
Spin Polarization Properties of Two Dimensional GaP3 Induced by 3d Transition-Metal Doping

Abstract
:1. Introduction
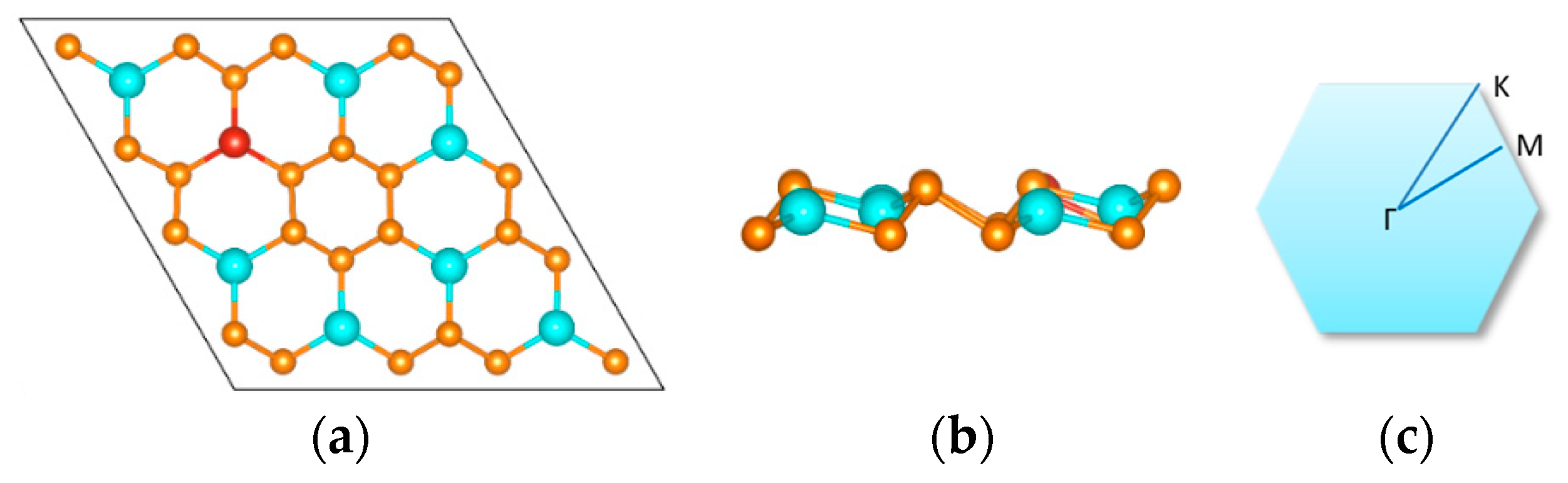
2. Materials and Methods
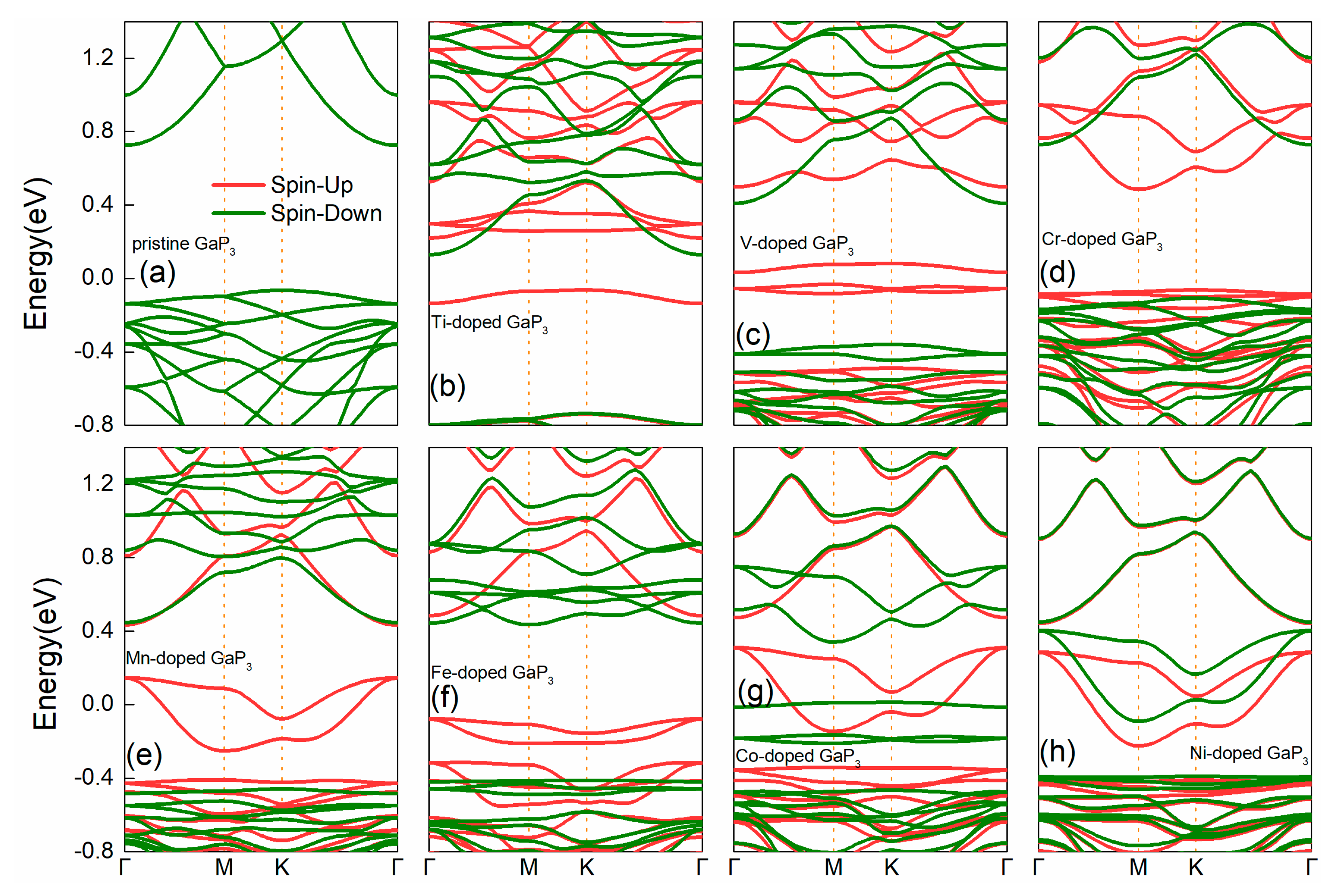
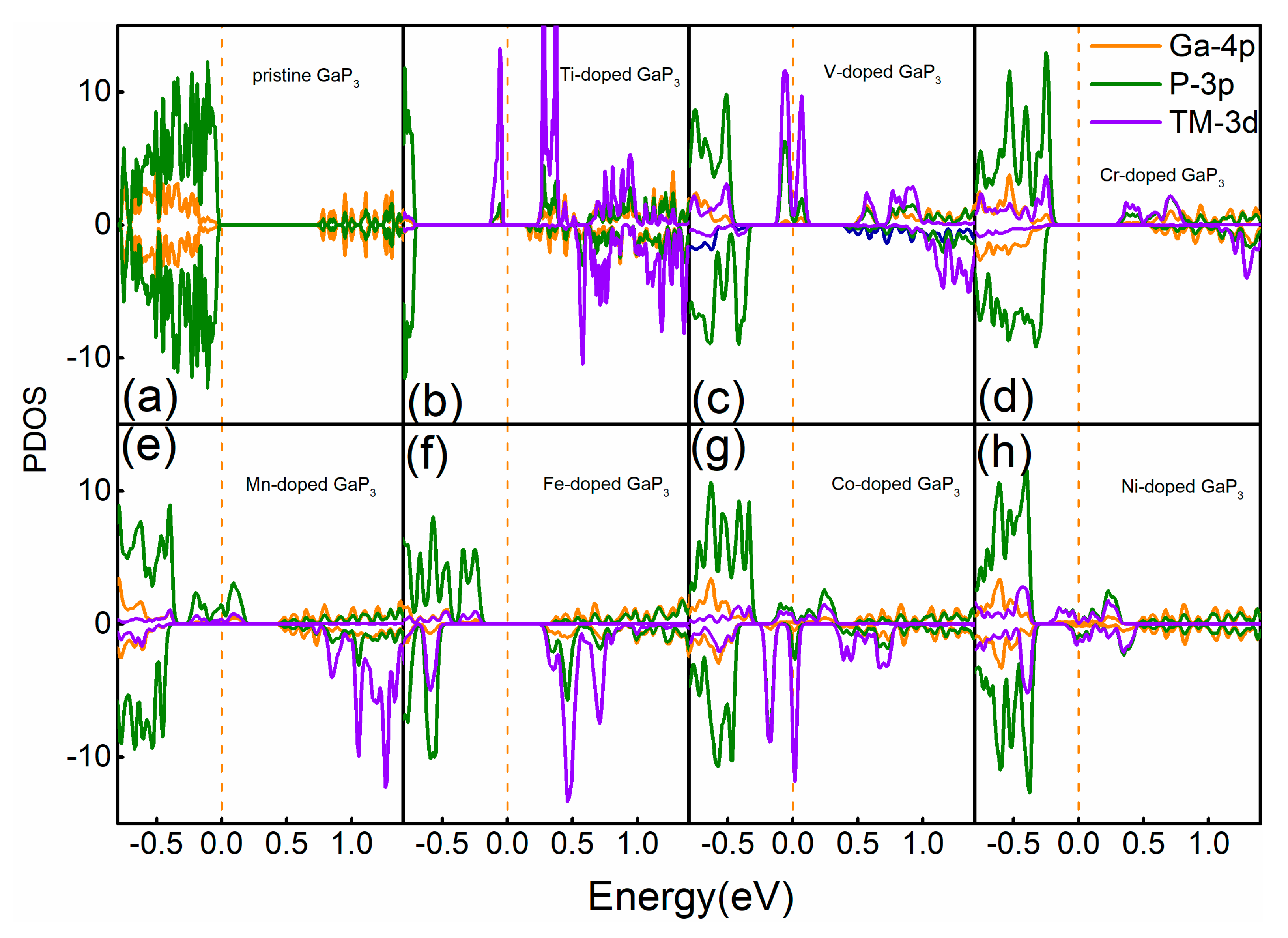
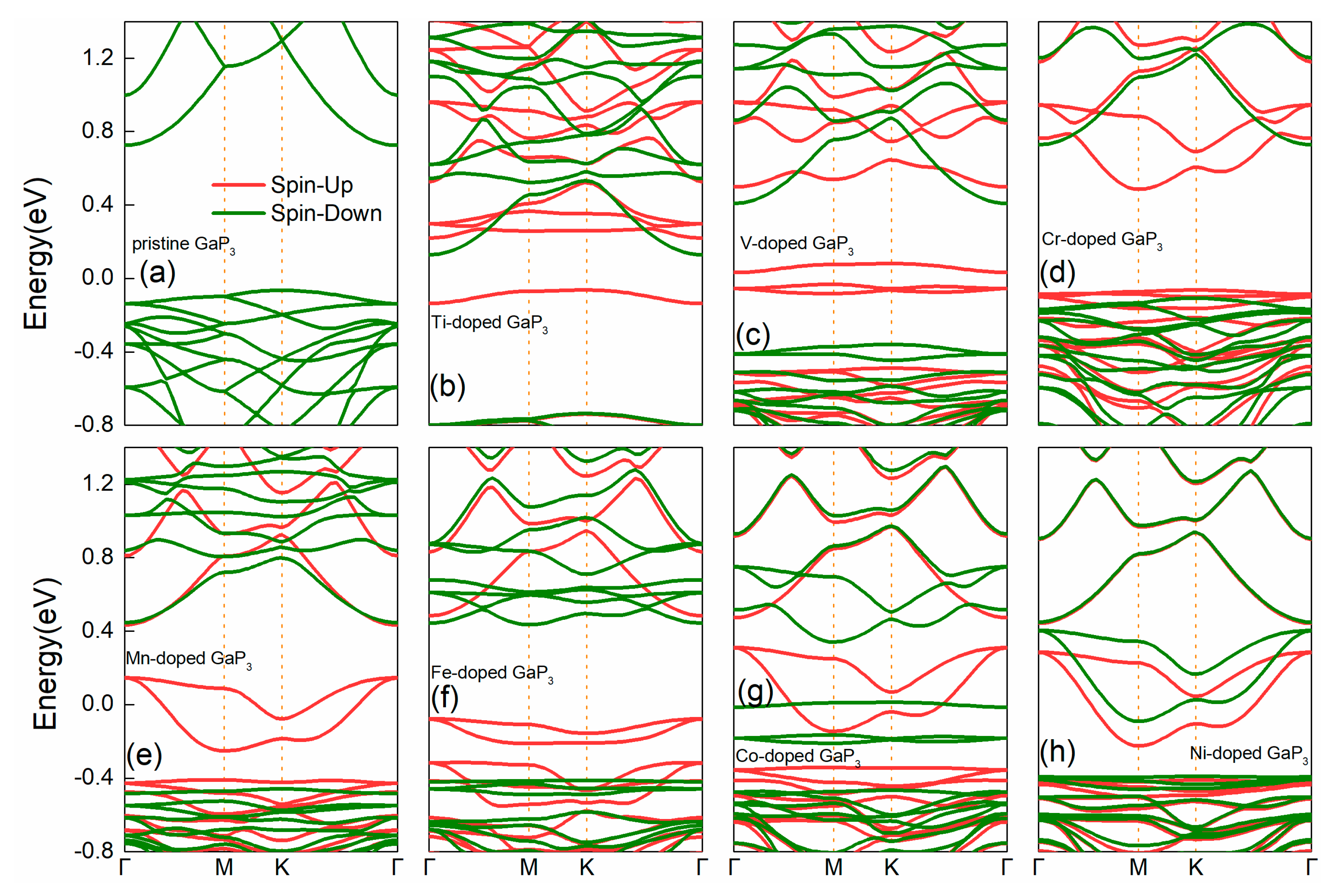
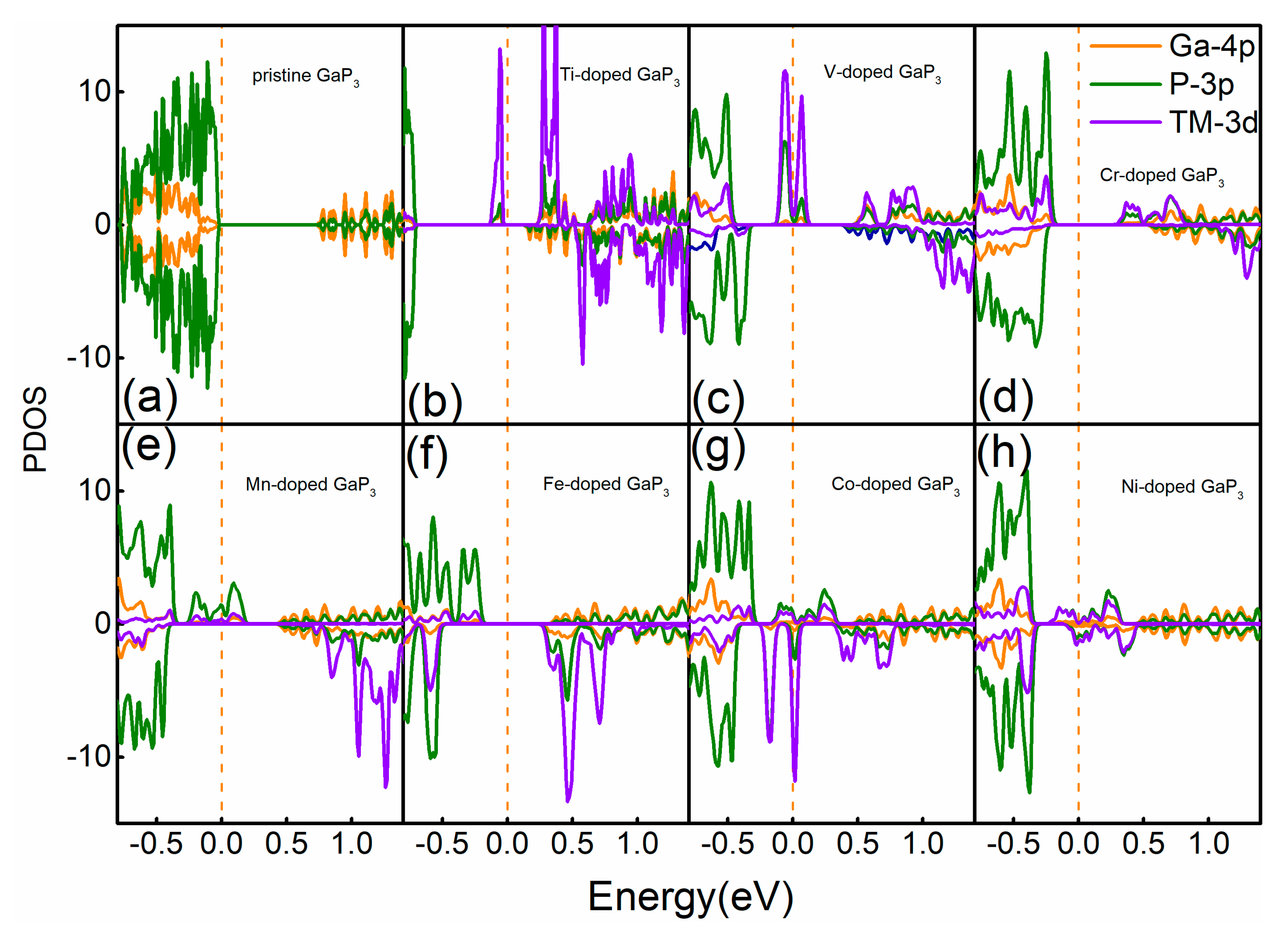
3. Results
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric field effect in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- AGeim, A.K.; Novoselov, K. The rise of graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar]
- Neto, A.H.C.; Guinea, F.; Peres, N.M.R.; Novoselov, K.S.; Geim, A.K. The electronic properties of graphene. Rev. Mod. Phys. 2009, 81, 109. [Google Scholar] [CrossRef] [Green Version]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.; Katsnelson, M.I.; Grigorieva, I.V.; Dubonos, S.V.; Firsov, A.A. Two-dimensional gas of massless Dirac fermions in graphene. Nature 2005, 438, 197–200. [Google Scholar] [CrossRef] [PubMed]
- Stankovich, S.; Dikin, D.A.; Dommett, G.H.B.; Kohlhaas, K.M.; Zimney, E.J.; Stach, E.A.; Piner, R.D.; Nguyen, S.; Ruoff, R.S. Graphene-based composite materials. Nat. Cell Biol. 2006, 442, 282–286. [Google Scholar] [CrossRef]
- Zhu, H.; Zhao, L.; Liu, J.; Xu, S.; Cai, W.; Jiang, S.; Zheng, L.; Su, L.; Xu, J. Monolayer graphene saturable absorber with sandwich structure for ultrafast solid-state laser. Opt. Eng. 2015, 55, 081304. [Google Scholar] [CrossRef]
