
Design and Manufacture of a Low-Cost Microfluidic System for the Synthesis of Giant Liposomes for the Encapsulation of Yeast Homologues: Applications in the Screening of Membrane-Active Peptide Libraries
 , , ,
, , ,  ,
,  and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
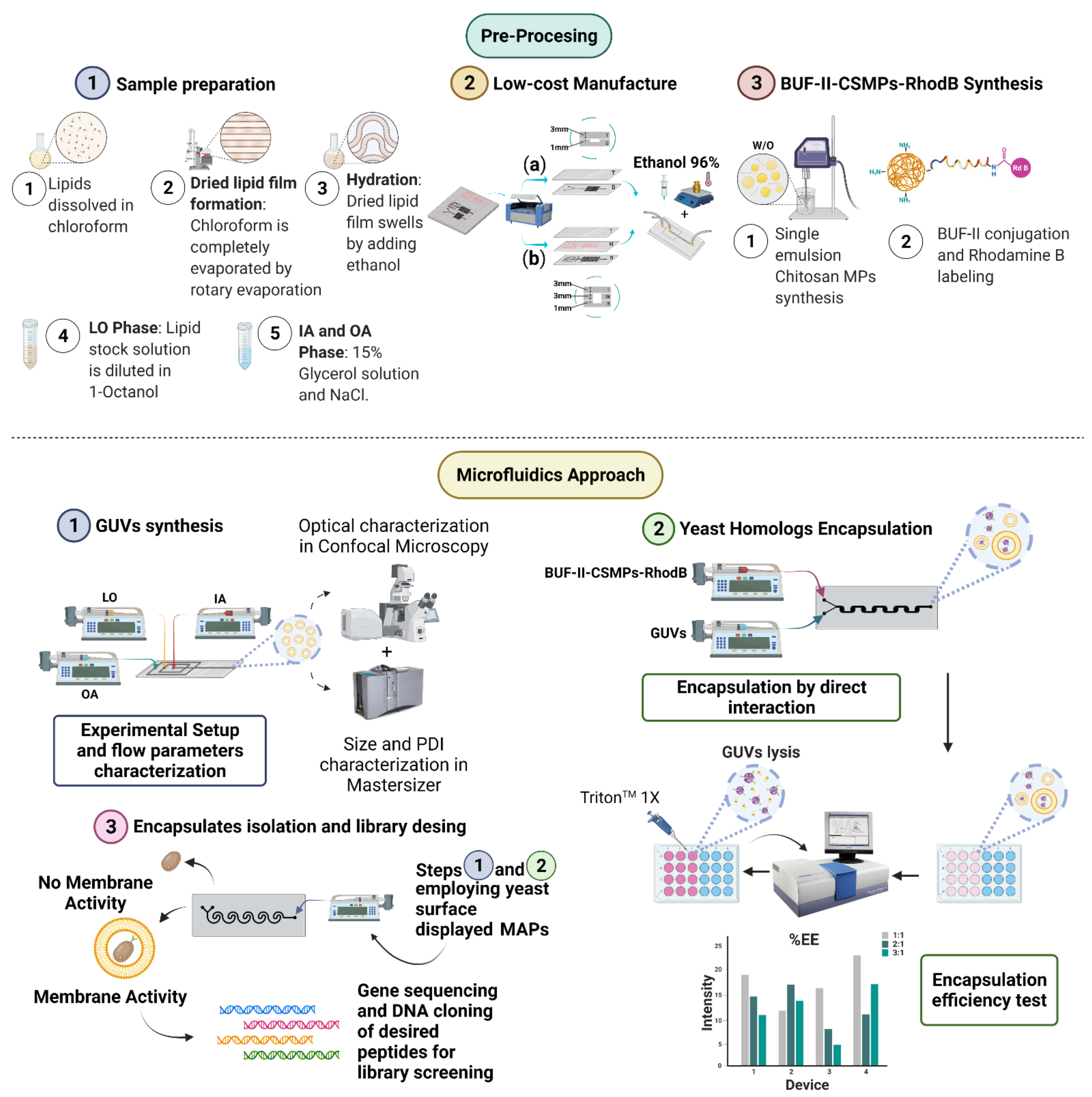
2.2. Microfluidic Systems Design and Multiphysics Simulations
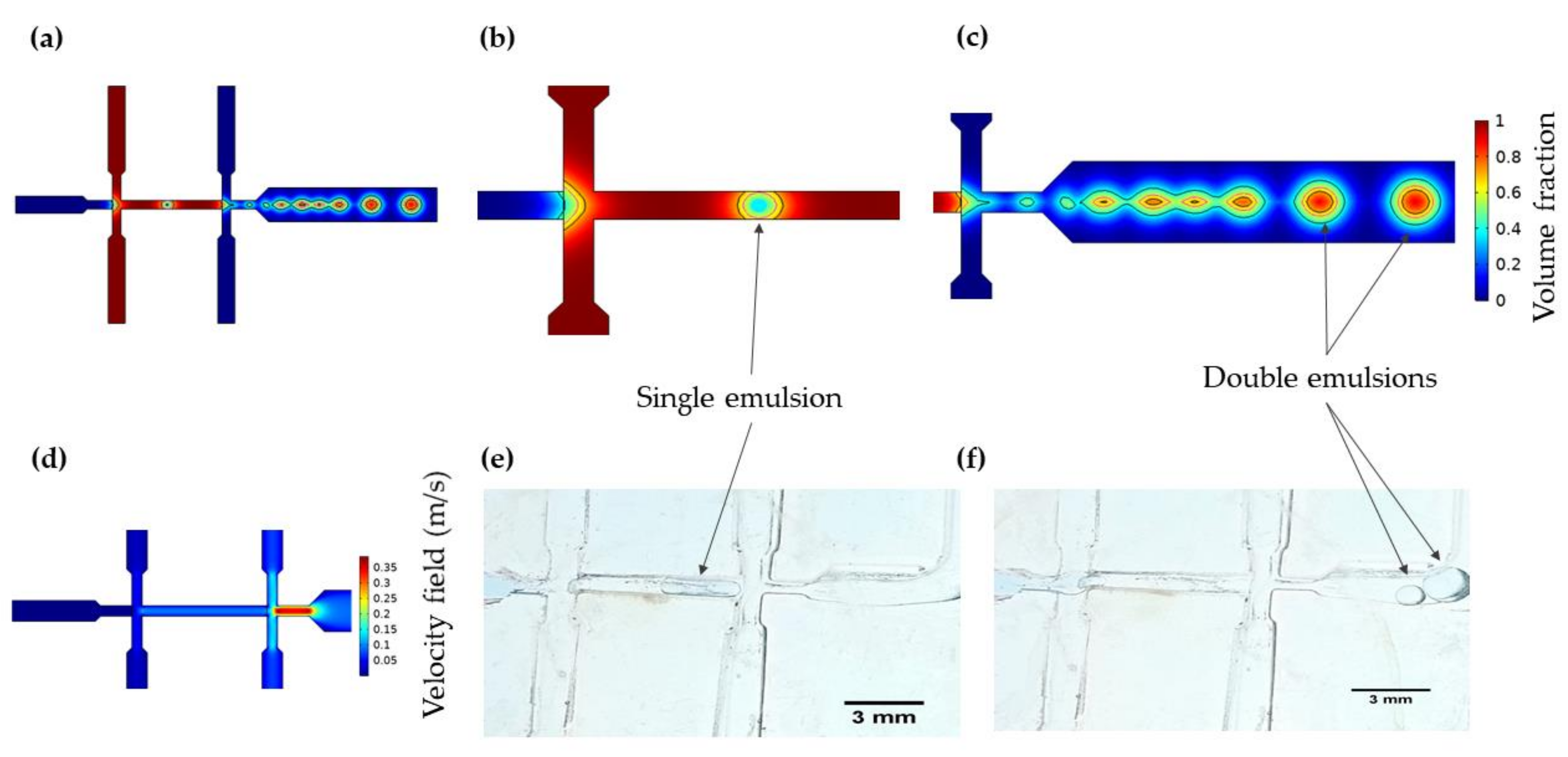
2.2.1. Double Emulsion Template for GUV Synthesis
2.2.2. Mixing-Induced Encapsulation by Direct Interaction
2.3. Device Fabrication and Manufacture
2.4. Low-Cost Octanol-Assisted Liposomes Assembly (OLA)
2.4.1. Sample Preparation
2.4.2. Experimental Setup
2.5. GUVs Characterization
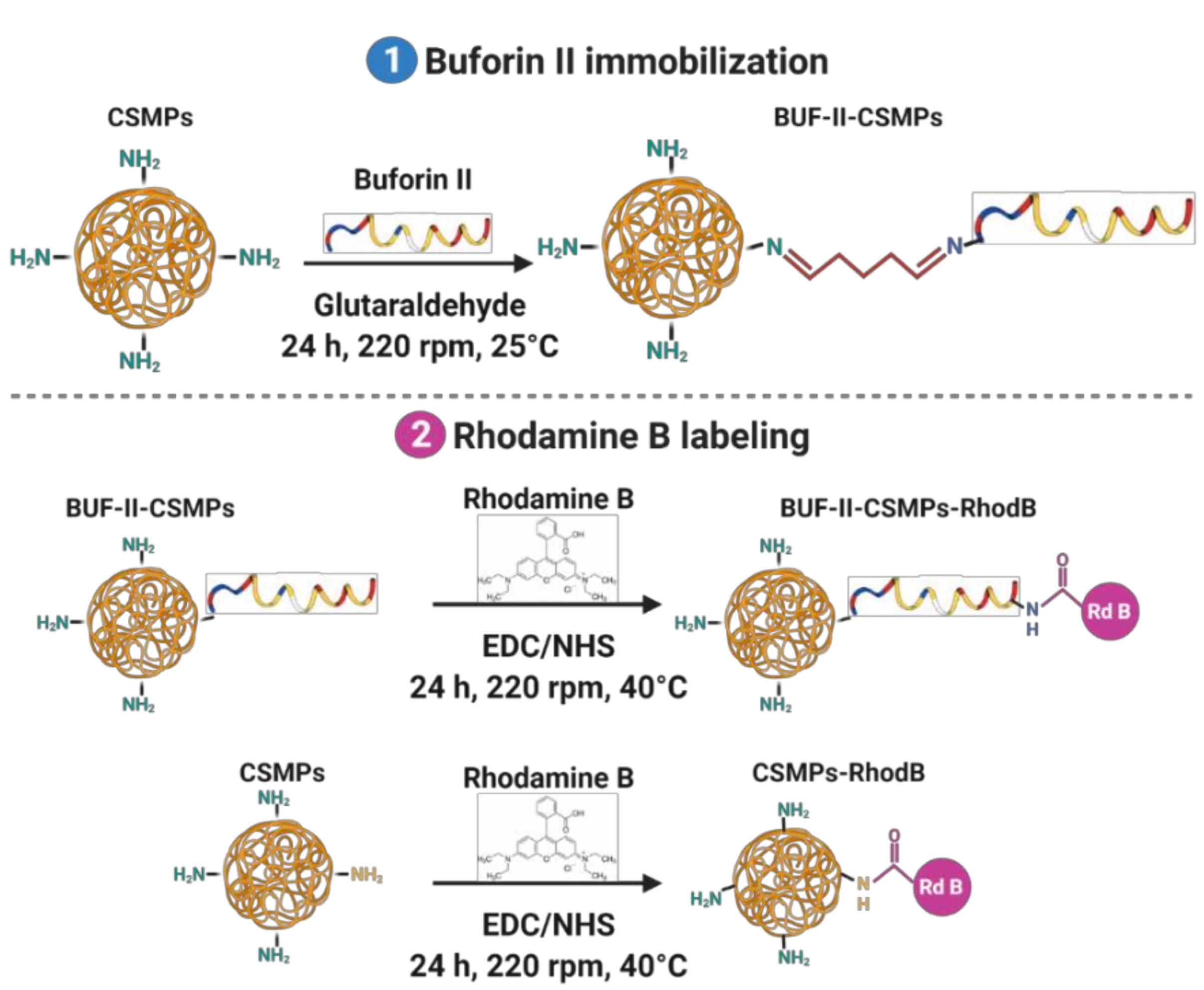
2.6. Synthesis of BUF-II-CSMPs Conjugates and Labeling with Rhodamine B
2.7. Characterization of CSMPs Bioconjugates and Encapsulates
2.8. Colorimetric Mixing Efficiency Test
2.9. Encapsulation of CSMPs Bioconjugates as Homologues of MAPs
3. Results and Discussion
3.1. Multiphysics Simulations
3.1.1. Double Emulsion Generation
3.1.2. Encapsulation by Direct Interaction
3.2. Low-Cost Octanol-Assisted Liposomes Assembly
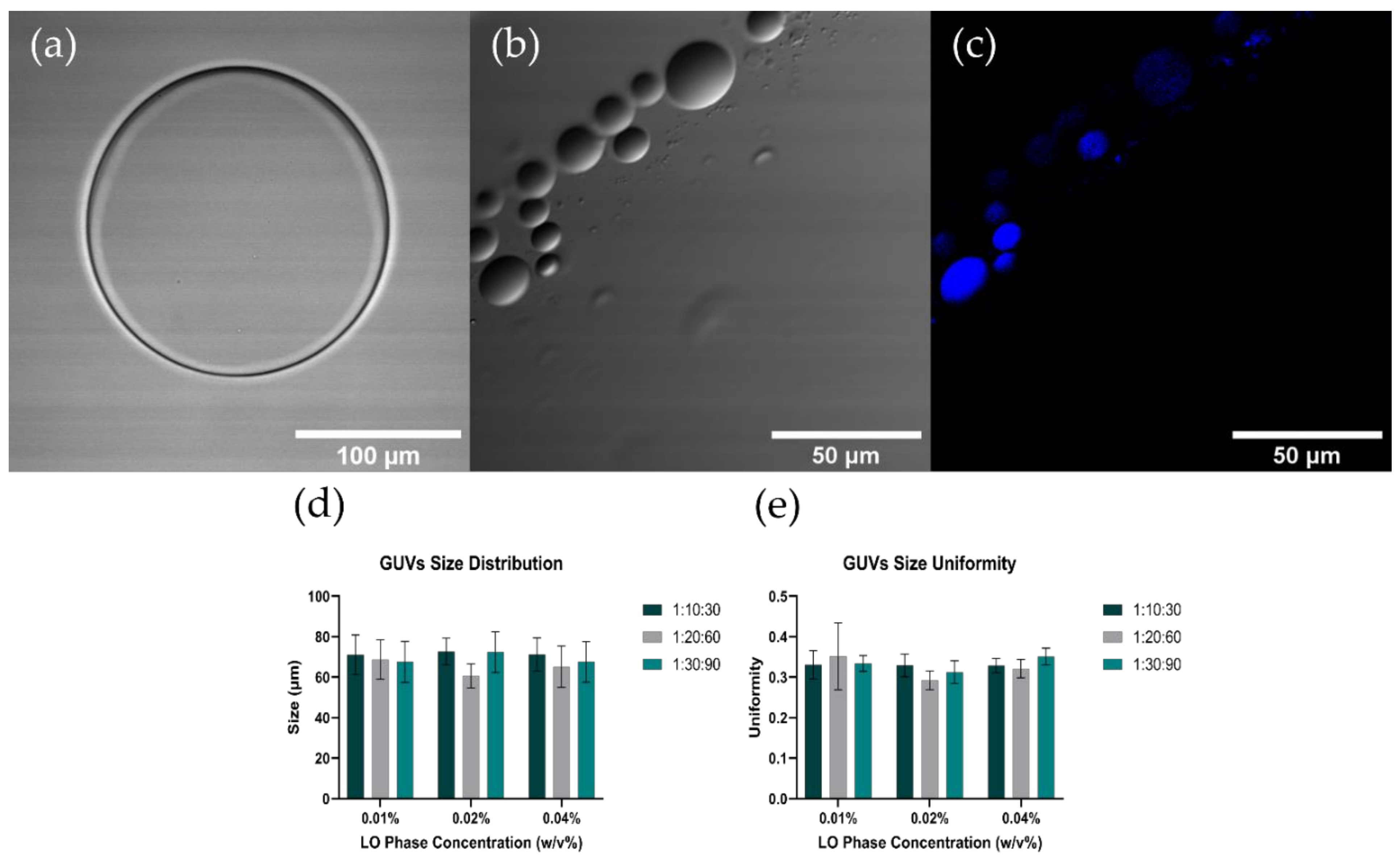
3.3. GUVs Characterization
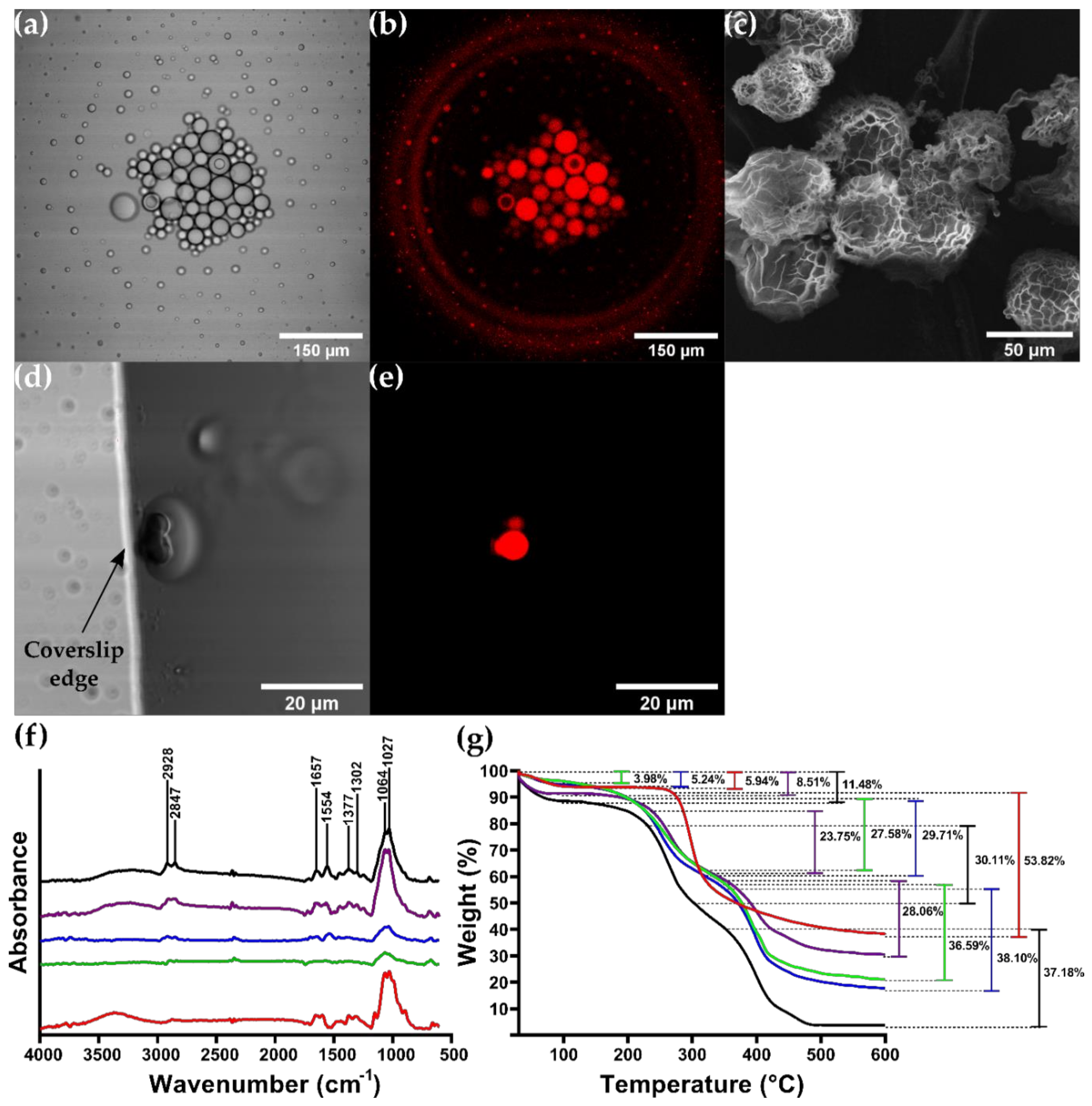
3.4. CSMPs and Bionconjugates Characterization
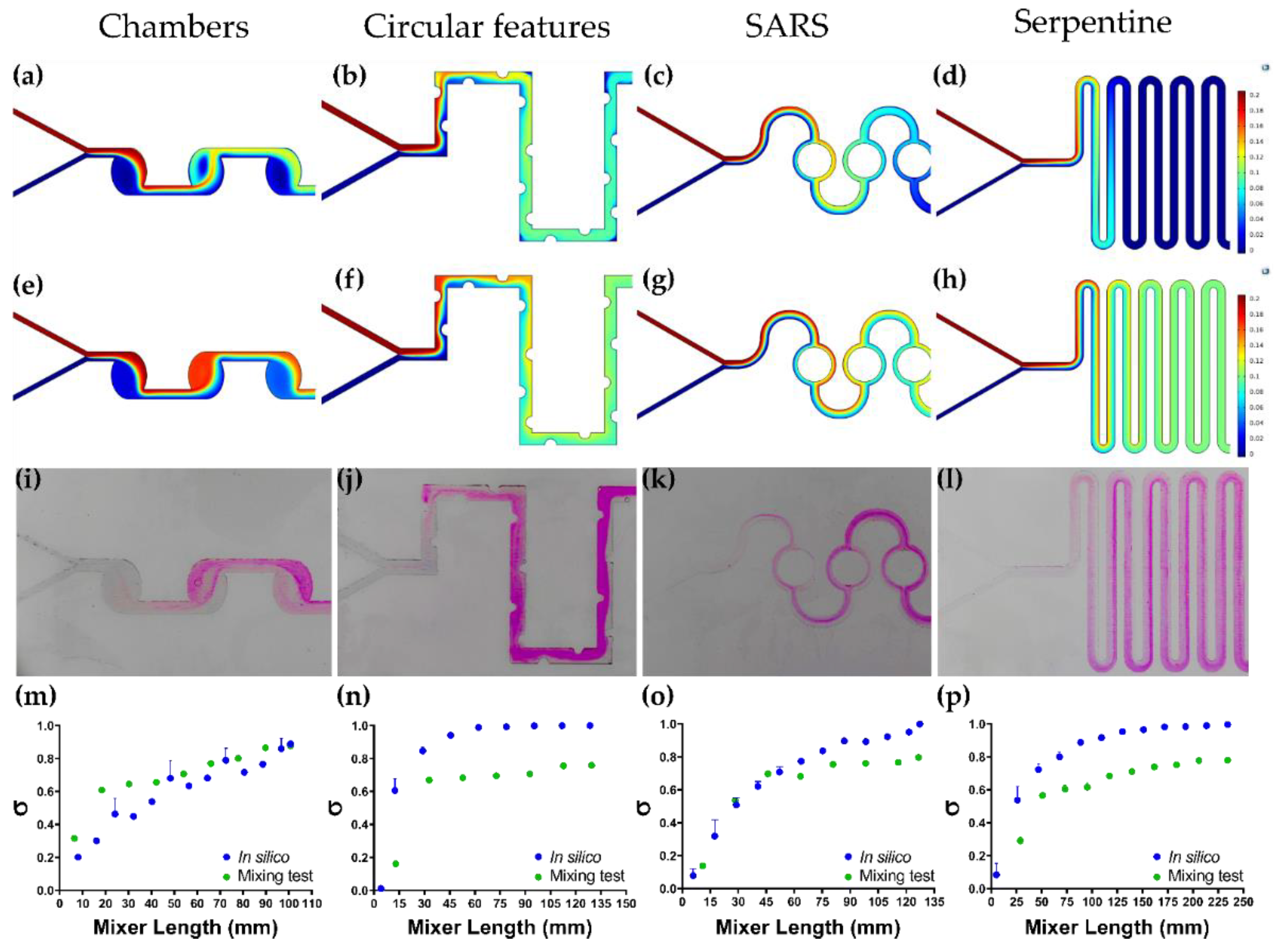
3.5. Colorimetric Mixing Efficiency Test vs. Mixing Efficiency In Silico
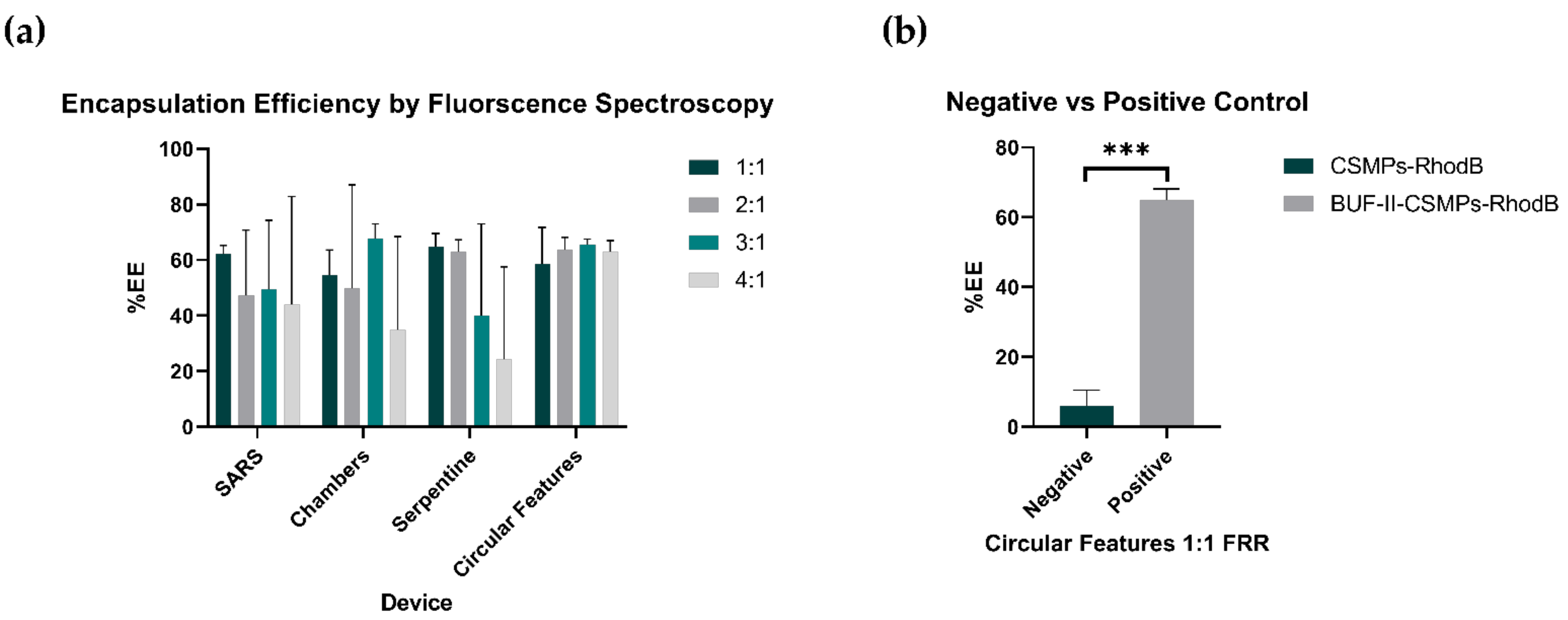
3.6. Encapsulation Bioconjugate Homologues
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Derakhshankhah, H.; Jafari, S. Cell Penetrating Peptides: A Concise Review with Emphasis on Biomedical Applications. Biomed. Pharmacother. 2018, 108, 1090–1096. [Google Scholar] [CrossRef]
- Henriques, S.T.; Melo, M.N.; Castanho, M.A.R.B. Cell-Penetrating Peptides and Antimicrobial Peptides: How Different Are They? Biochem. J. 2006, 399, 1–7. [Google Scholar] [CrossRef]
- Avci, F.G.; Akbulut, B.S.; Ozkirimli, E. Membrane Active Peptides and Their Biophysical Characterization. Biomolecules 2018, 8, 77. [Google Scholar] [CrossRef]
- Kmeck, A.; Tancer, R.J.; Ventura, C.R.; Wiedman, G.R. Synergies with and Resistance to Membrane-Active Peptides. Antibiotics 2020, 9, 620. [Google Scholar] [CrossRef]
- Zaman, S.B.; Hussain, M.A.; Nye, R.; Mehta, V.; Mamun, K.T.; Hossain, N. A Review on Antibiotic Resistance: Alarm Bells Are Ringing. Cureus 2017, 9, e1403. [Google Scholar] [CrossRef]
- Kaur, I. Novel Strategies to Combat Antimicrobial Resistance. J. Infect. Dis. Ther. 2016, 4, 292. [Google Scholar] [CrossRef]
- Mandal, S.M.; Roy, A.; Ghosh, A.K.; Hazra, T.K.; Basak, A.; Franco, O.L. Challenges and Future Prospects of Antibiotic Therapy: From Peptides to Phages Utilization. Front. Pharmacol. 2014, 5, 105. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Mumtaz, S.; Li, C.H.; Hussain, I.; Rotello, V.M. Combatting Antibiotic-Resistant Bacteria Using Nanomaterials. Chem. Soc. Rev. 2019, 48, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Sakeena, M.H.F.; Bennett, A.A.; McLachlan, A.J. Enhancing Pharmacists’ Role in Developing Countries to Overcome the Challenge of Antimicrobial Resistance: A Narrative Review. Antimicrob. Resist. Infect. Control 2018, 7, 63. [Google Scholar] [CrossRef] [PubMed]
