.B.; Phillips, G.R.; Qiao, R.F.; Norton, L.; Aaronson, S.A. Exogenous expression of N-cadherin in breast cancer cells induces cell migration, invasion, and metastasis. J. Cell Biol. 2000, 148, 779–790. [Google Scholar] [CrossRef] [PubMed]
- Costa, L.C.; Leite, C.F.; Cardoso, S.V.; Loyola, A.M.; Faria, P.R.; Souza, P.E.; Horta, M.C. Expression of epithelial-mesenchymal transition markers at the invasive front of oral squamous cell carcinoma. J. Appl. Oral Sci. 2015, 23, 169–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tania, M.; Khan, M.A.; Fu, J. Epithelial to mesenchymal transition inducing transcription factors and metastatic cancer. Tumour Biol. 2014, 35, 7335–7342. [Google Scholar] [CrossRef] [PubMed]
- Ruff, M.; Leyme, A.; Le Cann, F.; Bonnier, D.; Le Seyec, J.; Chesnel, F.; Fattet, L.; Rimokh, R.; Baffet, G.; Theret, N. The Disintegrin and Metalloprotease ADAM12 Is Associated with TGF-beta-Induced Epithelial to Mesenchymal Transition. PLoS ONE 2015, 10, e0139179. [Google Scholar] [CrossRef] [PubMed]
- Wendt, M.K.; Balanis, N.; Carlin, C.R.; Schiemann, W.P. STAT3 and epithelial-mesenchymal transitions in carcinomas. JAKSTAT 2014, 3, e28975. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Yang, G.; Jiang, T.; Zhu, G.; Li, H.; Qiu, Z. The effects and mechanisms of blockage of STAT3 signaling pathway on IL-6 inducing EMT in human pancreatic cancer cells in vitro. Neoplasma 2011, 58, 396–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.C.; Zhu, L.F.; Xu, X.H.; Ning, T.Y.; Ye, J.H.; Liu, L.K. Membrane Type 1 Matrix Metalloproteinase induces an epithelial to mesenchymal transition and cancer stem cell-like properties in SCC9 cells. BMC Cancer 2013, 13, e171. [Google Scholar] [CrossRef] [PubMed]
- Artacho-Cordon, F.; Rios-Arrabal, S.; Lara, P.C.; Artacho-Cordon, A.; Calvente, I.; Nunez, M.I. Matrix metalloproteinases: Otential therapy to prevent the development of second malignancies after breast radiotherapy. Surg. Oncol. 2012, 21, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Deryugina, E.I.; Quigley, J.P. Matrix metalloproteinases and tumor metastasis. Cancer Metastasis Rev. 2006, 25, 9–34. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Liang, B.; Reddy, S.T.; Farias-Eisner, R.; Su, X. Role of inflammation-associated microenvironment in tumorigenesis and metastasis. Curr. Cancer Drug Targets 2014, 14, 30–45. [Google Scholar] [CrossRef] [PubMed]
- Deryugina, E.I.; Quigley, J.P. Tumor angiogenesis: MMP-mediated induction of intravasation- and metastasis-sustaining neovasculature. Matrix. Biol. 2015, 44, 94–112. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Thayele Purayil, H.; Black, J.B.; Fetto, F.; Lynch, L.D.; Masannat, J.N.; Daaka, Y. Prostaglandin E2 receptor 4 mediates renal cell carcinoma intravasation and metastasis. Cancer Lett. 2017, 391, 50–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karnezis, T.; Shayan, R.; Fox, S.; Achen, M.G.; Stacker, S.A. The connection between lymphangiogenic signalling and prostaglandin biology: A missing link in the metastatic pathway. Oncotarget 2012, 3, 893–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasic, G.J.; Gasic, T.B.; Stewart, C.C. Antimetastatic effects associated with platelet reduction. Proc. Natl. Acad. Sci. USA 1968, 61, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Dovizio, M.; Alberti, S.; Sacco, A.; Guillem-Llobat, P.; Schiavone, S.; Maier, T.J.; Steinhilber, D.; Patrignani, P. Novel insights into the regulation of cyclooxygenase-2 expression by platelet-cancer cell cross-talk. Biochem. Soc. Trans. 