- Mas-Ballesté, R.; Gómez-Navarro, C.; Gomez-Herrero, J.; Zamora, F. 2D materials: To graphene and beyond. Nanoscale 2010, 3, 20–30. [Google Scholar] [CrossRef]
- Liu, L.; Feng, Y.P.; Shen, Z.X. Structural and electronic properties of h-BN. Phys. Rev. B 2003, 68, 104102. [Google Scholar] [CrossRef]
- Golberg, D.; Bando, Y.; Huang, Y.; Terao, T.; Mitome, M.; Tang, C.; Zhi, C. Boron Nitride Nanotubes and Nanosheets. ACS Nano 2010, 4, 2979–2993. [Google Scholar] [CrossRef]
- Warner, J.H.; Rümmeli, M.H.; Bachmatiuk, A.; Büchner, B. Atomic Resolution Imaging and Topography of Boron Nitride Sheets Produced by Chemical Exfoliation. ACS Nano 2010, 4, 1299–1304. [Google Scholar] [CrossRef]
- Manzeli, S.; Ovchinnikov, D.; Pasquier, D.; Yazyev, O.; Kis, A. 2D transition metal dichalcogenides. Nat. Rev. Mater. 2017, 2, 17033. [Google Scholar] [CrossRef]
- Wang, Q.-H.; Zadeh, K.K.; Kis, A.; Coleman, J.N.; Strano, M.S. Electronics and optoelectronics of two-dimensional transition metal dichalcogenides. Nat. Nanotechol. 2012, 7, 699–712. [Google Scholar] [CrossRef] [PubMed]
- Jariwala, D.; Sangwan, V.K.; Lauhon, L.; Marks, T.J.; Hersam, M.C. Emerging Device Applications for Semiconducting Two-Dimensional Transition Metal Dichalcogenides. ACS Nano 2014, 8, 1102–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Yao, W.; Xiao, D.; Heinz, T.F. Spin and pseudospins in layered transition metal dichalcogenides. Nat. Phys. 2014, 10, 343–350. [Google Scholar] [CrossRef]