- Post, K.W. Overview of Bacteria. In Diseases of Swine; Wiley: Hoboken, NJ, USA, 2019; pp. 743–748. [Google Scholar]
- Laxminarayan, R.; Brown, G.M. Economics of Antibiotic Resistance: A Theory of Optimal Use. J. Environ. Econ. Manag. 2001, 42, 183–206. [Google Scholar] [CrossRef]
- Pärn, K.; Eriste, E.; Langel, Ü. The Antimicrobial and Antiviral Applications of Cell-Penetrating Peptides. Cell-Penetrating Pept. 2015, 1324, 223–245. [Google Scholar]
- Wang, T.-Y.; Pellois, J.-P. Peptide Translocation through the Plasma Membrane of Human Cells: Can Oxidative Stress Be Exploited to Gain Better Intracellular Access? Commun. Integr. Biol. 2016, 9, e1205771. [Google Scholar] [CrossRef][Green Version]
- Kristensen, M.; Birch, D.; Mørck Nielsen, H. Applications and Challenges for Use of Cell-Penetrating Peptides as Delivery Vectors for Peptide and Protein Cargos. Int. J. Mol. Sci. 2016, 17, 185. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, T.A.E.; Hammami, R. Recent Insights into Structure-Function Relationships of Antimicrobial Peptides. J. Food Biochem. 2019, 43, e12546. [Google Scholar] [CrossRef] [PubMed]
- Biswaro, L.S.; da Costa Sousa, M.G.; Rezende, T.M.B.; Dias, S.C.; Franco, O.L. Antimicrobial Peptides and Nanotechnology, Recent Advances and Challenges. Front. Microbiol. 2018, 9, 855. [Google Scholar] [CrossRef] [PubMed]
- Burdukiewicz, M.; Sidorczuk, K.; Rafacz, D.; Pietluch, F.; Chilimoniuk, J.; Rödiger, S.; Gagat, P. Proteomic Screening for Prediction and Design of Antimicrobial Peptides with AmpGram. Int. J. Mol. Sci. 2020, 21, 4310. [Google Scholar] [CrossRef]
- Kumar, P.; Kizhakkedathu, J.; Straus, S. Antimicrobial Peptides: Diversity, Mechanism of Action and Strategies to Improve the Activity and Biocompatibility In Vivo. Biomolecules 2018, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Magana, M.; Pushpanathan, M.; Santos, A.L.; Leanse, L.; Fernandez, M.; Ioannidis, A.; Giulianotti, M.A.; Apidianakis, Y.; Bradfute, S.; Ferguson, A.L.; et al. The Value of Antimicrobial Peptides in the Age of Resistance. Lancet Infect. Dis. 2020, 20, e216–e230. [Google Scholar] [CrossRef]
- Travkova, O.G.; Moehwald, H.; Brezesinski, G. The Interaction of Antimicrobial Peptides with Membranes. Adv. Colloid Interface Sci. 2017, 247, 521–532. [Google Scholar] [CrossRef] [PubMed]
- Mookherjee, N.; Hancock, R.E.W. Cationic Host Defence Peptides: Innate Immune Regulatory Peptides as a Novel Approach for Treating Infections. Cell. Mol. Life Sci. 2007, 64, 922–933. [Google Scholar] [CrossRef]
- Li, J.; Koh, J.-J.; Liu, S.; Lakshminarayanan, R.; Verma, C.S.; Beuerman, R.W. Membrane Active Antimicrobial Peptides: Translating Mechanistic Insights to Design. Front. Neurosci. 2017, 11, 73. [Google Scholar] [CrossRef] [PubMed]
- Marquette, A.; Bechinger, B. Biophysical Investigations Elucidating the Mechanisms of Action of Antimicrobial Peptides and Their Synergism. Biomolecules 2018, 8, 18. [Google Scholar] [CrossRef] [PubMed]
- Le, C.F.; Fang, C.M.; Sekaran, S.D. Intracellular Targeting Mechanisms by Antimicrobial Peptides. Antimicrob. Agents Chemother. 2017, 61, e02340-16. [Google Scholar] [CrossRef]