2015, 43, 707–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dovizio, M.; Maier, T.J.; Alberti, S.; Di Francesco, L.; Marcantoni, E.; Munch, G.; John, C.M.; Suess, B.; Sgambato, A.; Steinhilber, D.; et al. Pharmacological inhibition of platelet-tumor cell cross-talk prevents platelet-induced overexpression of cyclooxygenase-2 in HT29 human colon carcinoma cells. Mol. Pharmacol. 2013, 84, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, J.S.; Talmage, K.E.; Massari, J.V.; La Jeunesse, C.M.; Flick, M.J.; Kombrinck, K.W.; Jirouskova, M.; Degen, J.L. Platelets and fibrin(ogen) increase metastatic potential by impeding natural killer cell-mediated elimination of tumor cells. Blood 2005, 105, 178–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, D.X.; Bos, P.D.; Massague, J. Metastasis: From dissemination to organ-specific colonization. Nat. Rev. Cancer 2009, 9, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Ben-Baruch, A. Organ selectivity in metastasis: Regulation by chemokines and their receptors. Clin. Exp. Metastasis 2008, 25, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Battinelli, E.M.; Markens, B.A.; Kulenthirarajan, R.A.; Machlus, K.R.; Flaumenhaft, R.; Italiano, J.E.J. Anticoagulation inhibits tumor cell-mediated release of platelet angiogenic proteins and diminishes platelet angiogenic response. Blood 2014, 123, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Aguirre-Ghiso, J.A. Models, mechanisms and clinical evidence for cancer dormancy. Nat. Rev. Cancer 2007, 7, 834–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coghlin, C.; Murray, G.I. The role of gene regulatory networks in promoting cancer progression and metastasis. Future Oncol. 2014, 10, 735–748. [Google Scholar] [CrossRef] [PubMed]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curran, S.; Murray, G.I. Matrix metalloproteinases: Molecular aspects of their roles in tumour invasion and metastasis. Eur. J. Cancer 2000, 36, 1621–1630. [Google Scholar] [CrossRef]
- Brown, G.T.; Murray, G.I. Current mechanistic insights into the roles of matrix metalloproteinases in tumour invasion and metastasis. J. Pathol. 2015, 237, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Liotta, L.A.; Kohn, E.C. The microenvironment of the tumour-host interface. Nature 2001, 411, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Visse, R.; Nagase, H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: Structure, function, and biochemistry. Circ. Res. 2003, 92, 827–839. [Google Scholar] [CrossRef] [PubMed]
- Murphy, D.A.; Courtneidge, S.A. The ‘ins’ and ‘outs’ of podosomes and invadopodia: Characteristics, formation and function. Nat. Rev. Mol. Cell Biol. 2011, 12, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Jacob, A.; Prekeris, R. The regulation of MMP targeting to invadopodia during cancer metastasis. Front. Cell Dev. Biol. 2015, 3, e4. [Google Scholar] [CrossRef] [PubMed]
- Paz, H.; Pathak, N.; Yang, J. Invading one step at a time: The role of invadopodia in tumor metastasis. Oncogene 2014, 33, 4193–4202. [Google Scholar] [CrossRef] [PubMed]
- Bates, A.L.; Pickup, M.W.; Hallett, M.A.; Dozier, E.A.; Thomas, S.; Fingleton, B. Stromal matrix metalloproteinase 2 regulates collagen expression and promotes the outgrowth of experimental metastases. J. Pathol. 2015, 235, 773–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendonsa, A.M.; VanSaun, M.N.; Ustione, A.; Piston, D.W.; Fingleton, B.M.; Gorden, D.L. Host and tumor derived MMP13 regulate extravasation and establishment of colorectal metastases in the liver. Mol. Cancer 2015, 14, e49. [Google Scholar] [CrossRef] [PubMed]