- Qian, X.; Liu, J.; Fu, L.; Liu, J. Quantum spin Hall effect in two-dimensional transition metal dichalcogenides. Science 2014, 346, 1344–1347. [Google Scholar] [CrossRef] [Green Version]
- Kou, L.; Chen, C.; Smith, S.C. Phosphorene: Fabrication, Properties, and Applications. J. Phys. Chem. Lett. 2015, 6, 2794–2805. [Google Scholar] [CrossRef] [Green Version]
- Carvalho, A.; Wang, M.; Zhu, X.; Rodin, A.S.; Su, H.; Neto, A.H.C. Phosphorene: From theory to applications. Nat. Rev. Mater. 2016, 1, 16061. [Google Scholar] [CrossRef]
- Liu, H.; Neal, A.; Zhu, Z.; Luo, Z.; Xu, X.; Tománek, D.; Ye, P.D. Phosphorene: An Unexplored 2D Semiconductor with a High Hole Mobility. ACS Nano 2014, 8, 4033–4041. [Google Scholar] [CrossRef] [Green Version]
- Xiao, P.; Sk, M.A.; Thia, L.; Ge, X.; Lim, R.J.; Wang, J.-Y.; Lim, K.H.; Wang, X. Molybdenum phosphide as an efficient electrocatalyst for the hydrogen evolution reaction. Energy Environ. Sci. 2014, 7, 2624–2629. [Google Scholar] [CrossRef] [Green Version]
- Popczun, E.J.; McKone, J.R.; Read, C.G.; Biacchi, A.J.; Wiltrout, A.M.; Lewis, N.S.; Schaak, R.E. Nanostructured Nickel Phosphide as an Electrocatalyst for the Hydrogen Evolution Reaction. J. Am. Chem. Soc. 2013, 135, 9267–9270. [Google Scholar] [CrossRef]
