
A Programmable Digital Microfluidic Assay for the Simultaneous Detection of Multiple Anti-Microbial Resistance Genes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
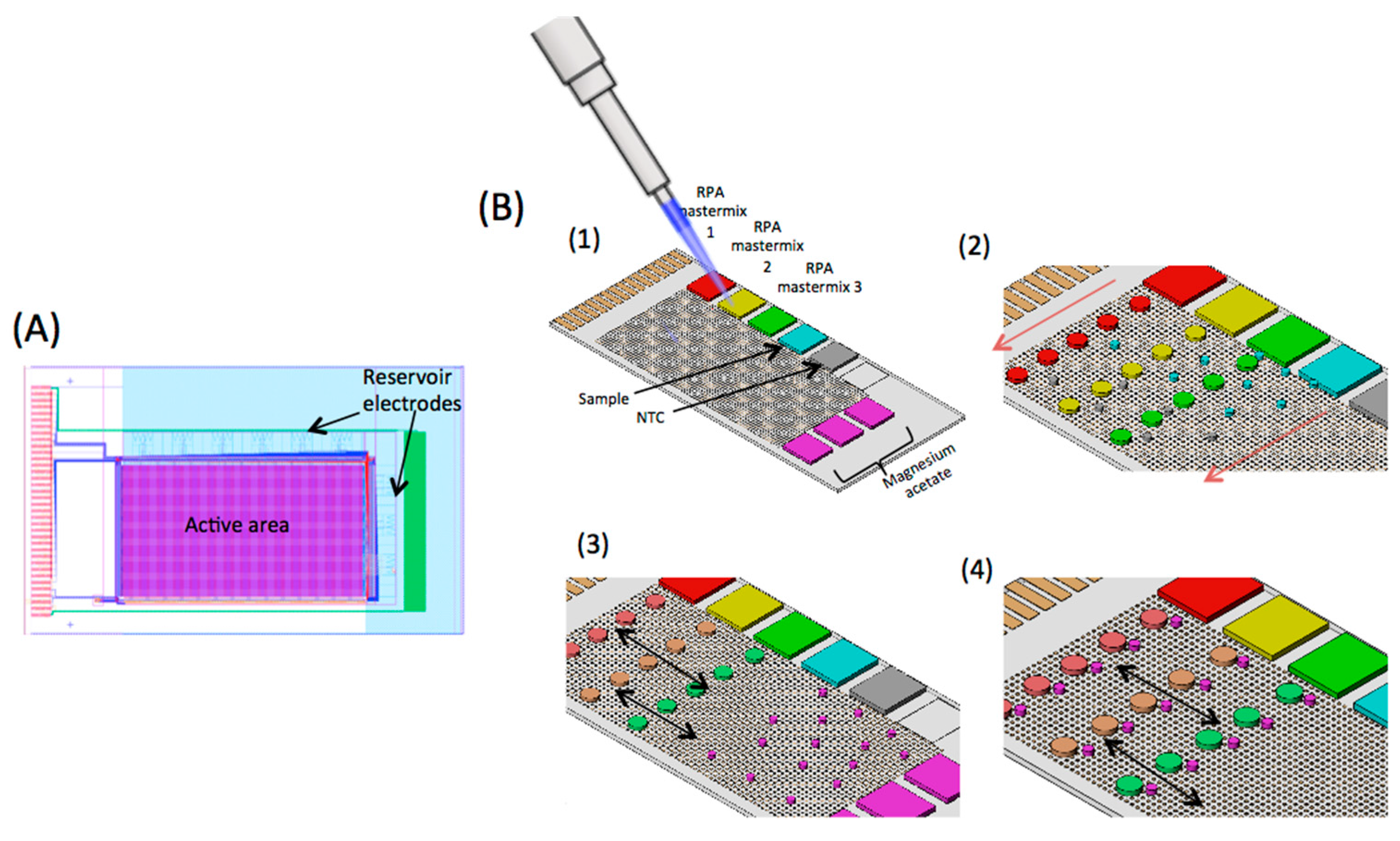
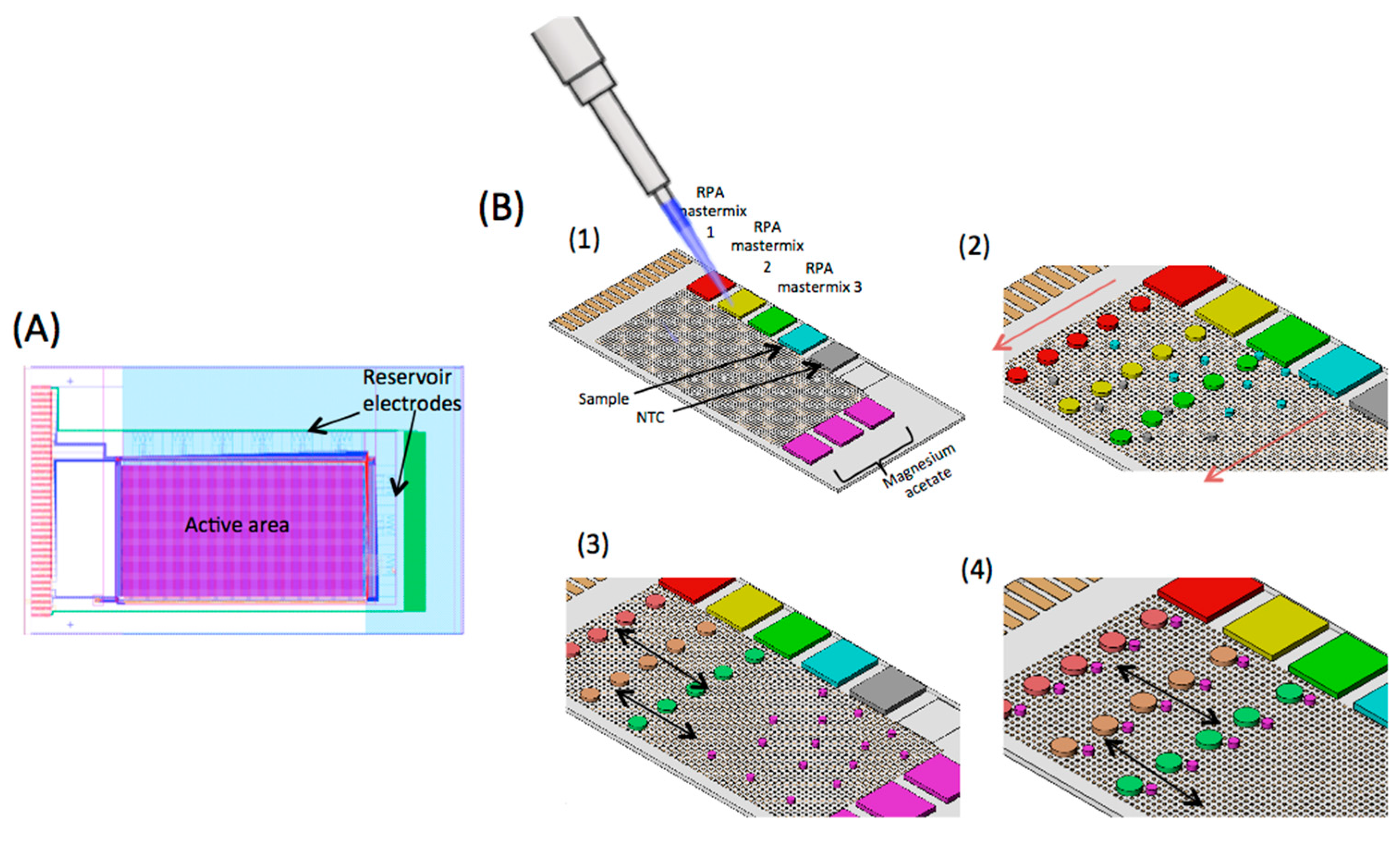
2.1. Digital Microfluidic (DMF) Device
2.2. Control Electronics and Software
2.3. Bacterial Culture and Plasmid Extraction
2.4. Benchtop RPA Assay
2.5. DMF RPA Assay
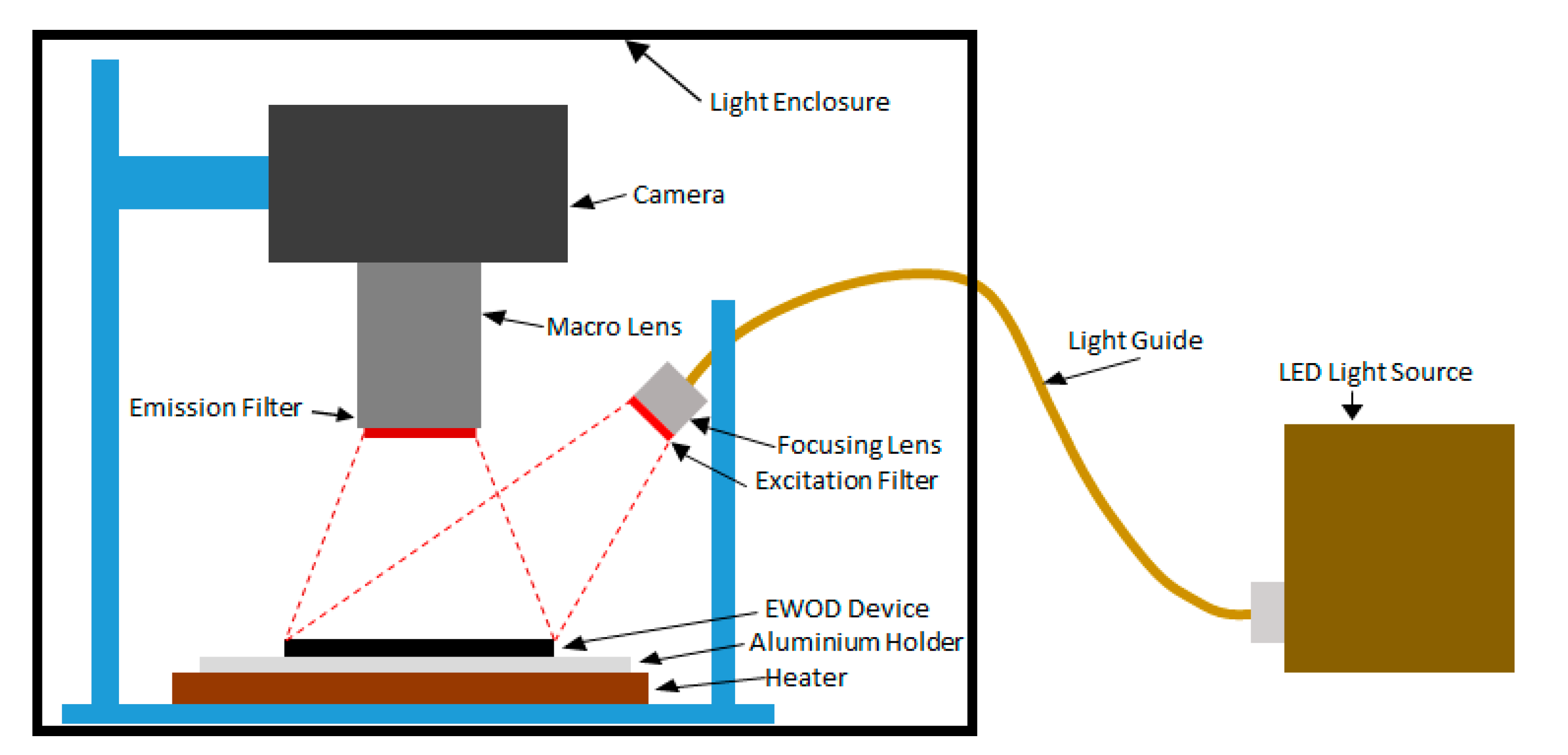