- Makinen, L.K.; Hayry, V.; Hagstrom, J.; Sorsa, T.; Passador-Santos, F.; Keski-Santti, H.; Haukka, J.; Makitie, A.A.; Haglund, C.; Atula, T. Matrix metalloproteinase-7 and matrix metalloproteinase-25 in oral tongue squamous cell carcinoma. Head Neck 2014, 36, 1783–1788. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Stamenkovic, I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-β and promotes tumor invasion and angiogenesis. Genes Dev. 2000, 14, 163–176. [Google Scholar] [PubMed]
- Lee, A.Y.; Fan, C.C.; Chen, Y.A.; Cheng, C.W.; Sung, Y.J.; Hsu, C.P.; Kao, T.Y. Curcumin Inhibits Invasiveness and Epithelial-Mesenchymal Transition in Oral Squamous Cell Carcinoma Through Reducing Matrix Metalloproteinase 2, 9 and Modulating p53-E-Cadherin Pathway. Integr. Cancer Ther. 2015, 14, 484–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdem, N.F.; Carlson, E.R.; Gerard, D.A.; Ichiki, A.T. Characterization of 3 oral squamous cell carcinoma cell lines with different invasion and/or metastatic potentials. J. Oral Maxillofac. Surg. 2007, 65, 1725–1733. [Google Scholar] [CrossRef] [PubMed]
- De Vicente, J.C.; Fresno, M.F.; Villalain, L.; Vega, J.A.; Hernandez Vallejo, G. Expression and clinical significance of matrix metalloproteinase-2 and matrix metalloproteinase-9 in oral squamous cell carcinoma. Oral Oncol. 2005, 41, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Zhou, M.; Peng, L.; Kong, S.; Miao, R.; Shi, Y.; Sheng, H.; Li, L. Upregulation of CD147 promotes cell invasion, epithelial-to-mesenchymal transition and activates MAPK/ERK signaling pathway in colorectal cancer. Int. J. Clin. Exp. Pathol. 2014, 7, 7432–7441. [Google Scholar] [PubMed]
- Tian, X.; Ye, C.; Yang, Y.; Guan, X.; Dong, B.; Zhao, M.; Hao, C. Expression of CD147 and matrix metalloproteinase-11 in colorectal cancer and their relationship to clinicopathological features. J. Transl. Med. 2015, 13, e337. [Google Scholar] [CrossRef] [PubMed]
- Biswas, C.; Zhang, Y.; DeCastro, R.; Guo, H.; Nakamura, T.; Kataoka, H.; Nabeshima, K. The human tumor cell-derived collagenase stimulatory factor (renamed EMMPRIN) is a member of the immunoglobulin superfamily. Cancer Res. 1995, 55, 434–439. [Google Scholar] [PubMed]
- Wu, J.; Hao, Z.W.; Zhao, Y.X.; Yang, X.M.; Tang, H.; Zhang, X.; Song, F.; Sun, X.X.; Wang, B.; Nan, G.; et al. Full-length soluble CD147 promotes MMP-2 expression and is a potential serological marker in detection of hepatocellular carcinoma. J. Transl. Med. 2014, 12, e190. [Google Scholar] [CrossRef] [PubMed]
- Kasinrerk, W.; Fiebiger, E.; Stefanova, I.; Baumruker, T.; Knapp, W.; Stockinger, H. Human leukocyte activation antigen M6, a member of the Ig superfamily, is the species homologue of rat OX-47, mouse basigin, and chicken HT7 molecule. J. Immunol. 1992, 149, 847–854. [Google Scholar] [PubMed]
- Fossum, S.; Mallett, S.; Barclay, A.N. The MRC OX-47 antigen is a member of the immunoglobulin superfamily with an unusual transmembrane sequence. Eur. J. Immunol. 1991, 21, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Seulberger, H.; Lottspeich, F.; Risau, W. The inducible blood—Brain barrier specific molecule HT7 is a novel immunoglobulin-like cell surface glycoprotein. EMBO J. 1990, 9, 2151–2158. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, L.S.; Delgado, M.L.; Ricardo, S.; Garcez, F.; do Amaral, B.; Pacheco, J.J.; Lopes, C.; Bousbaa, H. EMMPRIN expression in oral squamous cell carcinomas: Correlation with tumor proliferation and patient survival. BioMed Res. Int. 2014, 2014, e905680. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Zhang, X.; Guo, H.; Fan, C.; Chen, Z.; Zhu, P. Highly expressed CD147 on CD4(+) tumor infiltrating lymphocytes promotes the progress of breast cancer. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2015, 31, 961–964. [Google Scholar] [PubMed]
