Advances in Molecular Profiling and Categorisation of Pancreatic Adenocarcinoma and the Implications for Therapy

Abstract
1. Introduction
2. Genomic Studies in PDAC
3. The Role of Different Mutations in PDAC
3.1. KRAS Mutations in PDAC
3.2. DNA Damage Repair Mutations in PDAC
3.3. ATM Mutations in PDAC
3.4. TP53 Mutations in PDAC
3.5. SMAD4 Mutations in PDAC
3.6. CDKN2A Mutations in PDAC
3.7. BRAF Mutations in PDAC
3.8. Microsatellite Instability in PDAC
4. Mutation Burden in PDAC
5. Other Rare Alterations in PDAC
6. The Feasibility and Challenges of Clinical Molecular Profiling in PDAC
7. Discussion
8. Conclusions
Acknowledgments
Conflicts of Interest
References
- De Angelis, R.; Sant, M.; Coleman, M.P.; Francisci, S.; Baili, P.; Pierannunzio, D.; Trama, A.; Visser, O.; Brenner, H.; Ardanaz, E.; et al. Cancer survival in europe 1999–2007 by country and age: Results of EUROCARE-5—a population-based study. Lancet Oncol. 2014, 15, 23–34. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2017. CA: Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Cancer Research UK, Cancer Statistics, Pancreatic Cancer Statistics. Available online: http://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/pancreatic-cancer (accessed on 30 November 2017).
- Neoptolemos, J.P.; Palmer, D.H.; Ghaneh, P.; Psarelli, E.E.; Valle, J.W.; Halloran, C.M.; Faluyi, O.; O’Reilly, D.A.; Cunningham, D.; Wadsley, J.; et al. Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): A multicentre, open-label, randomised, phase 3 trial. Lancet 2017, 389, 1011–1024. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardiere, C.; et al. Folfirinox versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Goel, G.; Sun, W. Novel approaches in the management of pancreatic ductal adenocarcinoma: Potential promises for the future. J. Hematol. Oncol. 2015, 8, 44. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; van de Velde, H.; Karasek, P.; Oettle, H.; Vervenne, W.L.; Szawlowski, A.; Schoffski, P.; Post, S.; Verslype, C.; Neumann, H.; et al. Phase III trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer. J. Clin. 2004, 22, 1430–1438. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, C.S.; Azevedo, S.; Okusaka, T.; Van Laethem, J.L.; Lipton, L.R.; Riess, H.; Szczylik, C.; Moore, M.J.; Peeters, M.; Bodoky, G.; et al. A phase 3 randomized, double-blind, placebo-controlled trial of ganitumab or placebo in combination with gemcitabine as first-line therapy for metastatic adenocarcinoma of the pancreas: The gamma trial. Ann. Oncol. 2015, 26, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Middleton, G.; Palmer, D.H.; Greenhalf, W.; Ghaneh, P.; Jackson, R.; Cox, T.; Evans, A.; Shaw, V.E.; Wadsley, J.; Valle, J.W.; et al. Vandetanib plus gemcitabine versus placebo plus gemcitabine in locally advanced or metastatic pancreatic carcinoma (ViP): A prospective, randomised, double-blind, multicentre phase 2 trial. Lancet Oncol. 2017, 18, 486–499. [Google Scholar] [CrossRef]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the united states. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Salo-Mullen, E.E.; O’Reilly, E.M.; Kelsen, D.P.; Ashraf, A.M.; Lowery, M.A.; Yu, K.H.; Reidy, D.L.; Epstein, A.S.; Lincoln, A.; Saldia, A.; et al. Identification of germline genetic mutations in patients with pancreatic cancer. Cancer 2015, 121, 4382–4388. [Google Scholar] [CrossRef] [PubMed]
- Lal, G.; Liu, G.; Schmocker, B.; Kaurah, P.; Ozcelik, H.; Narod, S.A.; Redston, M.; Gallinger, S. Inherited predisposition to pancreatic adenocarcinoma: Role of family history and germ-line p16, BRCA1, and BRCA2 mutations. Cancer Res. 2000, 60, 409–416. [Google Scholar] [PubMed]
- Cowgill, S.M.; Muscarella, P. The genetics of pancreatic cancer. Am. J. Surg. 2003, 186, 279–286. [Google Scholar] [CrossRef]
- Gupta, C.; Mazzara, P.F. High-grade pancreatic intraepithelial neoplasia in a patient with familial adenomatous polyposis. Arch. Pathol. Lab. Med. 2005, 129, 1398–1400. [Google Scholar] [PubMed]
- Vasen, H.F.; Gruis, N.A.; Frants, R.R.; van Der Velden, P.A.; Hille, E.T.; Bergman, W. Risk of developing pancreatic cancer in families with familial atypical multiple mole melanoma associated with a specific 19 deletion of p16 (p16-Leiden). Int. J. Cancer 2000, 87, 809–811. [Google Scholar] [CrossRef]
- Lowenfels, A.B.; Maisonneuve, P.; DiMagno, E.P.; Elitsur, Y.; Gates, L.K., Jr.; Perrault, J.; Whitcomb, D.C. Hereditary pancreatitis and the risk of pancreatic cancer. International hereditary pancreatitis study group. J. Nat. Cancer Inst. 1997, 89, 442–446. [Google Scholar] [CrossRef] [PubMed]
- Shindo, K.; Yu, J.; Suenaga, M.; Fesharakizadeh, S.; Cho, C.; Macgregor-Das, A.; Siddiqui, A.; Witmer, P.D.; Tamura, K.; Song, T.J.; et al. Deleterious germline mutations in patients with apparently sporadic pancreatic adenocarcinoma. J. Clin. Oncol. 2017, 35, 3382–3390. [Google Scholar] [CrossRef] [PubMed]
- Waddell, N.; Pajic, M.; Patch, A.-M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Catalogue of Somatic Mutations in Cancer. Signatures of Mutational Processes in Human Cancer. Available online: http://cancer.sanger.ac.uk/cosmic/signatures (accessed on 30 November 2017).