2.6. Optical System and Data Analysis
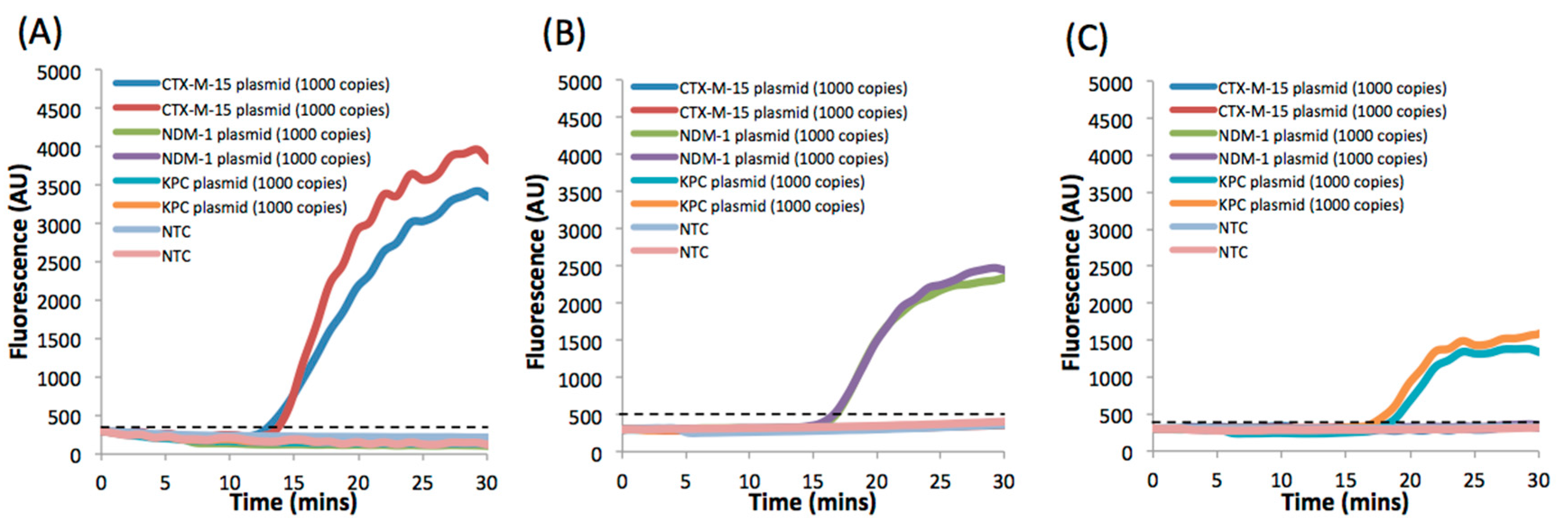
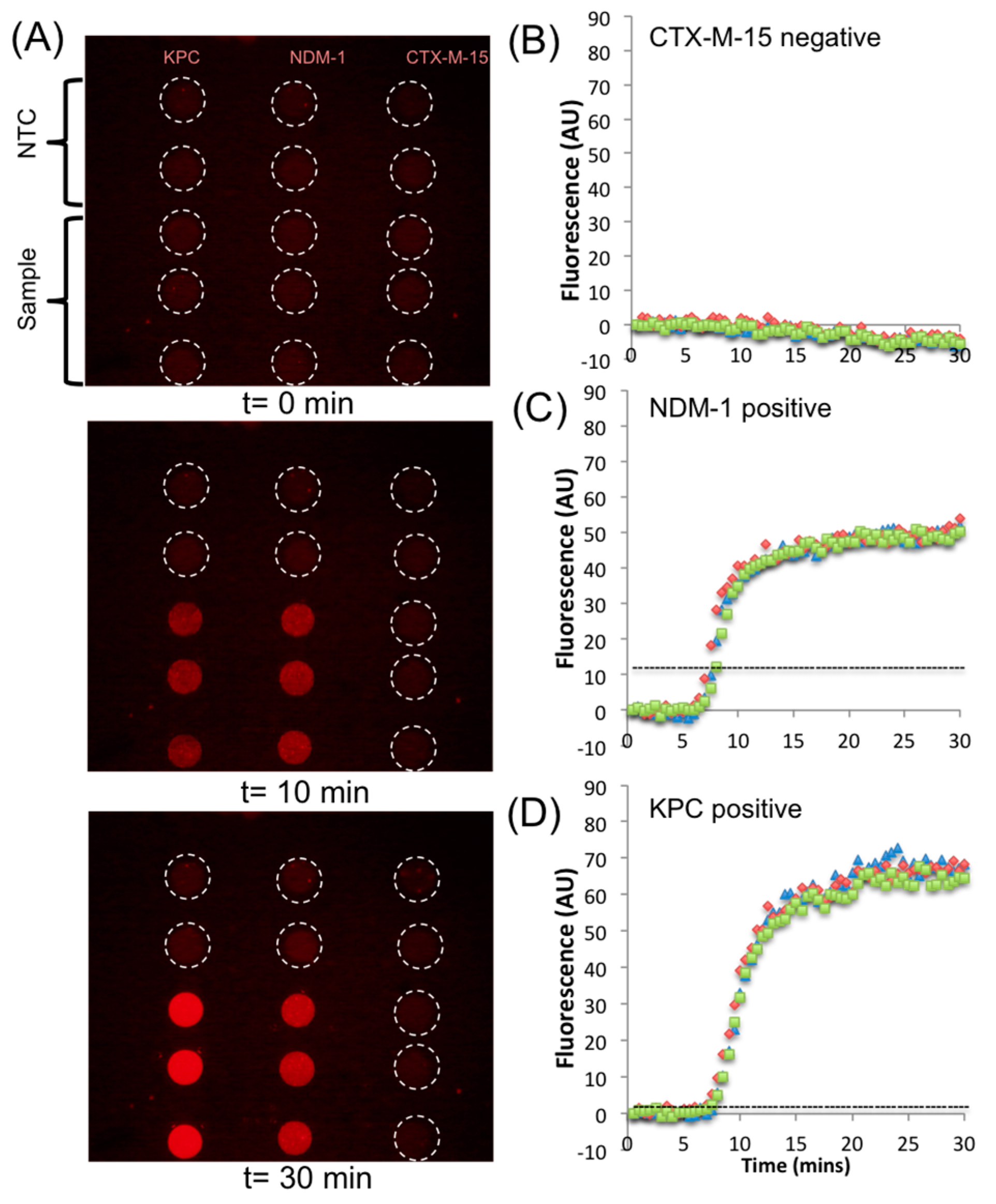
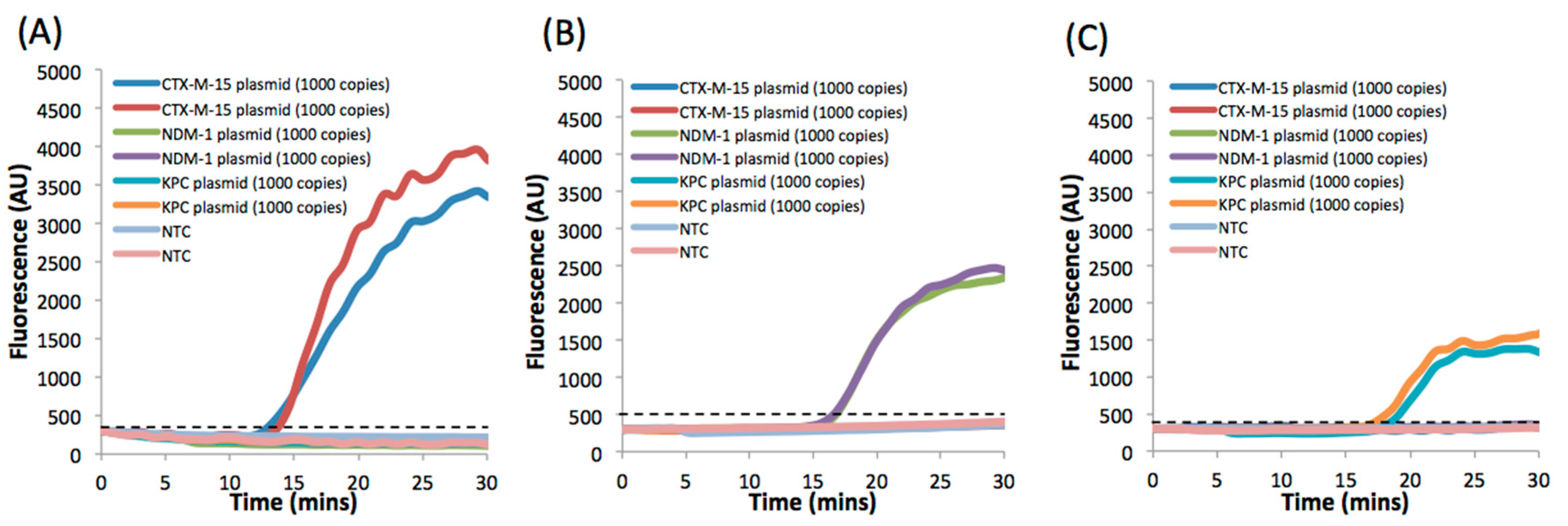
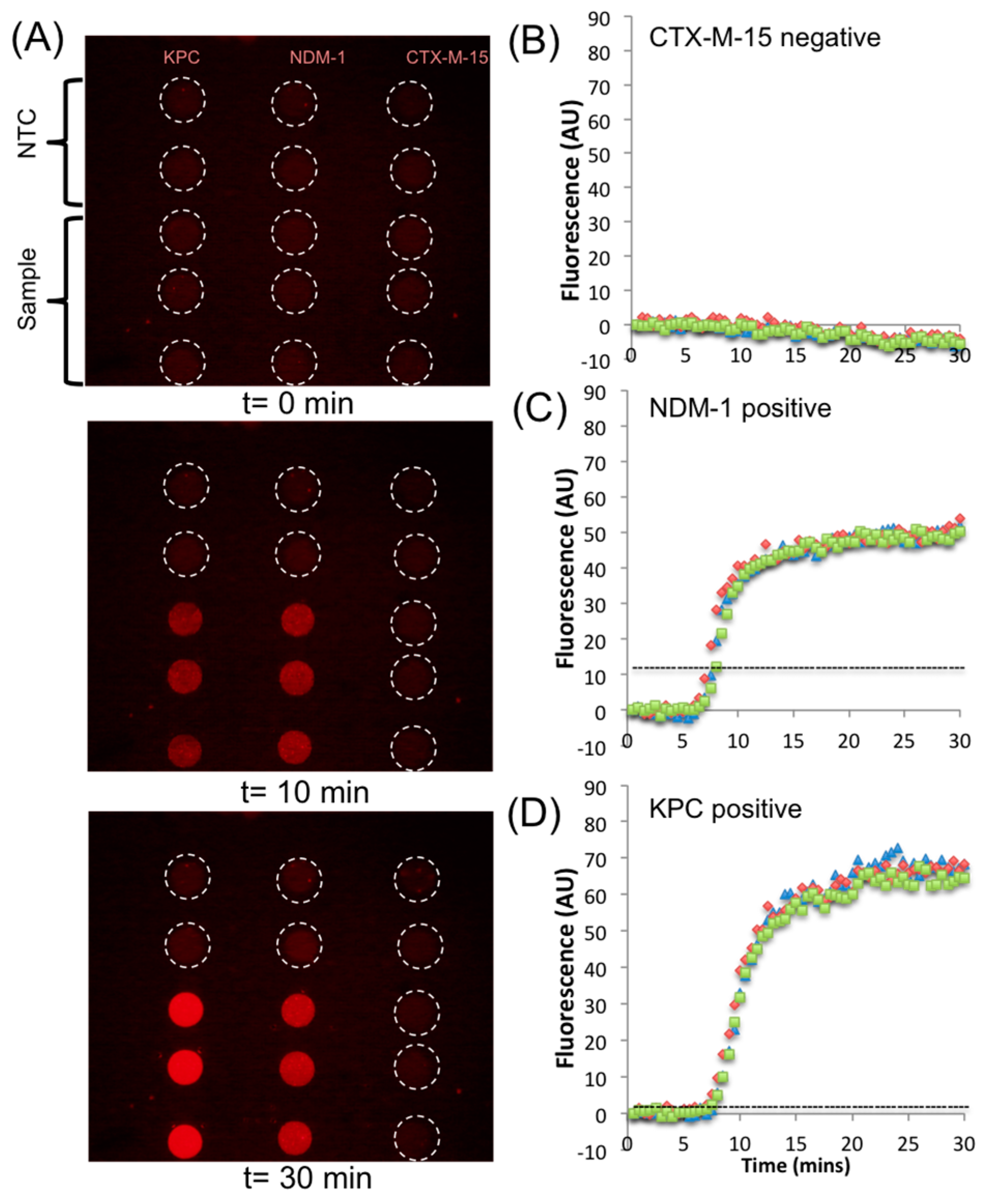
3. Results and Discussion
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Blair, J.M.A.; Webber, M.A.; Baylay, A.J.; Ogbolu, D.O.; Piddock, L.J.V. Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 2014, 13, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Pitout, J.D.; Laupland, K.B. Extended-spectrum