- Papadimitropoulou, A.; Mamalaki, A. The glycosylated IgII extracellular domain of EMMPRIN is implicated in the induction of MMP-2. Mol. Cell Biochem. 2013, 379, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Khayati, F.; Perez-Cano, L.; Maouche, K.; Sadoux, A.; Boutalbi, Z.; Podgorniak, M.P.; Maskos, U.; Setterblad, N.; Janin, A.; Calvo, F.; et al. EMMPRIN/CD147 is a novel coreceptor of VEGFR-2 mediating its activation by VEGF. Oncotarget 2015, 6, 9766–9780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Q.; Li, J.; Xing, J.; Li, W.; Li, H.; Ke, X.; Zhang, J.; Ren, T.; Shang, Y.; Yang, H.; et al. CD147 promotes reprogramming of glucose metabolism and cell proliferation in HCC cells by inhibiting the p53-dependent signaling pathway. J. Hepatol. 2014, 61, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Koch, C.; Staffler, G.; Huttinger, R.; Hilgert, I.; Prager, E.; Cerny, J.; Steinlein, P.; Majdic, O.; Horejsi, V.; Stockinger, H. T cell activation-associated epitopes of CD147 in regulation of the T cell response, and their definition by antibody affinity and antigen density. Int. Immunol. 1999, 11, 777–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, P.; Lu, N.; Shi, Z.G.; Zhou, J.; Wu, Z.B.; Yang, Y.; Ding, J.; Chen, Z.N. CD147 overexpression on synoviocytes in rheumatoid arthritis enhances matrix metalloproteinase production and invasiveness of synoviocytes. Arthritis Res. Ther. 2006, 8, R44. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, A.H.; Diaz, L.A., Jr; Bonish, B.; Antony, P.A.; Fox, D.A. The pattern of expression of CD147/neurothelin during human T-cell ontogeny as defined by the monoclonal antibody 8D6. Tissue Antigens 1997, 50, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Gwinn, W.M.; Damsker, J.M.; Falahati, R.; Okwumabua, I.; Kelly-Welch, A.; Keegan, A.D.; Vanpouille, C.; Lee, J.J.; Dent, L.A.; Leitenberg, D.; et al. Novel approach to inhibit asthma-mediated lung inflammation using anti-CD147 intervention. J. Immunol. 2006, 177, 4870–4879. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, D.K.; Hahn, J.N.; Yong, V.W. EMMPRIN, an upstream regulator of MMPs, in CNS biology. Matrix Biol. 2015, 44, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.F.; Ding, S.F.; Gao, Y.M.; Liang, Y.; Foda, H.D. Expression of various matrix metalloproteinases in mice with hyperoxia-induced acute lung injury. Zhongguo Wei Zhong Bing Ji Jiu Yi Xue 2006, 18, 449–451. [Google Scholar] [PubMed]
- Wang, Z.Z.; Wang, Y.; Li, J.M.; Mou, F.X.; Wu, H. Significance of serum MMP-3, TIMP-1, and monocyte CD147 in rheumatoid arthritis patients of damp-heat Bi-syndrome and of cold-damp Bi-syndrome. Zhongguo Zhong Xi Yi Jie He Za Zhi 2013, 33, 770–773. [Google Scholar] [PubMed]
- Maeda, K.; Kosugi, T.; Sato, W.; Kojima, H.; Sato, Y.; Kamimura, D.; Kato, N.; Tsuboi, N.; Yuzawa, Y.; Matsuo, S.; et al. CD147/basigin limits lupus nephritis and Th17 cell differentiation in mice by inhibiting the interleukin-6/STAT-3 pathway. Arthritis Rheumatol. 2015, 67, 2185–2195. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Zhou, H.; Walian, P.J.; Jap, B.K. Regulation of gamma-secretase activity in Alzheimer’s disease. Biochem. 2007, 46, 2553–2563. [Google Scholar] [CrossRef] [PubMed]
