
Rapid Detection of Salmonella enterica in Food Using a Compact Disc-Shaped Device

Abstract
:
1. Introduction
2. Experimental Section
2.1. Bacterial Cells
2.2. PCR Reagents and Real-Time PCR
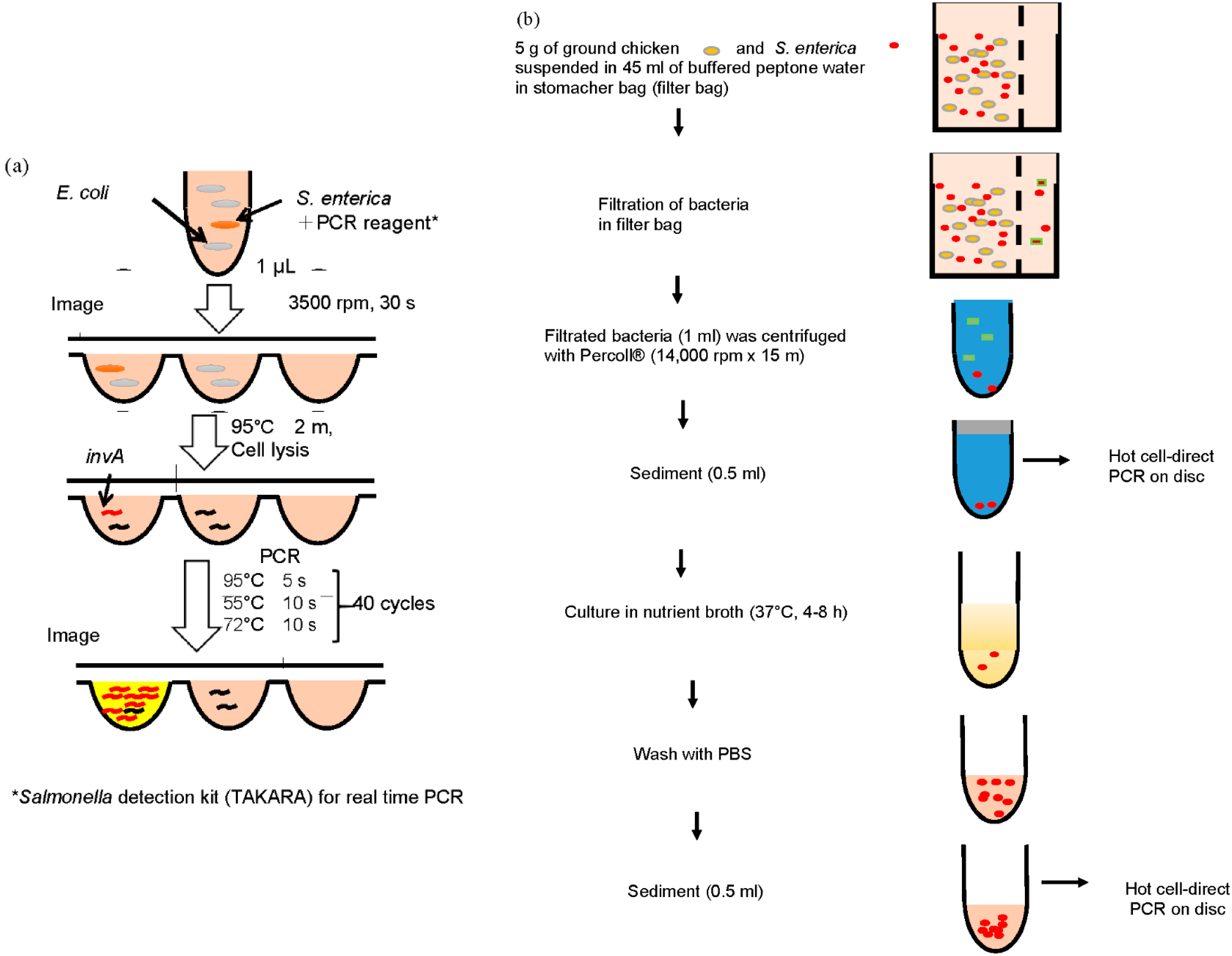