β-lactamase-producing Enterobacteriaceae: An emerging public-health concern. Lancet Infect. Dis. 2008, 8, 159–166. [Google Scholar] [CrossRef]
- Al Naiemi, N.; Duim, B.; Bart, A. A CTX-M extended-spectrum-lactamase in Pseudomonas aeruginosa and Stenotrophomonas maltophilia. J. Med. Microbiol. 2006, 55, 1607–1608. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.-H.; Hu, Z.-Q. Epidemiology and genetics of CTX-M extended-spectrum β-lactamases in Gram-negative bacteria. Crit. Rev. Microbiol. 2013, 39, 79–101. [Google Scholar] [CrossRef] [PubMed]
- Cantón, R.; González-Alba, J.M.; Galán, J.C. CTX-M Enzymes: Origin and Diffusion. Front. Microbiol. 2012, 3, 110. [Google Scholar] [CrossRef] [PubMed]
- Meletis, G. Carbapenem resistance: Overview of the problem and future perspectives. Ther. Adv. Infect. Dis. 2016, 3, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Queenan, A.M.; Bush, K. Carbapenemases: The versatile beta-lactamases. Clin. Microbiol. Rev. 2007, 20, 440–458. [Google Scholar] [CrossRef] [PubMed]
- Health Protection Scotland and Information Servics Division. Scottish Antimicrobial Use and Resistance in Humans in 2015; Health Protection Scotland: Glasgow, UK, 2015. [Google Scholar]
- Nordmann, P.; Naas, T.; Poirel, L. Global spread of Carbapenemase-producing Enterobacteriaceae. Emerg. Infect. Dis. 2011, 17, 1791–1798. [Google Scholar] [CrossRef] [PubMed]
- Siegel, J.D.; Rhinehart, E.; Jackson, M.; Chiarello, L. Management of Multidrug-Resistant Organisms in Healthcare Settings. Available online: https://www.cdc.gov/hicpac/pdf/MDRO/MDROGuideline2006.pdf (accessed on 7 March 2017).
- O’Neill, J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations. The Review on Antimicrobial Resistance. Available online: https://amr-review.org/sites/default/files/160518_Final%20paper_with%20cover.pdf (accessed on 7 March 2017).