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch-repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Frey, M.K.; Pothuri, B. Homologous recombination deficiency (hrd) testing in ovarian cancer clinical practice: A review of the literature. Gynecol. Oncol. Res. Pract. 2017, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Madan, B.; Ke, Z.; Harmston, N.; Ho, S.Y.; Frois, A.O.; Alam, J.; Jeyaraj, D.A.; Pendharkar, V.; Ghosh, K.; Virshup, I.H.; et al. Wnt addiction of genetically defined cancers reversed by porcn inhibition. Oncogene 2016, 35, 2197–2207. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Adams, R.P.; Swain, S.M. Does CDKN2A loss predict palbociclib benefit? Curr. Oncol. 2015, 22, e498–e501. [Google Scholar] [CrossRef] [PubMed]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the evolution of non–small-cell lung cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef] [PubMed]
- Wilentz, R.E.; Geradts, J.; Maynard, R.; Offerhaus, G.J.; Kang, M.; Goggins, M.; Yeo, C.J.; Kern, S.E.; Hruban, R.H. Inactivation of the p16 (INK4A) tumor-suppressor gene in pancreatic duct lesions: Loss of intranuclear expression. Cancer Res. 1998, 58, 4740–4744. [Google Scholar] [PubMed]
- Wilentz, R.E.; Iacobuzio-Donahue, C.A.; Argani, P.; McCarthy, D.M.; Parsons, J.L.; Yeo, C.J.; Kern, S.E.; Hruban, R.H. Loss of expression of DPC4 in pancreatic intraepithelial neoplasia: Evidence that DPC4 inactivation occurs late in neoplastic progression. Cancer Res. 2000, 60, 2002–2006. [Google Scholar] [PubMed]
- Moskaluk, C.A.; Hruban, R.H.; Kern, S.E. P16 and K-ras gene mutations in the intraductal precursors of human pancreatic adenocarcinoma. Cancer Res. 1997, 57, 2140–2143. [Google Scholar] [PubMed]
- Notta, F.; Chan-Seng-Yue, M.; Lemire, M.; Li, Y.; Wilson, G.W.; Connor, A.A.; Denroche, R.E.; Liang, S.B.; Brown, A.M.; Kim, J.C.; et al. A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature 2016, 538, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Makohon-Moore, A.P.; Zhang, M.; Reiter, J.G.; Bozic, I.; Allen, B.; Kundu, D.; Chatterjee, K.; Wong, F.; Jiao, Y.; Kohutek, Z.A.; et al. Limited heterogeneity of known driver gene mutations among the metastases of individual patients with pancreatic cancer. Nat. Genet. 2017, 49, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Raphael, B.J.; Hruban, R.H.; Aguirre, A.J.; Moffitt, R.A.; Yeh, J.J.; Stewart, C.; Robertson, A.G.; Cherniack, A.D.; Gupta, M.; Getz, G.; et al. Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell 2017, 32, 185–203.e113. [Google Scholar] [CrossRef] [PubMed]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef] [PubMed]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Bournet, B.; Buscail, C.; Muscari, F.; Cordelier, P.; Buscail, L. Targeting kras for diagnosis, prognosis, and treatment of pancreatic cancer: Hopes and realities. Eur. J. Cancer 2016, 54, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Bodoky, G.; Timcheva, C.; Spigel, D.R.; La Stella, P.J.; Ciuleanu, T.E.; Pover, G.; Tebbutt, N.C. A phase II open-label randomized study to assess the efficacy and safety of selumetinib (AZD6244 [ARRY-142886]) versus capecitabine in patients with advanced or metastatic pancreatic cancer who have failed first-line gemcitabine therapy. Investig. New Drugs 2012, 30, 1216–1223. [Google Scholar] [CrossRef] [PubMed]
- Lorusso, P.M.; Adjei, A.A.; Varterasian, M.; Gadgeel, S.; Reid, J.; Mitchell, D.Y.; Hanson, L.; DeLuca, P.; Bruzek, L.; Piens, J.; et al. Phase I and pharmacodynamic study of the oral MEK inhibitor CI-1040 in patients with advanced malignancies. J. Clin. Oncol. 2005, 23, 5281–5293. [Google Scholar] [CrossRef] [PubMed]
- Lowery, M.A.; Kelsen, D.P.; Stadler, Z.K.; Yu, K.H.; Janjigian, Y.Y.; Ludwig, E.; D’Adamo, D.R.; Salo-Mullen, E.; Robson, M.E.; Allen, P.J.; et al. An emerging entity: Pancreatic adenocarcinoma associated with a known brca mutation: Clinical descriptors, treatment implications, and future directions. Oncologist 2011, 16, 1397–1402. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Kanji, Z.S.; Epelbaum, R.; Devaud, N.; Dagan, E.; Holter, S.; Aderka, D.; Paluch-Shimon, S.; Kaufman, B.; Gershoni-Baruch, R.; et al. Overall survival and clinical characteristics of pancreatic cancer in brca mutation carriers. Br. J. Cancer 2014, 111, 1132–1138. [Google Scholar] [CrossRef] [PubMed]
- Aung, K.L.; Holter, S.; Borgida, A.; Connor, A.; Pintilie, M.; Dhani, N.C.; Hedley, D.W.; Knox, J.J.; Gallinger, S. Overall survival of patients with pancreatic adenocarcinoma and BRCA1 or BRCA2 germline mutation. J. Clin. Oncol. 2016, 34, 4123. [Google Scholar]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.L.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: A preplanned retrospective analysis of outcomes by brca status in a randomised phase 2 trial. Lancet Oncol. 2014, 15, 852–861. [Google Scholar] [CrossRef]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmana, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Riaz, N.; Blecua, P.; Lim, R.S.; Shen, R.; Higginson, D.S.; Weinhold, N.; Norton, L.; Weigelt, B.; Powell, S.N.; Reis-Filho, J.S. Pan-cancer analysis of BI-allelic alterations in homologous recombination DNA repair genes. Nat. Commun. 2017, 8, 857. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute (NCI). A Randomized Phase II Study of Gemcitabine, Cisplatin +/− Veliparib in Patients with Pancreas Adenocarcinoma and a Known BRCA/ PALB2 Mutation (Part I) and a Phase II Single Arm Study of Single-Agent Veliparib in Previously Treated Pancreas Adenocarcinoma (Part II). Available online: https://clinicaltrials.Gov/ct2/show/nct01585805 (accessed on 30 November 2017).