- Andersson, D.I.; Hughes, D.; Kubicek-Sutherland, J.Z. Mechanisms and Consequences of Bacterial Resistance to Antimicrobial Peptides. Drug Resist. Updat. 2016, 26, 43–57. [Google Scholar] [CrossRef]
- Jung, H.J.; Jeong, K.-S.; Lee, D.G. Effective Antibacterial Action of Tat (47–58) by Increased Uptake into Bacterial Cells in the Presence of Trypsin. J. Microbiol. Biotechnol. 2008, 18, 990–996. [Google Scholar] [PubMed]
- Zhu, W.L.; Shin, S.Y. Effects of Dimerization of the Cell-Penetrating Peptide Tat Analog on Antimicrobial Activity and Mechanism of Bactericidal Action. J. Pept. Sci. 2009, 15, 345–352. [Google Scholar] [CrossRef]
- Matsuzaki, K.; Murase, O.; Fujii, N.; Miyajima, K. Translocation of a Channel-Forming Antimicrobial Peptide, Magainin 2, across Lipid Bilayers by Forming a Pore. Biochemistry 1995, 34, 6521–6526. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Rozek, A.; Hancock, R.E.W. Interaction of Cationic Antimicrobial Peptides with Model Membranes. J. Biol. Chem. 2001, 276, 35714–35722. [Google Scholar] [CrossRef] [PubMed]
- Tucker, A.T.; Leonard, S.P.; DuBois, C.D.; Knauf, G.A.; Cunningham, A.L.; Wilke, C.O.; Trent, M.S.; Davies, B.W. Discovery of Next-Generation Antimicrobials through Bacterial Self-Screening of Surface-Displayed Peptide Libraries. Cell 2018, 172, 618–628.e13. [Google Scholar] [CrossRef]
- Attene-Ramos, M.S.; Austin, C.P.; Xia, M. High Throughput Screening, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2014; Volume 2, ISBN 9780123864543. [Google Scholar]
- Du, G.; Fang, Q.; den Toonder, J.M.J. Microfluidics for Cell-Based High Throughput Screening Platforms-A Review. Anal. Chim. Acta 2016, 903, 36–50. [Google Scholar] [CrossRef]
- Payne, E.M.; Holland-Moritz, D.A.; Sun, S.; Kennedy, R.T. High-Throughput Screening by Droplet Microfluidics: Perspective into Key Challenges and Future Prospects. Lab Chip 2020, 20, 2247–2262. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Luo, J.; Qin, J.; Yang, M. Screening Techniques for the Identification of Bioactive Compounds in Natural Products. J. Pharm. Biomed. Anal. 2019, 168, 189–200. [Google Scholar] [CrossRef]
- Holland-Moritz, D.A.; Wismer, M.K.; Mann, B.F.; Farasat, I.; Devine, P.; Guetschow, E.D.; Mangion, I.; Welch, C.J.; Moore, J.C.; Sun, S.; et al. Mass Activated Droplet Sorting (MADS) Enables High-Throughput Screening of Enzymatic Reactions at Nanoliter Scale. Angew. Chem. Int. Ed. 2020, 59, 4470–4477. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, X.; Liu, L.; Zhou, P.; Zhou, J.; Shi, X.; Wang, Y. A Microarray Platform Designed for High-Throughput Screening the Reaction Conditions for the Synthesis of Micro/Nanosized Biomedical Materials. Bioact. Mater. 2020, 5, 286–296. [Google Scholar] [CrossRef]
- Mashaghi, S.; Abbaspourrad, A.; Weitz, D.A.; van Oijen, A.M. Droplet Microfluidics: A Tool for Biology, Chemistry and Nanotechnology. TrAC Trends Anal. Chem. 2016, 82, 118–125. [Google Scholar] [CrossRef]
- Puentes, P.R.; Henao, M.C.; Torres, C.E.; Gómez, S.C.; Gómez, L.A.; Burgos, J.C.; Arbeláez, P.; Osma, J.F.; Muñoz-Camargo, C.; Reyes, L.H.; et al. Design, Screening, and Testing of Non-Rational Peptide Libraries with Antimicrobial Activity: In Silico and Experimental Approaches. Antibiotics 2020, 9, 854. [Google Scholar] [CrossRef]