- Boedicker, J.Q.; Li, L.; Kline, T.R.; Ismagilov, R.F. Detecting bacteria and determining their susceptibility to antibiotics by stochastic confinement in nanoliter droplets using plug-based microfluidics. Lab Chip 2008, 8, 1265–1272. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Yoo, J.; Lee, M.; Kim, E.-G.; Lee, J.S.; Lee, S.; Joo, S.; Song, S.H.; Kim, E.-C.; Lee, H.C.; et al. A rapid antimicrobial susceptibility test based on single-cell morphological analysis. Sci. Transl. Med. 2014, 6, 267ra174. [Google Scholar] [CrossRef] [PubMed]
- Cira, N.J.; Ho, J.Y.; Dueck, M.E.; Weibel, D.B. A self-loading microfluidic device for determining the minimum inhibitory concentration of antibiotics. Lab Chip 2012, 12, 1052–1059. [Google Scholar] [CrossRef] [PubMed]
- Schoepp, N.G.; Khorosheva, E.M.; Schlappi, T.S.; Curtis, M.S.; Humphries, R.M.; Hindler, J.A.; Ismagilov, R.F. Digital Quantification of DNA Replication and Chromosome Segregation Enables Determination of Antimicrobial Susceptibility after only 15 Minutes of Antibiotic Exposure. Angew. Chem. Int. Ed. 2016, 55, 9557–9561. [Google Scholar] [CrossRef] [PubMed]
- Kelley, S.O. New Technologies for Rapid Bacterial Identification and Antibiotic Resistance Profiling. J. Lab. Autom. 2016. [Google Scholar] [CrossRef] [PubMed]
- Pulido, M.R.; Garcia-Quintanilla, M.; Martin-Pena, R.; Cisneros, J.M.; McConnell, M.J. Progress on the development of rapid methods for antimicrobial susceptibility testing. J. Antimicrob. Chemother. 2013, 68, 2710–2717. [Google Scholar] [CrossRef] [PubMed]
- Fluit, A.C.; Visser, M.R.; Schmitz, F.J. Molecular detection of antimicrobial resistance. Clin. Microbiol. Rev. 2001, 14, 836–871. [Google Scholar] [CrossRef] [PubMed]
- Strommenger, B.; Kettlitz, C.; Werner, G.; Witte, W. Multiplex PCR assay for simultaneous detection of nine clinically relevant antibiotic resistance genes in Staphylococcus aureus. J. Clin. Microbiol. 2003, 41, 4089–4094. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, D.; Bhanumathi, R.; Singh, D.V. Multiplex PCR for detection of antibiotic resistance genes and the SXT element: Application in the characterization of Vibrio cholerae. J. Med. Microbiol. 2007, 56, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Xu, B.; Yang, Y.; Liu, D.; Yang, M.; Wang, J.; Shen, H.; Zhou, X.; Ma, X. A high throughput multiplex PCR assay for simultaneous detection of seven aminoglycoside-resistance genes in Enterobacteriaceae. BMC Microbiol. 2013, 13, 58. [Google Scholar] [CrossRef] [PubMed]
- Daher, R.K.; Stewart, G.; Boissinot, M.; Bergeron, M.G. Isothermal Recombinase Polymerase Amplification Assay Applied to the Detection of Group B Streptococci in Vaginal/Anal Samples. Clin. Chem. 2014, 60, 660–666. [Google Scholar] [CrossRef] [PubMed]
- Piepenburg, O.; Williams, C.H.; Stemple, D.L.; Armes, N.A. DNA Detection Using Recombination Proteins. PLoS Biol. 2006, 4, e204. [Google Scholar] [CrossRef] [PubMed]
- Czilwik, G.; Messinger, T.; Strohmeier, O.; Wadle, S.; von Stetten, F.; Paust, N.; Roth, G.; Zengerle, R.; Saarinen, P.; Niittymäki, J.; et al. Rapid and fully automated bacterial pathogen detection on a centrifugal-microfluidic LabDisk using highly sensitive nested PCR with integrated sample preparation. Lab Chip 2015, 15, 3749–3759. [Google Scholar] [CrossRef] [PubMed]
- Choi, G.; Jung, J.H.; Park, B.H.; Oh, S.J.; Seo, J.H.; Choi, J.S.; Seo, T.S. A centrifugal direct recombinase polymerase amplification (direct-RPA) microdevice for multiplex and real-time identification of food poisoning bacteria. Lab Chip 2016, 16, 2309–2316. [Google Scholar] [CrossRef] [PubMed]