- AstraZeneca. A Phase III, Randomised, Double Blind, Placebo Controlled, Multicentre Study of Maintenance Olaparib Monotherapy in Patients with gBRCA Mutated Metastatic Pancreatic Cancer Whose Disease Has Not Progressed on First Line Platinum Based Chemotherapy. Available online: https://clinicaltrials.Gov/ct2/show/nct02184195 (accessed on 30 November 2017).
- Perkhofer, L.; Schmitt, A.; Romero Carrasco, M.C.; Ihle, M.; Hampp, S.; Ruess, D.A.; Hessmann, E.; Russell, R.; Lechel, A.; Azoitei, N.; et al. ATM deficiency generating genomic instability sensitizes pancreatic ductal adenocarcinoma cells to therapy-induced DNA damage. Cancer Res. 2017, 77, 5576–5590. [Google Scholar] [CrossRef] [PubMed]
- McCabe, N.; Turner, N.C.; Lord, C.J.; Kluzek, K.; Bialkowska, A.; Swift, S.; Giavara, S.; O’Connor, M.J.; Tutt, A.N.; Zdzienicka, M.Z.; et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006, 66, 8109–8115. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. Brcaness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Polak, P.; Kim, J.; Braunstein, L.Z.; Karlic, R.; Haradhavala, N.J.; Tiao, G.; Rosebrock, D.; Livitz, D.; Kübler, K.; Mouw, K.W.; et al. A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat. Genet. 2017, 49, 1476. [Google Scholar] [CrossRef] [PubMed]
- MD Anderson Cancer Center. Olaparib for Brcaness Phenotype in Pancreatic Cancer: Phase II Study. Available online: https://clinicaltrials.Gov/ct2/show/nct02677038 (accessed on 30 November 2017).
- Weber, A.M.; Ryan, A.J. Atm and atr as therapeutic targets in cancer. Pharmacol.Ther. 2015, 149, 124–138. [Google Scholar] [CrossRef] [PubMed]
- Royal Marsden NHS Foundation Trust. A Phase I Study to Assess the Tolerability, Safety and Biological Effects of ATR Inhibitor (AZD6738) as a Single Agent and in Combination with Palliative Radiation Therapy in Patients with Solid Tumours. Available online: https://clinicaltrials.Gov/ct2/show/nct02223923 (accessed on 30 November 2017).
- AstraZeneca. A Modular Phase I, Open-Label, Multicentre Study to Assess the Safety, Tolerability, Pharmacokinetics and Preliminary Anti-Tumour Activity of AZD6738 in Combination with Cytotoxic Chemotherapy and/or DNA Damage Repair/Novel Anti-Cancer Agents in Patients with Advanced Solid Malignancies. Available online: https://clinicaltrials.Gov/ct2/show/nct02264678 (accessed on 30 November 2017).