- Guo, M.T.; Rotem, A.; Heyman, J.A.; Weitz, D.A. Droplet Microfluidics for High-Throughput Biological Assays. Lab Chip 2012, 12, 2146. [Google Scholar] [CrossRef]
- Wong, I.; Ho, C.-M. Surface Molecular Property Modifications for Poly(Dimethylsiloxane) (PDMS) Based Microfluidic Devices. Microfluid. Nanofluidics 2009, 7, 291. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, A.D.; Tawfik, D.S. Miniaturising the Laboratory in Emulsion Droplets. Trends Biotechnol. 2006, 24, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Thorsen, T.; Roberts, R.W.; Arnold, F.H.; Quake, S.R. Dynamic Pattern Formation in a Vesicle-Generating Microfluidic Device. Phys. Rev. Lett. 2001, 86, 4163–4166. [Google Scholar] [CrossRef] [PubMed]
- Safa, N.; Vaithiyanathan, M.; Sombolestani, S.; Charles, S.; Melvin, A.T. Population-Based Analysis of Cell-Penetrating Peptide Uptake Using a Microfluidic Droplet Trapping Array. Anal. Bioanal. Chem. 2019, 411, 2729–2741. [Google Scholar] [CrossRef] [PubMed]
- Yaginuma, K.; Aoki, W.; Miura, N.; Ohtani, Y.; Aburaya, S.; Kogawa, M.; Nishikawa, Y.; Hosokawa, M.; Takeyama, H.; Ueda, M. High-Throughput Identification of Peptide Agonists against GPCRs by Co-Culture of Mammalian Reporter Cells and Peptide-Secreting Yeast Cells Using Droplet Microfluidics. Sci. Rep. 2019, 9, 10920. [Google Scholar] [CrossRef] [PubMed]
- Beneyton, T.; Thomas, S.; Griffiths, A.D.; Nicaud, J.-M.; Drevelle, A.; Rossignol, T. Droplet-Based Microfluidic High-Throughput Screening of Heterologous Enzymes Secreted by the Yeast Yarrowia Lipolytica. Microb. Cell Fact. 2017, 16, 18. [Google Scholar] [CrossRef]
- Sjostrom, S.L.; Bai, Y.; Huang, M.; Liu, Z.; Nielsen, J.; Joensson, H.N.; Andersson Svahn, H. High-Throughput Screening for Industrial Enzyme Production Hosts by Droplet Microfluidics. Lab Chip 2014, 14, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Pottel, J.; Moitessier, N. Customizable Generation of Synthetically Accessible, Local Chemical Subspaces. J. Chem. Inf. Model. 2017, 57, 454–467. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Bai, Z.; Lam, K.; Pan, T. Microfluidics-Enabled Combinatorial Peptide Library for High Throughput Screening. In Proceedings of the 18th International Conference on Miniaturized Systems for Chemistry and Life Sciences MicroTAS 2014, San Antonio, TX, USA, 26–30 October 2014; pp. 1506–1508. [Google Scholar]
- Carney, R.P.; Thillier, Y.; Kiss, Z.; Sahabi, A.; Heleno Campos, J.C.; Knudson, A.; Liu, R.; Olivos, D.; Saunders, M.; Tian, L.; et al. Combinatorial Library Screening with Liposomes for Discovery of Membrane Active Peptides. ACS Comb. Sci. 2017, 19, 299–307. [Google Scholar] [CrossRef]
- Wang, W.; Wei, Z.; Zhang, D.; Ma, H.; Wang, Z.; Bu, X.; Li, M.; Geng, L.; Lausted, C.; Hood, L.; et al. Rapid Screening of Peptide Probes through In Situ Single-Bead Sequencing Microarray. Anal. Chem. 2014, 86, 11854–11859. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhao, S.; Yang, G.; Liu, R.; Xiao, W.; Disano, P.; Lam, K.S.; Pan, T. Combinatorial Peptide Microarray Synthesis Based on Microfluidic Impact Printing. ACS Comb. Sci. 2019, 21, 6–10. [Google Scholar] [CrossRef]