2.3. Fabrication of CD-Shaped Device
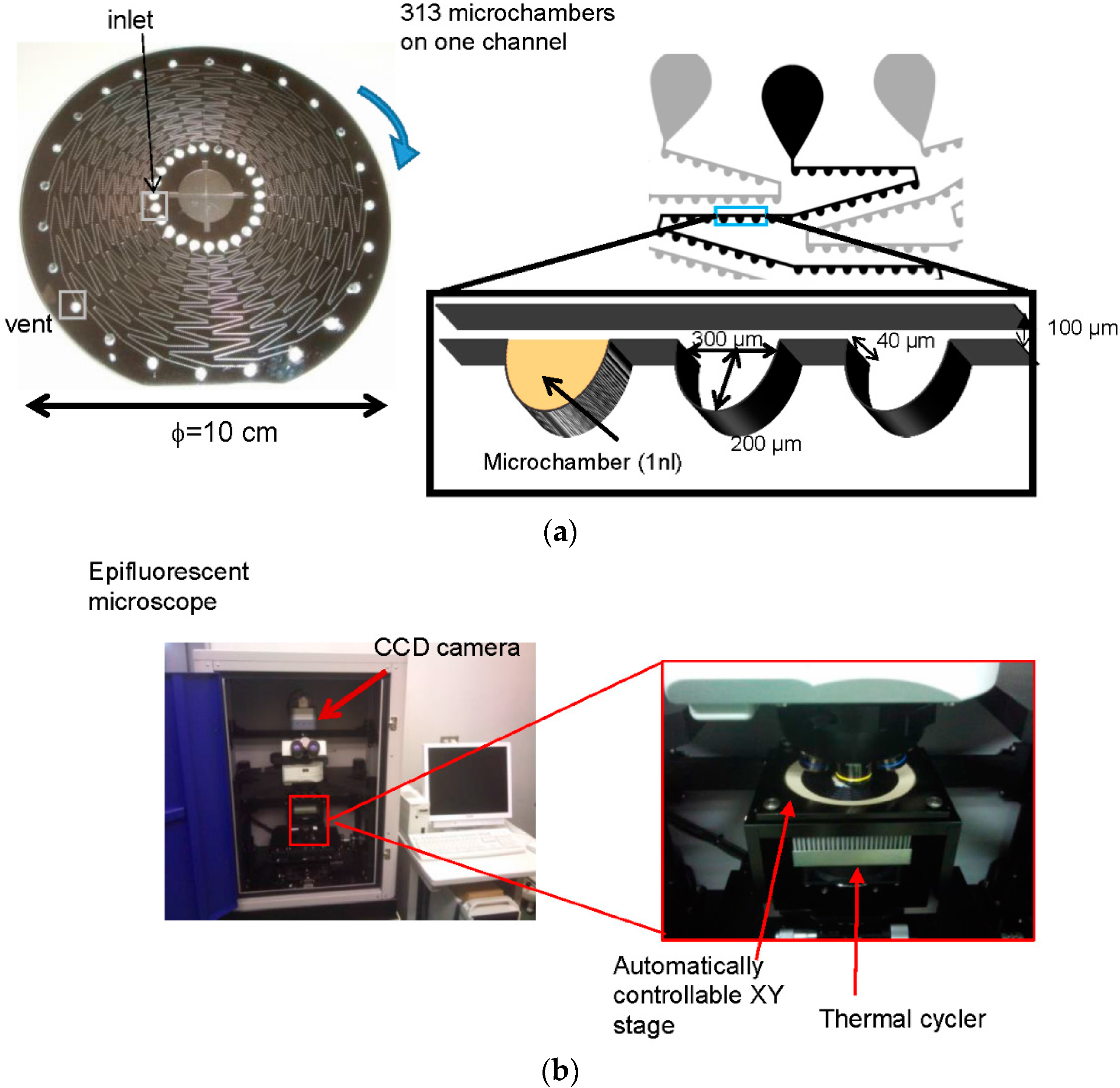
2.4. Detection of S. Enterica Using a CD Shaped Device
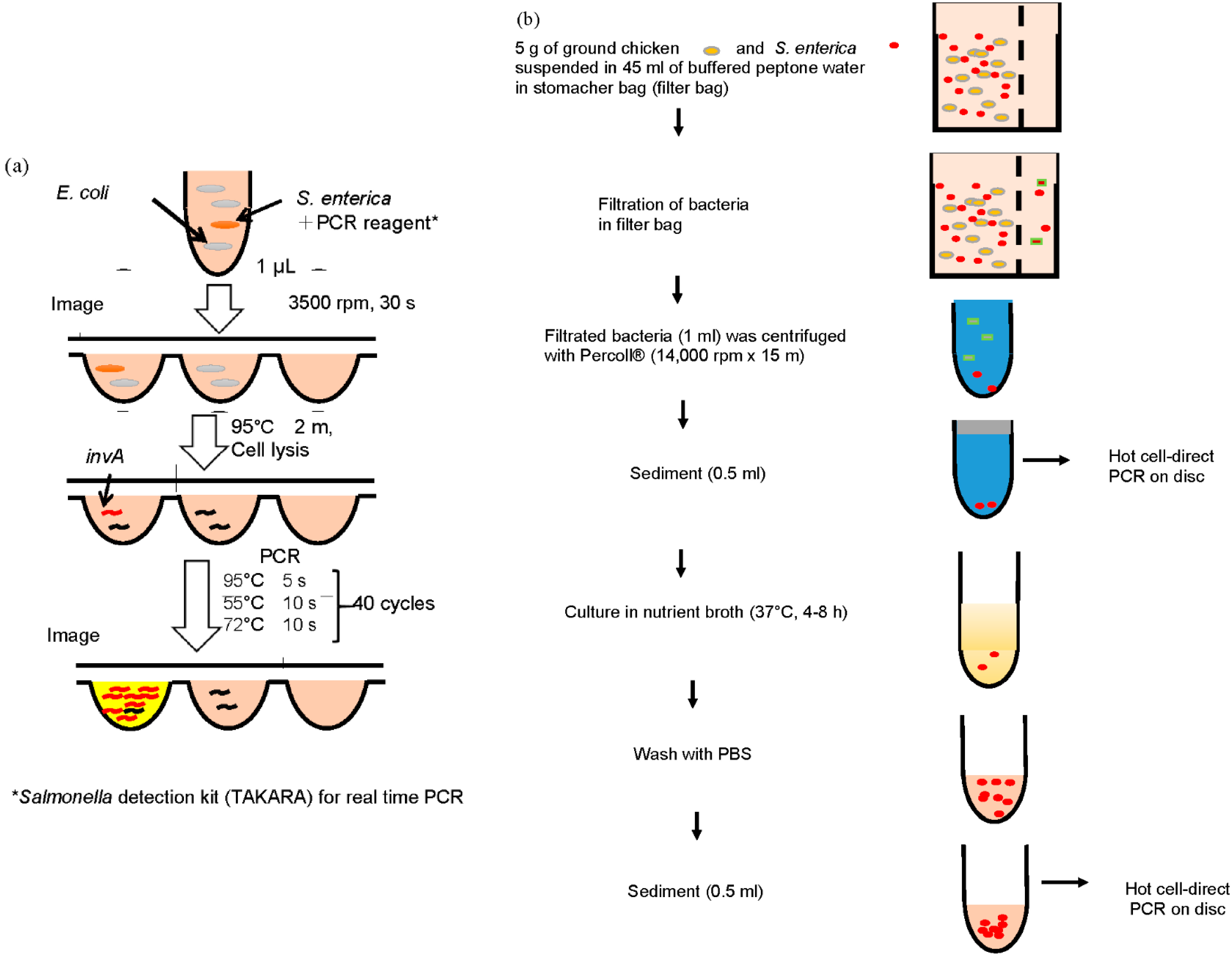
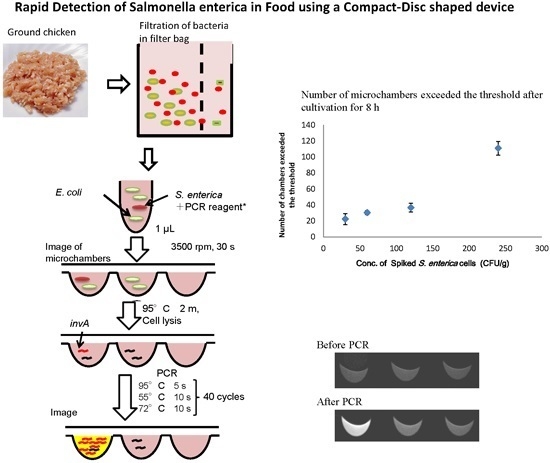
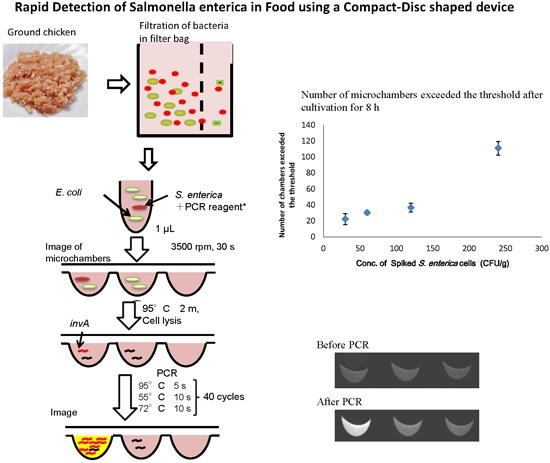