- Kim, H.; Saka, B.; Knight, S.; Borges, M.; Childs, E.; Klein, A.; Wolfgang, C.; Herman, J.; Adsay, V.N.; Hruban, R.H.; et al. Having pancreatic cancer with tumoral loss of ATM and normal TP53 protein expression is associated with a poorer prognosis. Clin. Cancer Res. 2014, 20, 1865–1872. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.; Perkhofer, L.; Liebau, S.; Lin, Q.; Lechel, A.; Feld, F.M.; Hessmann, E.; Gaedcke, J.; Guthle, M.; Zenke, M.; et al. Loss of ATM accelerates pancreatic cancer formation and epithelial-mesenchymal transition. Nat. Commun. 2015, 6, 7677. [Google Scholar] [CrossRef] [PubMed]
- Drosos, Y.; Escobar, D.; Chiang, M.Y.; Roys, K.; Valentine, V.; Valentine, M.B.; Rehg, J.E.; Sahai, V.; Begley, L.A.; Ye, J.; et al. Atm-deficiency increases genomic instability and metastatic potential in a mouse model of pancreatic cancer. Sci. Rep. 2017, 7, 11144. [Google Scholar] [CrossRef] [PubMed]
- Ayars, M.; Eshleman, J.; Goggins, M. Susceptibility of ATM-deficient pancreatic cancer cells to radiation. Cell Cycle 2017, 16, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Morton, J.P.; Timpson, P.; Karim, S.A.; Ridgway, R.A.; Athineos, D.; Doyle, B.; Jamieson, N.B.; Oien, K.A.; Lowy, A.M.; Brunton, V.G.; et al. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Weissmueller, S.; Manchado, E.; Saborowski, M.; Morris, J.P.; Wagenblast, E.; Davis, C.A.; Moon, S.H.; Pfister, N.T.; Tschaharganeh, D.F.; Kitzing, T.; et al. Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor β signaling. Cell 2014, 157, 382–394. [Google Scholar] [CrossRef] [PubMed]
- Dhayat, S.A.; Mardin, W.A.; Seggewiss, J.; Strose, A.J.; Matuszcak, C.; Hummel, R.; Senninger, N.; Mees, S.T.; Haier, J. Microrna profiling implies new markers of gemcitabine chemoresistance in mutant p53 pancreatic ductal adenocarcinoma. PLoS ONE 2015, 10, e0143755. [Google Scholar] [CrossRef] [PubMed]
- Oza, A.M.; Weberpals, J.I.; Provencher, D.M.; Grischke, E.-M.; Hall, M.; Uyar, D.; Estevez-Diz, M.D.P.; Marmé, F.; Kuzmin, A.; Rosenberg, P.; et al. An international, biomarker-directed, randomized, phase II trial of AZD1775 plus paclitaxel and carboplatin (p/C) for the treatment of women with platinum-sensitive, TP53-mutant ovarian cancer. J. Clin. Oncol. 2015, 33, 5506. [Google Scholar]
- Gourley, C.; Green, J.; Gabra, H.; Vergote, I.; Basu, B.; Brenton, J.D.; Björklund, U.; Smith, A.M.; Euler, M.V. PISARRO: A EUTROC phase Ib study of APR-246 in combination with carboplatin (C) and pegylated liposomal doxorubicin (PLD) in platinum sensitive relapsed high grade serous ovarian cancer (HGSOC). J. Clin. Oncol. 2016, 34, 5571. [Google Scholar]
- A Phase Ib/2 Study Evaluating the Efficacy of APR-246, a First-In-Class Agent Targeting Mutant p53 in the Treatment of Platinum Resistant Advanced and Metastatic Oesophageal or Gastro-Oesophageal Junction Cancers. Available online: https://clinicaltrials.Gov/show/nct02999893 (accessed on 30 November 2017).
- Phase II, Single-Arm Study of AZD1775 Monotherapy in Relapsed Small Cell Lung Cancer Patients with MYC Family Amplification or CDKN2A Mutation Combined with TP53 Mutation. Available online: https://clinicaltrials.Gov/show/nct02688907 (accessed on 30 November 2017).
- A Multicentre Phase II Study of AZD1775 Plus Chemotherapy in Patients with Platinum-Resistant Epithelial Ovarian, Fallopian Tube, or Primary Peritoneal Cancer. Available online: https://clinicaltrials.Gov/show/nct02272790 (accessed on 30 November 2017).
- Ormanns, S.; Haas, M.; Remold, A.; Kruger, S.; Holdenrieder, S.; Kirchner, T.; Heinemann, V.; Boeck, S. The impact of SMAD4 loss on outcome in patients with advanced pancreatic cancer treated with systemic chemotherapy. Int. J. Mol. Sci. 2017, 18, 1094. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Wu, W.; Huang, C.; Cen, G.; Jiang, T.; Cao, J.; Huang, K.; Qiu, Z. SMAD4 and its role in pancreatic cancer. Tumour Boil. 2015, 36, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Crane, C.H.; Varadhachary, G.R.; Yordy, J.S.; Staerkel, G.A.; Javle, M.M.; Safran, H.; Haque, W.; Hobbs, B.D.; Krishnan, S.; Fleming, J.B.; et al. Phase II trial of cetuximab, gemcitabine, and oxaliplatin followed by chemoradiation with cetuximab for locally advanced (T4) pancreatic adenocarcinoma: Correlation of SMAD4(DPC4) immunostaining with pattern of disease progression. J. Clin. Oncol. 2011, 29, 3037–3043. [Google Scholar] [CrossRef] [PubMed]
- Ottenhof, N.A.; Morsink, F.H.M.; ten Kate, F.; van Noorden, C.J.F.; Offerhaus, G.J.A. Multivariate analysis of immunohistochemical evaluation of protein expression in pancreatic ductal adenocarcinoma reveals prognostic significance for persistent SMAD4 expression only. Cell. Oncol. 2012, 35, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.H.; Kim, H.J.; Hwang, D.W.; Lee, J.H.; Song, K.B.; Jun, E.; Shim, I.K.; Hong, S.M.; Kim, H.J.; Park, K.M.; et al. The DPC4/SMAD4 genetic status determines recurrence patterns and treatment outcomes in resected pancreatic ductal adenocarcinoma: A prospective cohort study. Oncotarget 2017, 8, 17945–17959. [Google Scholar] [PubMed]
- Shugang, X.; Hongfa, Y.; Jianpeng, L.; Xu, Z.; Jingqi, F.; Xiangxiang, L.; Wei, L. Prognostic value of SMAD4 in pancreatic cancer: A meta-analysis. Trans. Oncol. 2016, 9, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Dey, P.; Baddour, J.; Muller, F.; Wu, C.C.; Wang, H.; Liao, W.-T.; Lan, Z.; Chen, A.; Gutschner, T.; Kang, Y.; et al. Genomic deletion of malic enzyme 2 confers collateral lethality in pancreatic cancer. Nature 2017, 542, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Soura, E.; Eliades, P.; Shannon, K.; Stratigos, A.; Tsao, H. Hereditary melanoma: Update on syndromes and management—Genetics of familial atypical multiple mole melanoma syndrome. J. Am. Acad. Der. 2016, 74, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Ghiorzo, P.; Fornarini, G.; Sciallero, S.; Battistuzzi, L.; Belli, F.; Bernard, L.; Bonelli, L.; Borgonovo, G.; Bruno, W.; De Cian, F.; et al. CDKN2A is the main susceptibility gene in italian pancreatic cancer families. J. Med. Genet. 2012, 49, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Oshima, M.; Okano, K.; Muraki, S.; Haba, R.; Maeba, T.; Suzuki, Y.; Yachida, S. Immunohistochemically detected expression of 3 major genes (CDKN2A/p16, TP53, and SMAD4/DPC4) strongly predicts survival in patients with resectable pancreatic cancer. Ann. Surg. 2013, 258, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Phase II Study of Ilorasertib (ABT348) in Patients with CDKN2A Deficient Solid Tumors. Available online: https://clinicaltrials.Gov/show/nct02478320 (accessed on 30 November 2017).