- Schaich, M.; Cama, J.; Al Nahas, K.; Sobota, D.; Sleath, H.; Jahnke, K.; Deshpande, S.; Dekker, C.; Keyser, U.F. An Integrated Microfluidic Platform for Quantifying Drug Permeation across Biomimetic Vesicle Membranes. Mol. Pharm. 2019, 16, 2494–2501. [Google Scholar] [CrossRef]
- Hall, K.; Aguilar, M.-I. Surface Plasmon Resonance Spectroscopy for Studying the Membrane Binding of Antimicrobial Peptides. In Surface Plasmon Resonance; Humana Press: Totowa, NJ, USA, 2010; pp. 213–223. [Google Scholar]
- Šakanovič, A.; Hodnik, V.; Anderluh, G. Surface Plasmon Resonance for Measuring Interactions of Proteins with Lipids and Lipid Membranes. In Lipid-Protein Interactions; Humana: New York, NY, USA, 2019; pp. 53–70. [Google Scholar]
- Deshpande, S.; Caspi, Y.; Meijering, A.E.C.; Dekker, C. Octanol-Assisted Liposome Assembly on Chip. Nat. Commun. 2016, 7, 1–9. [Google Scholar] [CrossRef]
- Al Nahas, K.; Cama, J.; Schaich, M.; Hammond, K.; Deshpande, S.; Dekker, C.; Ryadnov, M.G.; Keyser, U.F. A Microfluidic Platform for the Characterisation of Membrane Active Antimicrobials. Lab Chip 2019, 19, 837–844. [Google Scholar] [CrossRef]
- Kuhn, P.; Eyer, K.; Allner, S.; Lombardi, D.; Dittrich, P.S. A Microfluidic Vesicle Screening Platform: Monitoring the Lipid Membrane Permeability of Tetracyclines. Anal. Chem. 2011, 83, 8877–8885. [Google Scholar] [CrossRef]
- Kolesinska, B.; Eyer, K.; Robinson, T.; Dittrich, P.S.; Beck, A.K.; Seebach, D.; Walde, P. Interaction of Β3/Β2-Peptides, Consisting of Val-Ala-Leu Segments, with POPC Giant Unilamellar Vesicles (GUVs) and White Blood Cancer Cells (U937)—A New Type of Cell-Penetrating Peptides, and a Surprising Chain-Length Dependence of Their Vesicle- and Ce. Chem. Biodivers. 2015, 12, 697–732. [Google Scholar] [CrossRef]
- Muijlwijk, K. Microfluidic Methods to Study Emulsion Formation; Wageningen University: Wageningen, The Netherlands, 2017; ISBN 9789463430715. [Google Scholar]
- Michelon, M.; Huang, Y.; de la Torre, L.G.; Weitz, D.A.; Cunha, R.L. Single-Step Microfluidic Production of W/O/W Double Emulsions as Templates for Β-Carotene-Loaded Giant Liposomes Formation. Chem. Eng. J. 2019, 366, 27–32. [Google Scholar] [CrossRef]
- Campaña, A.L.; Sotelo, D.C.; Oliva, H.A.; Aranguren, A.; Ornelas-Soto, N.; Cruz, J.C.; Osma, J.F. Fabrication and Characterization of a Low-Cost Microfluidic System for the Manufacture of Alginate-Lacasse Microcapsules. Polymers 2020, 12, 1158. [Google Scholar] [CrossRef]
- Wang, K.; Luo, G. Microflow Extraction: A Review of Recent Development. Chem. Eng. Sci. 2017, 169, 18–33. [Google Scholar] [CrossRef]
- Bayareh, M. Artificial Diffusion in the Simulation of Micromixers: A Review. Proc. Inst. Mech. Eng. Part C J. Mech. Eng. Sci. 2020. [Google Scholar] [CrossRef]
- Bermudez, J.F.; Saldarriaga, J.F.; Osma, J.F. Portable and Low-Cost Respirometric Microsystem for the Static and Dynamic Respirometry Monitoring of Compost. Sensors 2019, 19, 4132. [Google Scholar] [CrossRef] [PubMed]
- Aranguren, A.; Torres, C.E.; Muñoz-Camargo, C.; Osma, J.F.; Cruz, J.C. Synthesis of Nanoscale Liposomes via Low-Cost Microfluidic Systems. Micromachines 2020, 11, 1050. [Google Scholar] [CrossRef]
- Mothilal, M.; Nagalakshmi, M.; Swati, P.S.; Damodharan, N.; Lakshmi, K.S. Optimisation and Characetrisation of Chitosan microspheres of aceclofenac. Int. J. Pharma Sci. Res. 2012, 3, 305–315. [Google Scholar]