- Shahzad, F.; Alhabeb, M.; Hatter, C.B.; Anasori, B.; Hong, S.M.; Koo, C.M.; Gogotsi, Y. Electromagnetic interference shielding with 2D transition metal carbides (MXenes). Science 2016, 353, 1137–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naguib, M.; Mochalin, V.; Barsoum, M.W.; Gogotsi, Y. 25th Anniversary Article: MXenes: A New Family of Two-Dimensional Materials. Adv. Mater. 2014, 26, 992–1005. [Google Scholar] [CrossRef] [PubMed]
- Anasori, B.; Lukatskaya, M.R.; Gogotsi, Y. 2D metal carbides and nitrides (MXenes) for energy storage. Nat. Rev. Mater. 2017, 2, 16098. [Google Scholar] [CrossRef]
- Sun, S.; Meng, F.; Wang, H.; Wang, H.; Ni, Y. Novel two-dimensional semiconductor SnP3: High stability, tunable bandgaps and high carrier mobility explored using first-principles calculations. J. Mater. Chem. A 2018, 6, 11890–11897. [Google Scholar] [CrossRef]
- Slassi, A.; Gali, S.M.; Pershin, A.; Gali, A.; Cornil, J.; Beljonne, D. Interlayer Bonding in Two-Dimensional Materials: The Special Case of SnP3 and GeP3. J. Phys. Chem. Lett. 2020, 11, 4503–4510. [Google Scholar] [CrossRef]
- Miao, N.; Xu, B.; Bristowe, N.C.; Zhou, J.; Sun, Z. Tunable Magnetism and Extraordinary Sunlight Absorbance in Indium Triphosphide Monolayer. J. Am. Chem. Soc. 2017, 139, 11125–11131. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, B.; Puri, S.; Agarwal, A.; Bhowmick, S. SnP3: A Previously Unexplored Two-Dimensional Material. J. Phys. Chem. C 2018, 122, 18185–18191. [Google Scholar] [CrossRef]