- Seizer, P.; Ochmann, C.; Schonberger, T.; Zach, S.; Rose, M.; Borst, O.; Klingel, K.; Kandolf, R.; MacDonald, H.R.; Nowak, R.A.; et al. Disrupting the EMMPRIN (CD147)-cyclophilin A interaction reduces infarct size and preserves systolic function after myocardial ischemia and reperfusion. Arterioscler Thromb. Vasc. Biol. 2011, 31, 1377–1386. [Google Scholar] [CrossRef] [PubMed]
- Pushkarsky, T.; Yurchenko, V.; Laborico, A.; Bukrinsky, M. CD147 stimulates HIV-1 infection in a signal-independent fashion. Biochem. Biophys. Res. Commun. 2007, 363, 495–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, N.; Osanai, T.; Fujiwara, T.; Kameda, K.; Matsunaga, T.; Okumura, K. C-reactive protein-induced upregulation of extracellular matrix metalloproteinase inducer in macrophages: Inhibitory effect of fluvastatin. Life Sci. 2006, 78, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Pennings, G.J.; Yong, A.S.; Kritharides, L. Expression of EMMPRIN (CD147) on circulating platelets in vivo. J. Thromb. Haemost. 2010, 8, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.; Bultmann, A.; Fischel, S.; Gillitzer, A.; Cullen, P.; Walch, A.; Jost, P.; Ungerer, M.; Tolley, N.D.; Lindemann, S.; et al. Extracellular matrix metalloproteinase inducer (CD147) is a novel receptor on platelets, activates platelets, and augments nuclear factor kappaB-dependent inflammation in monocytes. Circ. Res. 2008, 102, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Cipollone, F.; Prontera, C.; Pini, B.; Marini, M.; Fazia, M.; De Cesare, D.; Iezzi, A.; Ucchino, S.; Boccoli, G.; Saba, V.; et al. Overexpression of functionally coupled cyclooxygenase-2 and prostaglandin E synthase in symptomatic atherosclerotic plaques as a basis of prostaglandin E(2)-dependent plaque instability. Circulation 2001, 104, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Ye, J.; Guo, R.; Liu, H.; Wang, X.; Qi, F.; Guo, C. The effect of the expression of angiotensin II on extracellular matrix metalloproteinase inducer (EMMPRIN) in macrophages is mediated via the AT1/COX-2/PGE.sub.2 pathway. Inflamm.Res. 2010, 1033. [Google Scholar] [CrossRef] [PubMed]
- Dean, N.R.; Newman, J.R.; Helman, E.E.; Zhang, W.; Safavy, S.; Weeks, D.M.; Cunningham, M.; Snyder, L.A.; Tang, Y.; Yan, L.; et al. Anti-EMMPRIN monoclonal antibody as a novel agent for therapy of head and neck cancer. Clin. Cancer Res. 2009, 15, 4058–4065. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Liu, Y.; Huang, Y.; Huang, D.; Li, Y.; Wu, J.; Duan, M. Expression of COX-2, CD44v6 and CD147 and relationship with invasion and lymph node metastasis in hypopharyngeal squamous cell carcinoma. PLoS ONE 2013, 8, e71048. [Google Scholar] [CrossRef] [PubMed]
- Cathcart, J.; Pulkoski-Gross, A.; Cao, J. Targeting Matrix Metalloproteinases in Cancer: Bringing New Life to Old Ideas. Genes Dis. 2015, 2, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Tlatli, R.; El Ayeb, M. MMP inhibitors and cancer treatment trials, limitations and hopes for the future. Arch. Inst. Pasteur. Tunis. 2013, 90, 3–21. [Google Scholar] [PubMed]
- Basudhar, D.; Cheng, R.C.; Bharadwaj, G.; Ridnour, L.A.; Wink, D.A.; Miranda, K.M. Chemotherapeutic potential of diazeniumdiolate-based aspirin prodrugs in breast Cancer. Free Radic. Biol. Med. 2015, 83, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Thorat, M.A.; Cuzick, J. Role of aspirin in cancer prevention. Curr. Oncol. Rep. 2013, 15, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Jia-Jun, T.; Su-Mei, L.; Liang, Y.; Ju-Ke, M.; Ya-Kui, M.; Hai-Bo, W.; Wei, X. Nimesulide inhibited the growth of hypopharyngeal carcinoma cells via suppressing Survivin expression. Head Neck Oncol. 2012, 4, e7. [Google Scholar]