- Perez, J.; Cifuentes, J.; Cuellar, M.; Suarez-Arnedo, A.; Cruz, J.C.; Muñoz-Camargo, C. Cell-Penetrating and Antibacterial BUF-II Nanobioconjugates: Enhanced Potency via Immobilization on Polyetheramine-Modified Magnetite Nanoparticles. Int. J. Nanomed. 2019, 14, 8483–8497. [Google Scholar] [CrossRef] [PubMed]
- Huanming, X.; Jiawei, W.; Zhiping, W. A Comparative Discussion of Different Designs of Passive Micromixers: Specific Sensitivities of Mixing Efficiency on Reynolds Numbers and Fluid Properties. Microsyst. Technol. 2018, 24, 1253–1263. [Google Scholar] [CrossRef]
- Vladisavljević, G.T.; Al Nuumani, R.; Nabavi, S.A. Microfluidic Production of Multiple Emulsions. Micromachines 2017, 8, 75. [Google Scholar] [CrossRef]
- Li, J.; Barrow, D.A. A New Droplet-Forming Fluidic Junction for the Generation of Highly Compartmentalised Capsules. Lab Chip 2017, 17, 2873–2881. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Lee, R.J.; Lee, L.J. Microfluidic Methods for Production of Liposomes. Methods Enzymol. 2009, 465, 129–141. [Google Scholar] [CrossRef]
- Conchouso, D.; Rawashdeh, E.; Arevalo, A.; Castro, D.; Foulds, I.G. Simulation of a 3D Flow-Focusing Capillary-Based Droplet Generator. In Proceedings of the COMSOL Conference, Rotterdam, The Netherlands, 23–25 October 2013; pp. 1–5. [Google Scholar]
- Ansari, M.A.; Kim, K.-Y. A Numerical Study of Mixing in a Microchannel with Circular Mixing Chambers. AIChE J. 2009, 55, 2217–2225. [Google Scholar] [CrossRef]
- Chung, Y.C.; Hsu, Y.L.; Jen, C.P.; Lu, M.C.; Lin, Y.C. Design of Passive Mixers Utilizing Microfluidic Self-Circulation in the Mixing Chamber. Lab Chip 2004, 4, 70–77. [Google Scholar] [CrossRef]
- Usefian, A.; Bayareh, M. Numerical and Experimental Investigation of an Efficient Convergent–Divergent Micromixer. Meccanica 2020, 55, 1025–1035. [Google Scholar] [CrossRef]
- Wangikar, S.S.; Patowari, P.K.; Misra, R.D. Numerical and Experimental Investigations on the Performance of a Serpentine Microchannel with Semicircular Obstacles. Microsyst. Technol. 2018, 24, 3307–3320. [Google Scholar] [CrossRef]
- Shanko, E.S.; van de Burgt, Y.; Anderson, P.D.; den Toonder, J.M.J. Microfluidic Magnetic Mixing at Low Reynolds Numbers and in Stagnant Fluids. Micromachines 2019, 10, 731. [Google Scholar] [CrossRef] [PubMed]
- Bayareh, M.; Ashani, M.N.; Usefian, A. Active and Passive Micromixers: A Comprehensive Review. Chem. Eng. Process. Process Intensif. 2020, 147, 107771. [Google Scholar] [CrossRef]
- Raza, W.; Kim, K.Y. Asymmetrical Split-and-Recombine Micromixer with Baffles. Micromachines 2019, 10, 844. [Google Scholar] [CrossRef]
- Juraeva, M.; Kang, D.J. Mixing Performance of a Cross-Channel Split-and-Recombine Micro-Mixer Combined with Mixing Cell. Micromachines 2020, 11, 685. [Google Scholar] [CrossRef]
- Karthikeyan, K.; Sujatha, L. Study of Permissible Flow Rate and Mixing Efficiency of the Micromixer Devices. Int. J. Chem. React. Eng. 2019, 17, 1–15. [Google Scholar] [CrossRef]
- Plevniak, K.; Campbell, M.; He, M. 3D Printed Microfluidic Mixer for Point-of-Care Diagnosis of Anemia. In Proceedings of the 2016 38th Annual International Conference of the IEEE Engineering in Medicine and Biology Society (EMBC), Orlando, FL, USA, 16–20 August 2016; Volume 2016, pp. 267–270. [Google Scholar] [CrossRef]