2.5. Separation of S. Enterica from Chicken
3. Results and Discussion
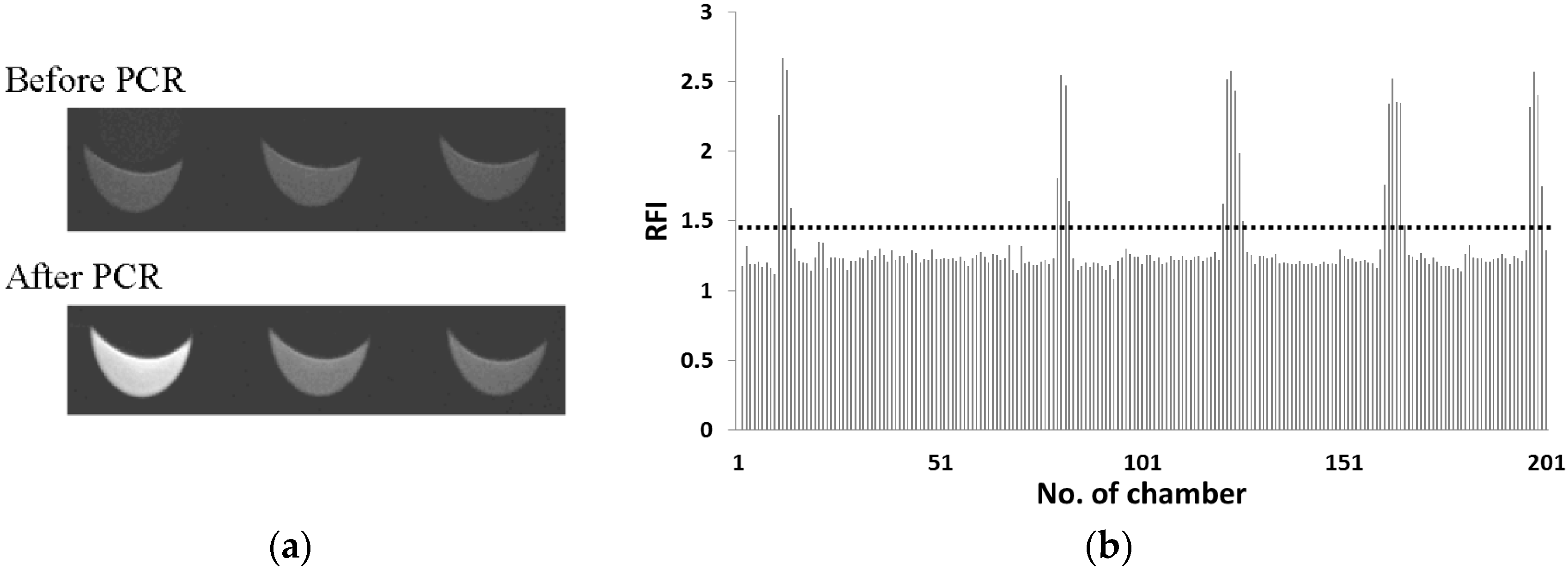
3.1. Detection of S. Enterica in Reaction Mixture with E. coli on CD-Shaped Device


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Concentration