- Shojaei, F.; Kang, H.S. Partially planar BP3 with high electron mobility as a phosphorene analog. J. Mater. Chem. C 2017, 5, 11267–11274. [Google Scholar] [CrossRef]
- Lu, N.; Zhuo, Z.; Guo, H.; Wu, P.; Fa, W.; Wu, X.; Zeng, X.-C. CaP3: A New Two-Dimensional Functional Material with Desirable Bandgap and Ultrahigh Carrier Mobility. J. Phys. Chem. Lett. 2018, 9, 1728–1733. [Google Scholar] [CrossRef] [Green Version]
- Jing, Y.; Ma, Y.; Li, Y.; Heine, T. GeP3: A Small Indirect Band Gap 2D Crystal with High Carrier Mobility and Strong Interlayer Quantum Confinement. Nano Lett. 2017, 17, 1833–1838. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.-Y.; Yang, C.-L.; Wang, M.-S.; Ma, X.-G. Two-dimensional BiP3 with high carrier mobility and moderate band gap for hydrogen generation from water splitting. Appl. Surf. Sci. 2020, 501, 144263. [Google Scholar] [CrossRef]
- Yao, S.; Zhang, X.; Zhang, Z.; Chen, A.; Zhou, Z. 2D Triphosphides: SbP3 and GaP3 monolayer as promising photocatalysts for water splitting. Int. J. Hydrogen Energy 2019, 44, 5948–5954. [Google Scholar] [CrossRef]
- Sun, Z.; Yuan, K.; Chang, Z.; Bi, S.; Zhang, X.; Tang, D. Ultra-low thermal conductivity and high thermoelectric performance of two-dimensional triphosphides (InP3, GaP3, SbP3 and SnP3): A comprehensive first-principles study. Nanoscale 2020, 12, 3330–3342. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zou, X.; Crespi, V.H.; Yakobson, B.I. Intrinsic Magnetism of Grain Boundaries in Two-Dimensional Metal Dichalcogenides. ACS Nano 2013, 7, 10475–10481. [Google Scholar] [CrossRef] [Green Version]