- Elvin, J.A.; Gay, L.M.; Ort, R.; Shuluk, J.; Long, J.; Shelley, L.; Lee, R.; Chalmers, Z.R.; Frampton, G.M.; Ali, S.M.; et al. Clinical benefit in response to palbociclib treatment in refractory uterine leiomyosarcomas with a common CDKN2A alteration. Ooncologist 2017, 22, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Chou, A.; Froio, D.; Nagrial, A.M.; Parkin, A.; Murphy, K.J.; Chin, V.T.; Wohl, D.; Steinmann, A.; Stark, R.; Drury, A.; et al. Tailored first-line and second-line CDK4-targeting treatment combinations in mouse models of pancreatic cancer. Gut 2017. [Google Scholar] [CrossRef] [PubMed]
- An Open-Label Phase Ib Study of Palbociclib (Oral CDK 4/6 Inhibitor) Plus Abraxane (Registered) (Nab-Paclitaxel) in Patients with Metastatic Pancreatic Ductal Adenocarcinoma. Available online: https://clinicaltrials.Gov/show/nct02501902 (accessed on 30 November 2017).
- Phase I Study of the CDK4/6 Inhibitor Palbociclib (PD-0332991) in Combination with the PI3K/mTOR Inhibitor Gedatolisib (PF-05212384) for Patients with Advanced Squamous Cell Lung, Pancreatic, Head & Neck and Other Solid Tumors. Available online: https://clinicaltrials.Gov/show/nct03065062 (accessed on 30 November 2017).
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.C.; Mansour, J.; Mollaee, M.; Wagner, K.U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed]
- Humphris, J.L.; Patch, A.-M.; Nones, K.; Bailey, P.J.; Johns, A.L.; McKay, S.; Chang, D.K.; Miller, D.K.; Pajic, M.; Kassahn, K.S.; et al. Hypermutation in pancreatic cancer. Gastroenterology 2017, 152, 68–74.e62. [Google Scholar] [CrossRef] [PubMed]
- The US Food and Drug Administration News Release. FDA Approves First Cancer Treatment for Any Solid Tumor with a Specific Genetic Feature. Available online: https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm560167.htm (accessed on 30 November 2017).
- Diaz, L.A.; Marabelle, A.; Delord, J.-P.; Shapira-Frommer, R.; Geva, R.; Peled, N.; Kim, T.W.; Andre, T.; Cutsem, E.V.; Guimbaud, R.; et al. Pembrolizumab therapy for microsatellite instability high (MSI-H) colorectal cancer (CRC) and non-CRC. J. Clin. Oncol. 2017, 35, 3071. [Google Scholar]
- Cheng, D.T.; Mitchell, T.N.; Zehir, A.; Shah, R.H.; Benayed, R.; Syed, A.; Chandramohan, R.; Liu, Z.Y.; Won, H.H.; Scott, S.N.; et al. Memorial sloan kettering-integrated mutation profiling of actionable cancer targets (MSK-impact): A hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J. Mol. Diagn. 2015, 17, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Fox, A.J.; Hiemenz, M.C.; Lieberman, D.B.; Sukhadia, S.; Li, B.; Grubb, J.; Candrea, P.; Ganapathy, K.; Zhao, J.; Roth, D.; et al. Next generation sequencing for the detection of actionable mutations in solid and liquid tumors. J. Vis. Exp. 2016, 52758. [Google Scholar] [CrossRef] [PubMed]
- Eifert, C.; Pantazi, A.; Sun, R.; Xu, J.; Cingolani, P.; Heyer, J.; Russell, M.; Lvova, M.; Ring, J.; Tse, J.Y.; et al. Clinical application of a cancer genomic profiling assay to guide precision medicine decisions. Pers. Med. 2017, 14, 309–325. [Google Scholar] [CrossRef] [PubMed]
- Campbell, B.B.; Light, N.; Fabrizio, D.; Zatzman, M.; Fuligni, F.; de Borja, R.; Davidson, S.; Edwards, M.; Elvin, J.A.; Hodel, K.P.; et al. Comprehensive analysis of hypermutation in human cancer. Cell 2017, 171, 1042–1056. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar]
- Royal, R.E.; Levy, C.; Turner, K.; Mathur, A.; Hughes, M.; Kammula, U.S.; Sherry, R.M.; Topalian, S.L.; Yang, J.C.; Lowy, I.; et al. Phase 2 trial of single agent ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J. Immunother. 2010, 33, 828–833. [Google Scholar] [CrossRef] [PubMed]
- Aglietta, M.; Barone, C.; Sawyer, M.B.; Moore, M.J.; Miller, W.H., Jr.; Bagala, C.; Colombi, F.; Cagnazzo, C.; Gioeni, L.; Wang, E.; et al. A phase I dose escalation trial of tremelimumab (CP-675,206) in combination with gemcitabine in chemotherapy-naive patients with metastatic pancreatic cancer. Ann. Oncol. 2014, 25, 1750–1755. [Google Scholar] [CrossRef] [PubMed]
- Kunk, P.R.; Bauer, T.W.; Slingluff, C.L.; Rahma, O.E. From bench to bedside a comprehensive review of pancreatic cancer immunotherapy. J. Immunother. Cancer 2016, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- A Phase Ib/2, Open-Label, Multicenter, Randomized Umbrella Study Evaluating the Efficacy and Safety of Multiple Immunotherapy-Based Treatment Combinations in Patients with Metastatic Pancreatic Ductal Adenocarcinoma (Morpheus-Pancreatic Cancer). Available online: https://clinicaltrials.Gov/show/nct03193190 (accessed on 30 November 2017).