of S. enterica (cells·μL−1) | Number of Microchambers with RFI > 1.4 |
|---|---|
| 50 | 19 |
| 100 | 36 |
| 400 | 92 |
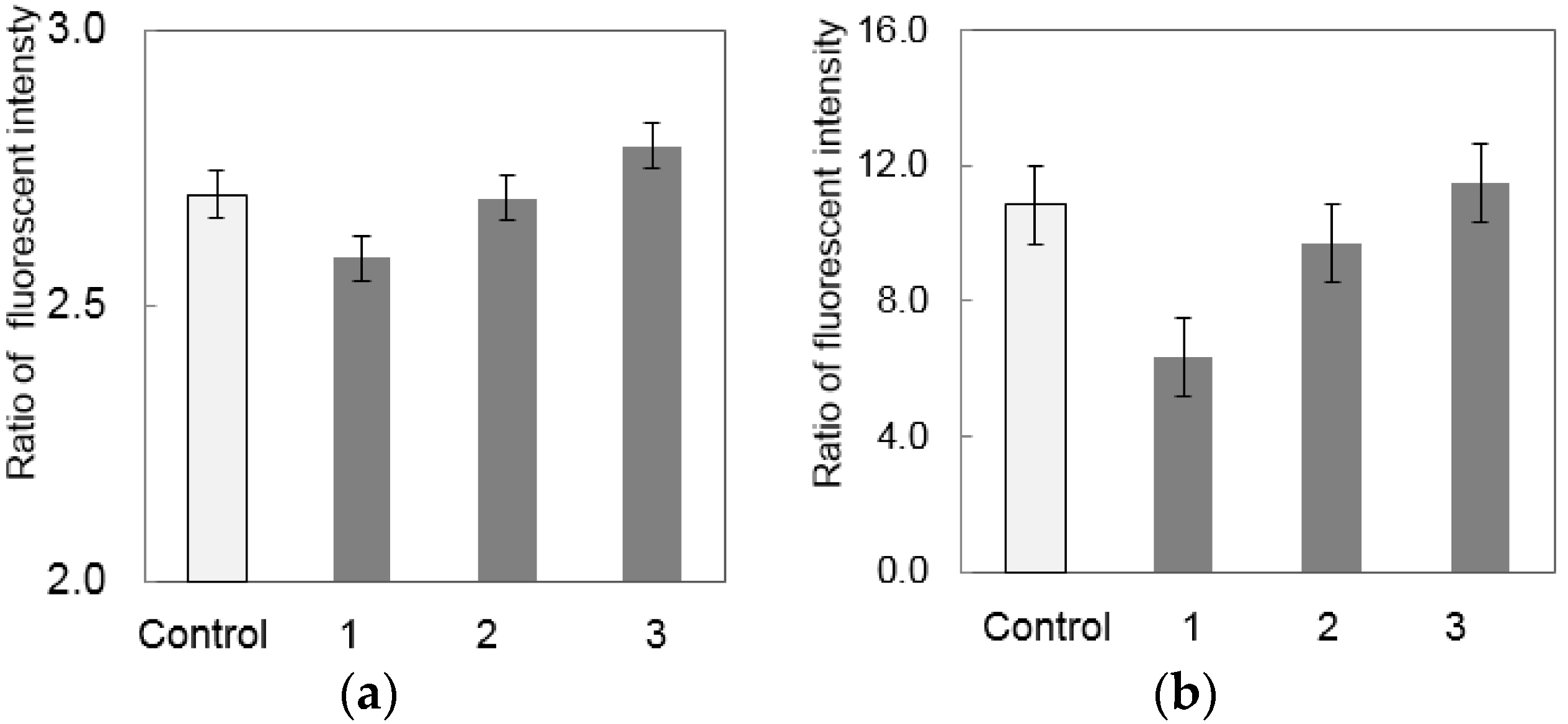
3.2. Separation of S. enterica from Chicken