- Guan, J.; Yu, G.; Ding, X.; Chen, W.; Shi, Z.; Huang, X.; Sun, C. The Effects of the Formation of Stone–Wales Defects on the Electronic and Magnetic Properties of Silicon Carbide Nanoribbons: A First-Principles Investigation. Chem. Phys. Chem. 2013, 14, 2841–2852. [Google Scholar] [CrossRef] [PubMed]
- Gao, G.; Ding, G.; Li, J.; Yao, K.; Wu, M.; Guangqian, D. Monolayer MXenes: Promising half-metals and spin gapless semiconductors. Nanoscale 2016, 8, 8986–8994. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Guo, M.; Zhang, G.; Li, W. Tuning the electronic and magnetic properties of porous graphene-like carbon nitride through 3d transition-metal doping. Carbon 2017, 117, 120–125. [Google Scholar] [CrossRef]
- Zhao, X.; Qiu, B.; Hu, G.; Yue, W.; Ren, J.; Yuan, X. Spin Polarization Properties of Pentagonal PdSe2 Induced by 3D Transition-Metal Doping: First-Principles Calculations. Materials 2018, 11, 2339. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Qiu, B.; Zhao, X.; Hu, G.; Yue, W.; Yuan, X.; Ren, J. Spin polarization properties of two-dimensional MoSeTe induced by transition-metal doping: First-principles calculations. Eur. Phys. J. B 2019, 92, 266. [Google Scholar] [CrossRef]