- A Phase 2 Study of Neoadjuvant Chemotherapy with and without Immunotherapy to CA125 (Oregovomab) Followed by Hypofractionated Stereotactic Radiotherapy and Concurrent HIV Protease Inhibitor Nelfinavir in Patients with Locally Advanced Pancreatic Cancer. Available online: https://clinicaltrials.Gov/show/nct01959672 (accessed on 30 November 2017).
- A Phase 2 Study of Cabiralizumab (BMS-986227, FPA008) Administered in Combination with Nivolumab (BMS-936558) with and without Chemotherapy in Patients with Advanced Pancreatic Cancer. Available online: https://clinicaltrials.Gov/show/nct03336216 (accessed on 30 November 2017).
- A Pilot Study of a GVAX Pancreas Vaccine (with Cyclophosphamide) in Combination with a PD-1 Blockade Antibody (Pembrolizumab) and a Macrophage Targeting Agent (CSF1R Inhibitor) for the Treatment of Patients with Borderline Resectable Adenocarcinoma of the Pancreas. Available online: https://clinicaltrials.Gov/show/nct03153410 (accessed on 30 November 2017).
- A Phase Ib Dose-Escalation and Cohort-Expansion Study of the Safety, Tolerability, and Efficacy of a Novel Transforming Growth Factor-β Receptor I Kinase Inhibitor (Galunisertib) Administered in Combination with the Anti-PD-l1 Antibody Durvalumab (MEDI4736) in Recurrent or Refractory Metastatic Pancreatic Cancer. In: Clinicaltrials. Available online: https://clinicaltrials.Gov/show/nct02734160 (accessed on 30 November 2017).
- A Phase 2 Study of GM-CSF Secreting Allogeneic Pancreatic Cancer Vaccine in Combination with PD-1 Blockade Antibody (Pembrolizumab) and Stereotactic Body Radiation Therapy (SBRT) for the Treatment of Patients with Locally Advanced Adenocarcinoma of the Pancreas. Available online: https://clinicaltrials.Gov/show/nct02648282 (accessed on 30 November 2017).
- A Phase 2 Clinical Trial of GVAX Pancreas Vaccine (with Cyclophosphamide) in Combination with Nivolumab and Stereotactic Body Radiation Therapy (SBRT) Followed by Definitive Resection for Patients with Borderline Resectable Pancreatic Adenocarcinoma. Available online: https://clinicaltrials.Gov/show/nct03161379 (accessed on 30 November 2017).
- A Randomized Multicenter Phase Ib/2 Study to Assess the Safety and the Immunological Effect of Chemoradiation Therapy (CRT) in Combination with Pembrolizumab (MK-3475) Compared to CRT Alone in Patients with Resectable or Borderline Resectable Pancreatic Cancer. Available online: https://clinicaltrials.Gov/show/nct02305186 (accessed on 30 November 2017).
- Nivolumab and Ipilimumab and Radiation Therapy in Microsatellite Stable (MSS) and Microsatellite Instability (MSI) High Colorectal and Pancreatic Cancer. Available online: https://clinicaltrials.Gov/show/nct03104439 (accessed on 30 November 2017).
- A Randomized Phase 2 Study of the Safety, Efficacy, and Immune Response of CRS-207, Nivolumab, and Ipilimumab with or without GVAX Pancreas Vaccine (with Cyclophosphamide) in Patients with Previously Treated Metastatic Pancreatic Adenocarcinoma. Available online: https://clinicaltrials.Gov/show/nct03190265 (accessed on 30 November 2017).
- Phase 2 Study of Epacadostat, Pembrolizumab, and CRS-207, with or without Cyclophosphamide and GVAX Pancreas Vaccine in Patients with Metastatic Pancreas Cancer. Available online: https://clinicaltrials.Gov/show/nct03006302 (accessed on 30 November 2017).
- Phase 1/2 Study to Evaluate the Safety and Preliminary Efficacy of Nivolumab Combined with Daratumumab in Participants with Advanced or Metastatic Solid Tumors. Available online: https://clinicaltrials.Gov/show/nct03098550 (accessed on 30 November 2017).
- A Phase 1 Immunotherapy Study of Evofosfamide in Combination with Ipilimumab in Patients with Advanced Solid Malignancies. Available online: https://clinicaltrials.Gov/show/nct03098160 (accessed on 30 November 2017).
- A Phase 1b and 2 Open-Label, Multi-Center Study of MEDI4736 Evaluated in Different Combinations in Patients with Metastatic Pancreatic Ductal Adenocarcinoma. Available online: https://clinicaltrials.Gov/show/nct02583477 (accessed on 30 November 2017).
- A Two-Part, Open-Label Phase 1/2 Study to Evaluate Pharmacodynamic Effects and Safety of Olaptesed Pegol Monotherapy and Safety and Efficacy of Olaptesed Pegol/Pembrolizumab Combination Therapy in Metastatic Colorectal and Pancreatic Cancer. Available online: https://clinicaltrials.Gov/show/nct03168139 (accessed on 30 November 2017).