- Liu, R.H.; Stremler, M.A.; Sharp, K.V.; Olsen, M.G.; Santiago, J.G.; Adrian, R.J.; Aref, H.; Beebe, D.J. Passive Mixing in a Three-Dimensional Serpentine Microchannel. J. Microelectromech. Syst. 2000, 9, 190–197. [Google Scholar] [CrossRef]
- Abate, A.R.; Thiele, J.; Weitz, D.A. One-Step Formation of Multiple Emulsions in Microfluidics. Lab Chip 2011, 11, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Askari, A.H.; Shams, M.; Sullivan, P.E. Numerical Simulation of Double Emulsion Formation in Cross-Junctional Flow-Focusing Microfluidic Device Using Lattice Boltzmann Method. J. Dispers. Sci. Technol. 2019, 40, 1451–1460. [Google Scholar] [CrossRef]
- Deshpande, S.; Dekker, C. On-Chip Microfluidic Production of Cell-Sized Liposomes. Nat. Protoc. 2018, 13, 856–874. [Google Scholar] [CrossRef] [PubMed]
- Bao, P.; Paterson, D.A.; Peyman, S.A.; Jones, J.C.; Sandoe, J.A.T.; Gleeson, H.F.; Evans, S.D.; Bushby, R.J. Production of Giant Unilamellar Vesicles and Encapsulation of Lyotropic Nematic Liquid Crystals. Soft Matter 2021, 17, 2234–2241. [Google Scholar] [CrossRef]
- Yang, C.H.; Huang, K.S.; Lin, P.W.; Lin, Y.C. Using a Cross-Flow Microfluidic Chip and External Crosslinking Reaction for Monodisperse TPP-Chitosan Microparticles. Sens. Actuators B Chem. 2007, 124, 510–516. [Google Scholar] [CrossRef]
- Moura, M.J.; Martins, S.P.; Duarte, B.P.M. Production of Chitosan Microparticles Cross-Linked with Genipin-Identification of Factors Influencing Size and Shape Properties. Biochem. Eng. J. 2015, 104, 82–90. [Google Scholar] [CrossRef]
- Ko, J.A.; Park, H.J.; Hwang, S.J.; Park, J.B.; Lee, J.S. Preparation and Characterization of Chitosan Microparticles Intended for Controlled Drug Delivery. Int. J. Pharm. 2002, 249, 165–174. [Google Scholar] [CrossRef]
- Hoemann, C.D.; Guzmán-Morales, J.; Tran-Khanh, N.; Lavallée, G.; Jolicoeur, M.; Lavertu, M. Chitosan Rate of Uptake in HEK293 Cells Is Influenced by Soluble versus Microparticle State and Enhanced by Serum-Induced Cell Metabolism and Lactate-Based Media Acidification. Molecules 2013, 18, 1015–1035. [Google Scholar] [CrossRef]
- Gracheva, I.; Konovalova, M.; Aronov, D.; Moiseeva, E.; Fedorov, A.; Svirshchevskaya, E. Size-Dependent Biodistribution of Fluorescent Furano-Allocolchicinoid-Chitosan Formulations in Mice. Polymers 2021, 13, 2045. [Google Scholar] [CrossRef]
- Sutirman, Z.A.; Sanagi, M.M.; Abd Karim, K.J.; Wan Ibrahim, W.A. Preparation of Methacrylamide-Functionalized Crosslinked Chitosan by Free Radical Polymerization for the Removal of Lead Ions. Carbohydr. Polym. 2016, 151, 1091–1099. [Google Scholar] [CrossRef]
- De Souza, T.C.R.; Parize, A.L.; Brighente, I.M.C.; Fávere, V.T.; Laranjeira, M.C.M. Chitosan Microspheres Containing the Natural Urucum Pigment. J. Microencapsul. 2005, 22, 511–520. [Google Scholar] [CrossRef]
- Cuellar, M.; Cifuentes, J.; Perez, J.; Suarez-Arnedo, A.; Serna, J.A.; Groot, H.; Muñoz-Camargo, C.; Cruz, J.C. Novel BUF2-Magnetite Nanobioconjugates with Cell-Penetrating Abilities. Int. J. Nanomed. 2018, 13, 8087–8094. [Google Scholar] [CrossRef]
- Gedam, A.H.; Dongre, R.S. Adsorption Characterization of Pb(Ii) Ions onto Iodate Doped Chitosan Composite: Equilibrium and Kinetic Studies. RSC Adv. 2015, 5, 54188–54201. [Google Scholar] [CrossRef]