- Kersting, S.; Rausch, V.; Bier, F.F.; von Nickisch-Rosenegk, M. Multiplex isothermal solid-phase recombinase polymerase amplification for the specific and fast DNA-based detection of three bacterial pathogens. Mikrochim. Acta 2014, 181, 1715–1723. [Google Scholar] [CrossRef] [PubMed]
- Crannell, Z.; Castellanos-Gonzalez, A.; Nair, G.; Mejia, R.; White, A.C.; Richards-Kortum, R. Multiplexed Recombinase Polymerase Amplification Assay to Detect Intestinal Protozoa. Anal. Chem. 2016, 88, 1610–1616. [Google Scholar] [CrossRef] [PubMed]
- Renner, L.D.; Zan, J.; Hu, L.I.; Martinez, M.; Resto, P.J.; Siegel, A.C.; Torres, C.; Hall, S.B.; Slezak, T.R.; Nguyen, T.H.; et al. Detection of ESKAPE Bacterial Pathogens at the Point of Care Using Isothermal DNA-Based Assays in a Portable Degas-Actuated Microfluidic Diagnostic Assay Platform. Appl. Environ. Microbiol. 2017, 83. AEM.02449-16. [Google Scholar] [CrossRef] [PubMed]
- Mugele, F.; Baret, J.-C. Electrowetting: From basics to applications. J. Phys. Condens. Matter 2005, 17, 705–774. [Google Scholar] [CrossRef]
- Choi, K.; Ng, A.H.C.; Fobel, R.; Wheeler, A.R. Digital Microfluidics. Annu. Rev. Anal. Chem. 2012, 5, 413–440. [Google Scholar] [CrossRef] [PubMed]
- Samiei, E.; Tabrizian, M.; Hoorfar, M. A review of digital microfluidics as portable platforms for lab-on a-chip applications. Lab Chip 2016, 16, 2376–2396. [Google Scholar] [CrossRef] [PubMed]
- Hua, Z.; Rouse, J.L.; Eckhardt, A.E.; Srinivasan, V.; Pamula, V.K.; Schell, W.A.; Benton, J.L.; Mitchell, T.G.; Pollack, M.G. Multiplexed real-time polymerase chain reaction on a digital microfluidic platform. Anal. Chem. 2010, 82, 2310–2316. [Google Scholar] [CrossRef] [PubMed]
- Prakash, R.; Pabbaraju, K.; Wong, S.; Wong, A.; Tellier, R.; Kaler, K.V. Multiplex, Quantitative, Reverse Transcription PCR Detection of Influenza Viruses Using Droplet Microfluidic Technology. Micromachines 2014, 6, 63–79. [Google Scholar] [CrossRef]
- Hadwen, B.; Broder, G.R.; Morganti, D.; Jacobs, A.; Brown, C.; Hector, J.R.; Kubota, Y.; Morgan, H. Programmable large area digital microfluidic array with integrated droplet sensing for bioassays. Lab Chip 2012, 12, 3305–3313. [Google Scholar] [CrossRef] [PubMed]
- Kalsi, S.; Valiadi, M.; Tsaloglou, M.N.; Parry-Jones, L.; Jacobs, A.; Watson, R.; Turner, C.; Amos, R.; Hadwen, B.; Buse, J.; et al. Rapid and sensitive detection of antibiotic resistance on a programmable digital microfluidic platform. Lab Chip 2015, 15, 3065–3075. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalsi, S.; Sellars, S.L.; Turner, C.; Sutton, J.M.; Morgan, H. A Programmable Digital Microfluidic Assay for the Simultaneous Detection of Multiple Anti-Microbial Resistance Genes. Micromachines 2017, 8, 111. https://doi.org/10.3390/mi8040111
Kalsi S, Sellars SL, Turner C, Sutton JM, Morgan H. A Programmable Digital Microfluidic Assay for the Simultaneous Detection of Multiple Anti-Microbial Resistance Genes. Micromachines. 2017; 8(4):111. https://doi.org/10.3390/mi8040111
Chicago/Turabian StyleKalsi, Sumit, Samuel L. Sellars, Carrie Turner, J. Mark Sutton, and Hywel Morgan. 2017. "A Programmable Digital Microfluidic Assay for the Simultaneous Detection of Multiple Anti-Microbial Resistance Genes" Micromachines 8, no. 4: 111. https://doi.org/10.3390/mi8040111
APA StyleKalsi, S., Sellars, S. L., Turner, C., Sutton, J. M., & Morgan, H. (2017). A Programmable Digital Microfluidic Assay for the Simultaneous Detection of Multiple Anti-Microbial Resistance Genes. Micromachines, 8(4), 111. https://doi.org/10.3390/mi8040111