- Chen, Q.; Ouyang, Y.; Yuan, S.; Li, R.; Wang, J. Uniformly Wetting Deposition of Co Atoms on MoS2 Monolayer: A Promising Two-Dimensional Robust Half-Metallic Ferromagnet. ACS Appl. Mater. Interfaces 2014, 6, 16835–16840. [Google Scholar] [CrossRef]
- Kan, E.J.; Xiang, H.; Wu, F.; Tian, C.; Lee, C.; Yang, J.L.; Whangbo, M.-H. Prediction for room-temperature half-metallic ferromagnetism in the half-fluorinated single layers of BN and ZnO. Appl. Phys. Lett. 2010, 97, 122503. [Google Scholar] [CrossRef]
- Yuan, X.; Yang, M.; Tian, Y.; Cai, L.; Ren, J. Spin polarization properties of thiophene molecule adsorbed to the edge of zigzag graphene nanoribbon. Synth. Met. 2017, 226, 46–49. [Google Scholar] [CrossRef]
- Yuan, X.; Tian, Y.; Zhao, X.; Yue, W.; Hu, G.; Ren, J. Spin polarization properties of benzene/graphene with transition metals as dopants: First principles calculations. Appl. Surf. Sci. 2018, 439, 1158–1162. [Google Scholar] [CrossRef]
- Hashmi, A.; Hong, J. Transition Metal Doped Phosphorene: First-Principles Study. J. Phys. Chem. C 2015, 119, 9198–9204. [Google Scholar] [CrossRef]
- Barone, V.; Peralta, J. Magnetic Boron Nitride Nanoribbons with Tunable Electronic Properties. Nano Lett. 2008, 8, 2210–2214. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.; Sanville, E.; Henkelman, G. A grid-based Bader analysis algorithm without lattice bias. J. Phys. Condens. Matter 2009, 21, 084204. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Ti-Doped | V-Doped | Cr-Doped | Mn-Doped | Fe-Doped | Co-Doped | Ni-Doped | Pure | |