- Rayburn, E.R.; Ezell, S.J.; Zhang, R. Anti-Inflammatory Agents for Cancer Therapy. Mol. Cell Pharmacol. 2009, 1, 29–43. [Google Scholar] [CrossRef] [PubMed]
- Albouy, B.; Tourani, J.M.; Allain, P.; Rolland, F.; Staerman, F.; Eschwege, P.; Pfister, C. Preliminary results of the Prostacox phase II trial in hormonal refractory prostate cancer. BJU Int. 2007, 100, 770–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javle, M.M.; Cao, S.; Durrani, F.A.; Pendyala, L.; Lawrence, D.D.; Smith, P.F.; Creaven, P.J.; Noel, D.C.; Iyer, R.V.; Rustum, Y.M. Celecoxib and mucosal protection: Translation from an animal model to a phase I clinical trial of celecoxib, irinotecan, and 5-fluorouracil. Clin. Cancer Res. 2007, 13, 965–971. [Google Scholar] [CrossRef] [PubMed]
- Salman, M.C.; Ayhan, A. Use of anti-thrombotic agents during chemotherapy for epithelial ovarian cancer. Med. Hypotheses 2006, 66, 1179–1181. [Google Scholar] [CrossRef] [PubMed]
- Kao, J.; Genden, E.M.; Chen, C.T.; Rivera, M.; Tong, C.C.; Misiukiewicz, K.; Gupta, V.; Gurudutt, V.; Teng, M.; Packer, S.H. Phase 1 trial of concurrent erlotinib, celecoxib, and reirradiation for recurrent head and neck cancer. Cancer 2011, 117, 3173–3181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwiecien, S.; Magierowska, K.; Sliwowski, Z.; Wojcik, D.; Magierowski, M.; Brzozowski, T. New insight into the mechanisms of gastroduodenal injury induced by nonsteroidal anti-inflammatory drugs: Practical implications. Pol. Arch. Med. Wewn. 2015, 125, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Batlouni, M. Nonsteroidal anti-inflammatory drugs: Cardiovascular, cerebrovascular and renal effects. Arq. Bras. Cardiol. 2010, 94, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Scheiman, J.M.; Hindley, C.E. Strategies to optimize treatment with NSAIDs in patients at risk for gastrointestinal and cardiovascular adverse events. Clin. Ther. 2010, 32, 667–677. [Google Scholar] [CrossRef] [PubMed]
- Veltri, K.T. Yosprala: A Fixed Dose Combination of Aspirin and Omeprazole. Cardiol. Rev. 2018, 26, 50–53. [Google Scholar] [CrossRef] [PubMed]
- Mathur, S.; Conway, D.I.; Worlledge-Andrew, H.; Macpherson, L.M.; Ross, A.J. Assessment and prevention of behavioural and social risk factors associated with oral cancer: Protocol for a systematic review of clinical guidelines and systematic reviews to inform Primary Care dental professionals. Syst. Rev. 2015, 4, e184. [Google Scholar] [CrossRef] [PubMed]
- Nagao, T.; Warnakulasuriya, S.; Nakamura, T.; Kato, S.; Yamamoto, K.; Fukano, H.; Suzuki, K.; Shimozato, K.; Hashimoto, S. Treatment of oral leukoplakia with a low-dose of beta-carotene and vitamin C supplements: A randomized controlled trial. Int. J. Cancer 2015, 136, 1708–1717. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nasry, W.H.S.; Rodriguez-Lecompte, J.C.; Martin, C.K. Role of COX-2/PGE2 Mediated Inflammation in Oral Squamous Cell Carcinoma. Cancers 2018, 10, 348. https://doi.org/10.3390/cancers10100348
Nasry WHS, Rodriguez-Lecompte JC, Martin CK. Role of COX-2/PGE2 Mediated Inflammation in Oral Squamous Cell Carcinoma. Cancers. 2018; 10(10):348. https://doi.org/10.3390/cancers10100348
Chicago/Turabian StyleNasry, Walaa Hamed Shaker, Juan Carlos Rodriguez-Lecompte, and Chelsea K. Martin. 2018. "Role of COX-2/PGE2 Mediated Inflammation in Oral Squamous Cell Carcinoma" Cancers 10, no. 10: 348. https://doi.org/10.3390/cancers10100348
APA StyleNasry, W. H. S., Rodriguez-Lecompte, J. C., & Martin, C. K. (2018). Role of COX-2/PGE2 Mediated Inflammation in Oral Squamous Cell Carcinoma. Cancers, 10(10), 348. https://doi.org/10.3390/cancers10100348