- Nant Pancreatic Cancer Vaccine: Combination Immunotherapy with High-Affinity Natural Killer (HANK) in Subjects with Pancreatic Cancer Who Have Progressed on or after Standard-of-Care Therapy. Available online: https://clinicaltrials.Gov/show/nct03329248 (accessed on 30 November 2017).
- Immunotherapy and Irreversible Electroporation in the Treatment of Advanced Pancreatic Adenocarcinoma. Available online: https://clinicaltrials.Gov/show/nct03080974 (accessed on 30 November 2017).
- Steinhart, Z.; Pavlovic, Z.; Chandrashekhar, M.; Hart, T.; Wang, X.; Zhang, X.; Robitaille, M.; Brown, K.R.; Jaksani, S.; Overmeer, R.; et al. Genome-wide CRISPR screens reveal a Wnt-FZD5 signaling circuit as a druggable vulnerability of RNF43-mutant pancreatic tumors. Nat. Med. 2017, 23, 60–68. [Google Scholar] [CrossRef] [PubMed]
- A Phase I, Open-Label, Dose Escalation Study of Oral LGK974 in Patients with Malignancies Dependent on Wnt Ligands. Available online: https://clinicaltrials.Gov/show/nct01351103 (accessed on 30 November 2017).
- Ross, J.S.; Slodkowska, E.A.; Symmans, W.F.; Pusztai, L.; Ravdin, P.M.; Hortobagyi, G.N. The HER-2 receptor and breast cancer: Ten years of targeted anti-HER-2 therapy and personalized medicine. Oncologist 2009, 14, 320–368. [Google Scholar] [CrossRef] [PubMed]
- Tadesse, S.; Yu, M.; Kumarasiri, M.; Le, B.T.; Wang, S. Targeting CDK6 in cancer: State of the art and new insights. Cell Cycle 2015, 14, 3220–3230. [Google Scholar] [CrossRef] [PubMed]
- Karakas, B.; Bachman, K.E.; Park, B.H. Mutation of the PIK3CA oncogene in human cancers. Br. J. Cancer 2006, 94, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Lowery, M.A.; Jordan, E.J.; Basturk, O.; Ptashkin, R.N.; Zehir, A.; Berger, M.F.; Leach, T.; Herbst, B.; Askan, G.; Maynard, H.; et al. Real-time genomic profiling of pancreatic ductal adenocarcinoma: Potential actionability and correlation with clinical phenotype. Clin. Cancer Res. 2017, 23, 6094–6100. [Google Scholar] [CrossRef] [PubMed]
- Johns, A.L.; McKay, S.H.; Humphris, J.L.; Pinese, M.; Chantrill, L.A.; Mead, R.S.; Tucker, K.; Andrews, L.; Goodwin, A.; Leonard, C.; et al. Lost in translation: Returning germline genetic results in genome-scale cancer research. Genome Med. 2017, 9, 41. [Google Scholar] [CrossRef] [PubMed]
- Dreyer, S.B.; Chang, D.K.; Bailey, P.; Biankin, A.V. Pancreatic cancer genomes: Implications for clinical management and therapeutic development. Clin. Cancer Res. 2017, 23, 1638–1646. [Google Scholar] [CrossRef] [PubMed]


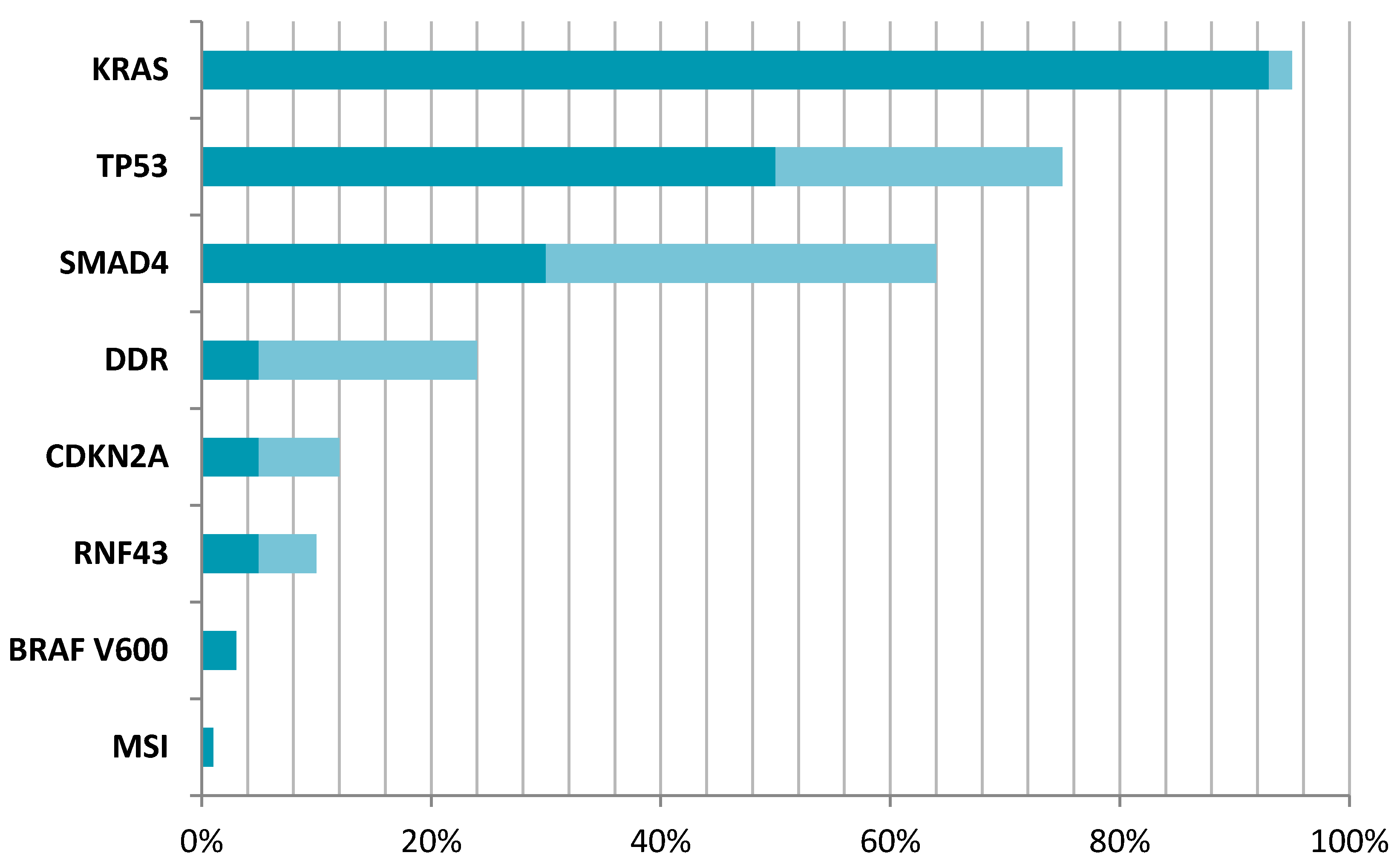{kind=link}
| Trial ID | Stage of Disease | Checkpoint Inhibitor | Combined with | Phase | Number of Patients | Countries Involved |
|---|---|---|---|---|---|---|
| NCT03193190 [95] | IV | Atezolizumab | Cobimetinib or PEGPH20 or BL-8040 | Ib–II | 185 | International |