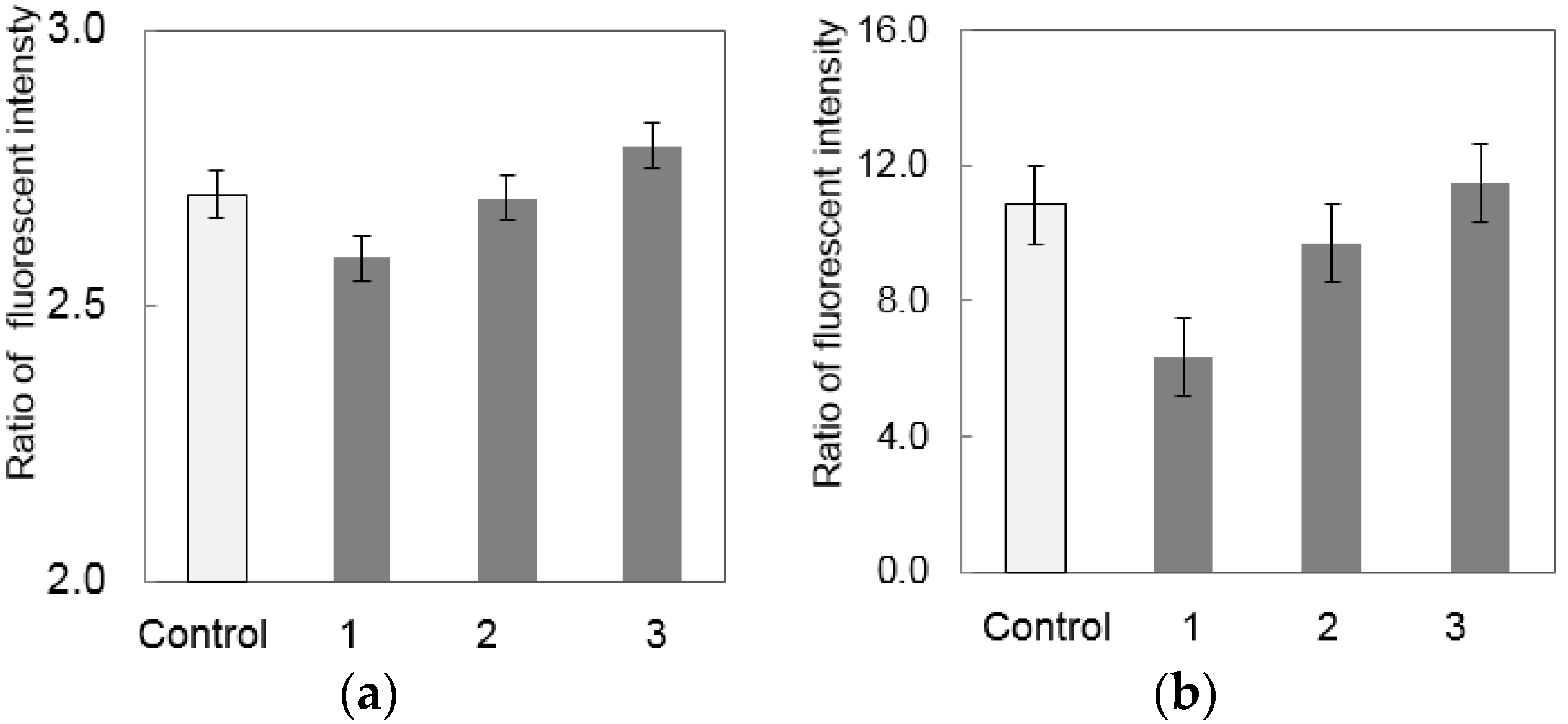
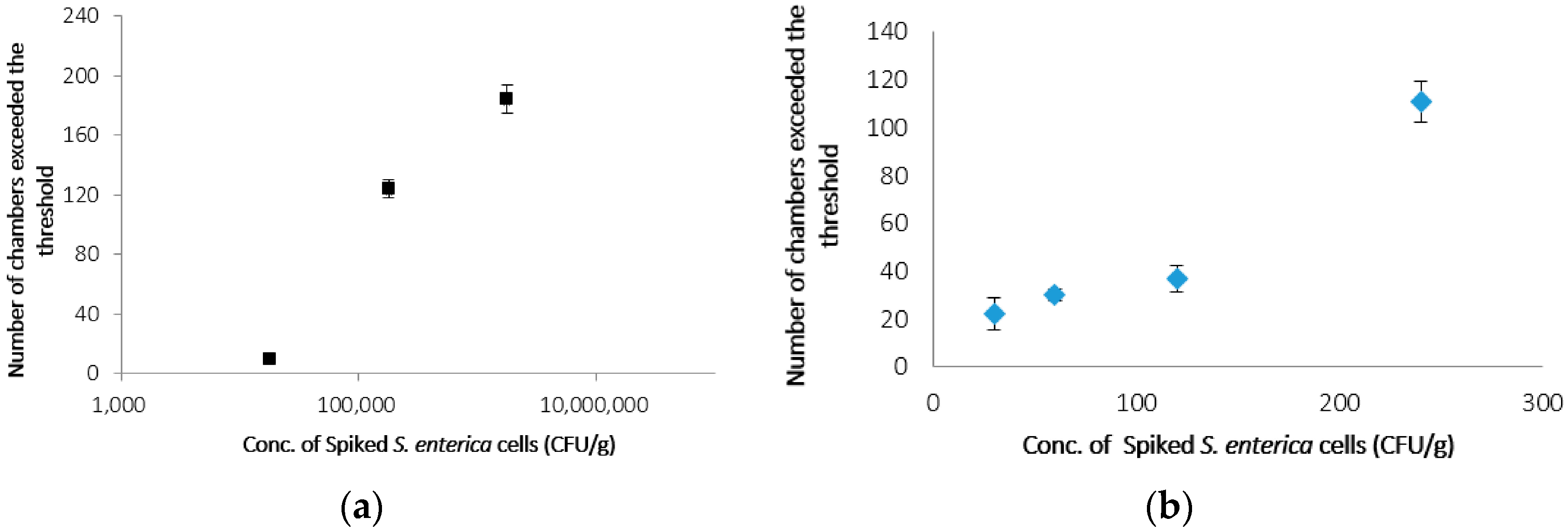
3.3. Detection of S. enterica from Chicken on CD-Shaped Device
| Concentration of S. enterica (CFUs·g−1) | Number of Microchambers Exceeded the Threshold |
|---|---|
| 4.6 × 104 | 0 |
| 4.6 × 105 | 6 |
| 4.6 × 106 | 24 |
| 4.6 × 107 | 170 |

4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hoelzer, K.; Switt, A.I.M.; Wiedmann, M. Animal contact as a source of human non-typhoidal salmonellosis. Vet. Res. 2011, 42, 34. [Google Scholar] [CrossRef] [PubMed]
- Japan Environmental Health Bureau, Ministry of Health and Welfare (Ed.) Standard Methods of Analysis in Food Safety Regulation—Biology; Japan Food Hygiene Association: Tokyo, Japan, 1990.