|---|---|---|---|---|---|---|---|---|---|
| Bond length (Å) | P-P | 2.23 | 2.24 | 2.25 | 2.22 | 2.21 | 2.25 | 2.25 | 2.23 |
| Ga-P | 2.36 | 2.35 | 2.35 | 2.37 | 2.36 | 2.34 | 2.35 | 2.36 | |
| TM-P | 2.42 | 2.37 | 2.38 | 2.32 | 2.23 | 2.23 | 2.24 | - | |
| Epol (eV) | 3.73 | 4.16 | 4.95 | 4.83 | 3.88 | 2.26 | 1.18 | - | |
| Δq (e) | 1.22 | 1.06 | 0.86 | 0.76 | 0.60 | 0.31 | 0.19 | - | |
| Magnetic moment (μB) | 1.00 | 2.00 | 3.00 | 4.00 | 5.00 | 1.44 | 0.44 | 0.00 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, H.; Guo, J.; Yuan, X.; Ren, J. Spin Polarization Properties of Two Dimensional GaP3 Induced by 3d Transition-Metal Doping. Micromachines 2021, 12, 743. https://doi.org/10.3390/mi12070743
Wei H, Guo J, Yuan X, Ren J. Spin Polarization Properties of Two Dimensional GaP3 Induced by 3d Transition-Metal Doping. Micromachines. 2021; 12(7):743. https://doi.org/10.3390/mi12070743
Chicago/Turabian StyleWei, Huihui, Jiatian Guo, Xiaobo Yuan, and Junfeng Ren. 2021. "Spin Polarization Properties of Two Dimensional GaP3 Induced by 3d Transition-Metal Doping" Micromachines 12, no. 7: 743. https://doi.org/10.3390/mi12070743
APA StyleWei, H., Guo, J., Yuan, X., & Ren, J. (2021). Spin Polarization Properties of Two Dimensional GaP3 Induced by 3d Transition-Metal Doping. Micromachines, 12(7), 743. https://doi.org/10.3390/mi12070743