| NCT01959672 [96] | I–III | Oregovomab | As single agent after chemotherapy and prior to Stereotactic Body Radiation Therapy | II | 66 | US |
| NCT03336216 [97] | III–IV | Nivolumab | Cabiralizumab or Cabiralizumab + chemotherapy | II | 160 | US |
| NCT03153410 [98] | II * | Pembrolizumab | Cyclophosphamide + GVAX Pancreas Vaccine + IMC-CS4 | I | 12 | US |
| NCT02734160 [99] | IV | Durvalumab | Galunisertib | Ib | 37 | International |
| NCT02648282 [100] | III | Pembrolizumab | Cyclophosphamide + GVAX Pancreas Vaccine + Stereotactic Body Radiation Therapy | II | 54 | US |
| NCT03161379 [101] | II * | Nivolumab | Cyclophosphamide + GVAX Pancreas Vaccine + Stereotactic Body Radiation Therapy | II | 50 | US |
| NCT02305186 [102] | I–II * | Pembrolizumab | Radiotherapy + capecitabine | Ib–II | 56 | US |
| NCT03104439 [103] | IV | Nivolumab and Ipilimumab | Radiotherapy | II | 80 | US |
| NCT03190265 [104] | IV | Nivolumab and Ipilimumab | Cyclophosphamide + GVAX Pancreas Vaccine + CRS-207 | II | 63 | US |
| NCT03006302 [105] | IV | Pembrolizumab | Cyclophosphamide + Epacadostat + CRS-207 + GVAX Pancreas Vaccine | II | 70 | US |
| NCT03098550 [106] | III–IV | Nivolumab | Daratumumab | I–II | 120 | International |
| NCT03098160 [107] | IV | Ipilimumab | Evofosfamide | I | 69 | US |
| NCT02583477 [108] | IV | Durvalumab | Nab-paclitaxel and gemcitabine or AZD5069 | Ib–II | 19 | US, UK |
| NCT03168139 [109] | IV | Pembrolizumab | Olaptesed pegol | I–II | 20 | Germany |
| NCT03329248 [110] | III–IV | Avelumab | ALT-803 + ETBX-011 + GI-4000 + haNK for infusion + bevacizumab + Capecitabine + Cyclophosphamide + Fluorouracil + Leucovorin + nab-Paclitaxel + lovaza + Oxaliplatin + SBRT | I–II | 80 | US |
| NCT03080974 [111] | III | Nivolumab | Irreversible Electroporation | II | 10 | US |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pihlak, R.; Weaver, J.M.J.; Valle, J.W.; McNamara, M.G. Advances in Molecular Profiling and Categorisation of Pancreatic Adenocarcinoma and the Implications for Therapy. Cancers 2018, 10, 17. https://doi.org/10.3390/cancers10010017
Pihlak R, Weaver JMJ, Valle JW, McNamara MG. Advances in Molecular Profiling and Categorisation of Pancreatic Adenocarcinoma and the Implications for Therapy. Cancers. 2018; 10(1):17. https://doi.org/10.3390/cancers10010017
Chicago/Turabian StylePihlak, Rille, Jamie M. J. Weaver, Juan W. Valle, and Mairéad G. McNamara. 2018. "Advances in Molecular Profiling and Categorisation of Pancreatic Adenocarcinoma and the Implications for Therapy" Cancers 10, no. 1: 17. https://doi.org/10.3390/cancers10010017
APA StylePihlak, R., Weaver, J. M. J., Valle, J. W., & McNamara, M. G. (2018). Advances in Molecular Profiling and Categorisation of Pancreatic Adenocarcinoma and the Implications for Therapy. Cancers, 10(1), 17. https://doi.org/10.3390/cancers10010017