- Salazar, J.K.; Wang, Y.; Yu, S.; Wang, H.; Zhang, W. Polymerase chain reaction-based serotyping of pathogenic bacteria in food. J. Microbiol. Methods 2015, 110, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Iida, K.; Abe, A.; Matsui, H.; Wakayama, S.; Kawahara, K. Rapid and sensitive method for detection of Salmonella strains using a combination of polymerase chain reaction and reverse dot-blot hybridization. FEMS Microbiol. Lett. 1993, 114, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Pahn, K.; de Grandis, S.A.; Clarke, R.C.; McEwen, S.A.; Galan, J.E.; Ginocchio, C.; Curtiss, R.; Gyles, C.L. Amplification of an invA gene sequence of Salmonella typhimurium by polymerase chain reaction as specific method of detection of Salmonella. Mol. Cell. Probes 1992, 6, 271–279. [Google Scholar]
- Way, J.S.; Josephson, K.L.; Pillai, S.D.; Abbaszadegan, M.; Gerba, C.P.; Pepper, I.L. Specific detection of Salmonella spp. by multiplex polymerase chain reaction. Appl. Environ. Microbiol. 1993, 59, 1473–1479. [Google Scholar] [PubMed]
- Kobayashi, K.; Taguchi, M.; Seto, K. Polymerase chain reaction (PCR) method for the detection of Salmonella by using Chelex resin after enrichment culture. J. Jpn. Assoc. Infect. Dis. 1994, 68, 1203–1210. [Google Scholar] [CrossRef]
- Cohen, H.J.; Mechanda, S.M.; Lin, W. PCR amplification of the fimA gene sequence of Salmonella typhimurium, a specific method for detection of Salmonella spp. Appl. Environ. Microbiol. 1996, 62, 4303–4308. [Google Scholar] [PubMed]
- Fachmann, M.S.R.; Josefsen, M.H.; Hoorfar, J.; Nielsen, M.T.; Lofstrom, C. Cost-effective optimization of real-time PCR based detection of Campylobacter and Salmonella with inhibitor tolerant DNA polymerases. J. Appl. Microbiol. 2015, 119, 1391–1402. [Google Scholar] [CrossRef] [PubMed]
- Strohmeier, O.; Keller, M.; Schwemmer, F.; Zehnle, S.; Mark, D.; von Stettn, F.; Zengerle, R.; Paust, N. Centrifugal microfluidic platforms: Advanced unit operations and applications. Chem. Soc. Rev. 2015, 44, 6187–6229. [Google Scholar] [CrossRef] [PubMed]
- Sundberg, S.O.; Wittwer, C.T.; Gao, C.; Gale, B.K. Spinning Disk Platform for Microfluidic Digital Polymerase Chain Reaction. Anal. Chem. 2010, 82, 1546–1550. [Google Scholar] [CrossRef] [PubMed]
- Schuler, F.; Trotter, M.; Geltman, M.; Schwemmer, F.; Wadle, S.; Domínguez-Garrido, E.; López, M.; Cervera-Acedo, C.; Santibáñez, P.; von Stetten, F.; et al. Digital droplet PCR on disk. Lab Chip 2016, 16, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Furutani, S.; Nagai, H.; Kubo, I. Single cell isolation on a centrifugal flow disk with integrated tandem microchambers. Sens. Lett. 2008, 6, 961–965. [Google Scholar] [CrossRef]
- Furutani, S.; Nagai, H.; Takamura, Y.; Kubo, I. Compact disk (CD)-shaped device for single cell isolation and PCR of the specific gene in the isolated cell. Anal. Bioanal. Chem. 2010, 398, 2997–3004. [Google Scholar] [CrossRef] [PubMed]
- Furutani, S.; Shozen, N.; Nagai, H.; Aoyama, Y.; Kubo, I. Development of a detection system for expressed genes in isolated single Jurkat cells. Sens. Mater. 2014, 26, 623–635. [Google Scholar]
- JFRL News. Salmonella Food Poisoning, the Importance and Difficulty of the Countermeasures. Available online: http://www.jfrl.or.jp/jfrlnews/files/news_no02.pdf (accessed on 24 December 2015). (In Japanese)
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Furutani, S.; Kajiya, M.; Aramaki, N.; Kubo, I. Rapid Detection of Salmonella enterica in Food Using a Compact Disc-Shaped Device. Micromachines 2016, 7, 10. https://doi.org/10.3390/mi7010010
Furutani S, Kajiya M, Aramaki N, Kubo I. Rapid Detection of Salmonella enterica in Food Using a Compact Disc-Shaped Device. Micromachines. 2016; 7(1):10. https://doi.org/10.3390/mi7010010
Chicago/Turabian StyleFurutani, Shunsuke, Mitsutoshi Kajiya, Narumi Aramaki, and Izumi Kubo. 2016. "Rapid Detection of Salmonella enterica in Food Using a Compact Disc-Shaped Device" Micromachines 7, no. 1: 10. https://doi.org/10.3390/mi7010010
APA StyleFurutani, S., Kajiya, M., Aramaki, N., & Kubo, I. (2016). Rapid Detection of Salmonella enterica in Food Using a Compact Disc-Shaped Device. Micromachines, 7(1), 10. https://doi.org/10.3390/mi7010010