- Swiecicki, J.-M.; Di Pisa, M.; Burlina, F.; Lécorché, P.; Mansuy, C.; Chassaing, G.; Lavielle, S. Accumulation of Cell-Penetrating Peptides in Large Unilamellar Vesicles: A Straightforward Screening Assay for Investigating the Internalization Mechanism. Biopolymers 2015, 104, 533–543. [Google Scholar] [CrossRef]
- Xu, W.; Han, L.; Lin, Z. Screening of Random Peptide Library of Hemagglutinin from Pandemic 2009 a(H1N1) Influenza Virus Reveals Unexpected Antigenically Important Regions. PLoS ONE 2011, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mesbah, K.; Thai, R.; Bregant, S.; Malloggi, F. DMF-MALDI: Droplet Based Microfluidic Combined to MALDI-TOF for Focused Peptide Detection. Sci. Rep. 2017, 7, 1–8. [Google Scholar] [CrossRef]
- Al Nahas, K.; Keyser, U.F. Standardizing Characterization of Membrane Active Peptides with Microfluidics. Biomicrofluidics 2021, 15, 041301. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Device | Mixing Time (MT) [s] | Mean Residence Time (MRT) [s] | Trajectory Length (TL) [mm] | Pressure Drop (ΔP) [Pa] | Mixing Efficiency (ME) | ME/ΔP [Pa−1] |
|---|---|---|---|---|---|---|
| Chambers | 12.76 ± 1.13 | 2.2323 ± 0.0004 | 90.7675 ± 0.0002 | 0.258 ± 0.003 | 87.60 ± 0.53% | 2.94 ± 0.08 |
| SARS | 4.61 ± 0.70 | 2.7123 ± 0.0007 | 122.0137 ± 0.0678 | 0.577 ± 0.043 | 79.70 ± 0.94% | 1.52 ± 0.16 |
| Serpentine | 7.95 ± 0.92 | 4.4304 ± 0.0001 | 216.6703 ± 0.0076 | 0.465 ± 0.030 | 77.96 ± 1.49% | 1.71 ± 0.06 |
| Circular Features | 3.85 ± 0.15 | 1.8139 ± 0.0002 | 127.2851 ± 0.0002 | 0.627 ± 0.031 | 75.86 ± 0.12% | 1.24 ± 0.03 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gómez, S.C.; Quezada, V.; Quiroz, I.; Muñoz-Camargo, C.; Osma, J.F.; Reyes, L.H.; Cruz, J.C. Design and Manufacture of a Low-Cost Microfluidic System for the Synthesis of Giant Liposomes for the Encapsulation of Yeast Homologues: Applications in the Screening of Membrane-Active Peptide Libraries. Micromachines 2021, 12, 1377. https://doi.org/10.3390/mi12111377
Gómez SC, Quezada V, Quiroz I, Muñoz-Camargo C, Osma JF, Reyes LH, Cruz JC. Design and Manufacture of a Low-Cost Microfluidic System for the Synthesis of Giant Liposomes for the Encapsulation of Yeast Homologues: Applications in the Screening of Membrane-Active Peptide Libraries. Micromachines. 2021; 12(11):1377. https://doi.org/10.3390/mi12111377
Chicago/Turabian StyleGómez, Saúl C., Valentina Quezada, Isabella Quiroz, Carolina Muñoz-Camargo, Johann F. Osma, Luis H. Reyes, and Juan C. Cruz. 2021. "Design and Manufacture of a Low-Cost Microfluidic System for the Synthesis of Giant Liposomes for the Encapsulation of Yeast Homologues: Applications in the Screening of Membrane-Active Peptide Libraries" Micromachines 12, no. 11: 1377. https://doi.org/10.3390/mi12111377
APA StyleGómez, S. C., Quezada, V., Quiroz, I., Muñoz-Camargo, C., Osma, J. F., Reyes, L. H., & Cruz, J. C. (2021). Design and Manufacture of a Low-Cost Microfluidic System for the Synthesis of Giant Liposomes for the Encapsulation of Yeast Homologues: Applications in the Screening of Membrane-Active Peptide Libraries. Micromachines, 12(11), 1377. https://doi.org/10.3390/mi12111377