1. Introduction
Nucleic acids are among the most important biomarkers of disease, and a large variety of diagnostic nucleic acid tests (NATs) have been developed to detect their presence in diagnostic settings, including polymerase chain reaction (PCR), loop-mediated isothermal amplification (LAMP), and rolling circle amplification [
1,
2,
3,
4]. Although very powerful, these tests rely on the use of enzymes, which poses several challenges for use at the point of care or in low-resource settings. For example, enzymes are typically the most expensive reagent in a NAT, and they require robust low-temperature storage conditions and labor-intensive sample preparation methods to achieve high sensitivity [
5,
6].
In response to these challenges, non-enzymatic NATs have been developed which amplify a target-induced signal using only thermodynamically-driven DNA hybridization reactions. Examples include hybridization chain reaction, entropy-driven catalysis, and catalyzed hairpin assembly [
7,
8,
9,
10]. These tests overcome the obstacles posed by enzymes, but typically have sub-optimal sensitivity due to their poor amplification. There have been several interesting demonstrations of non-enzymatic NATs that achieve increased sensitivity via exponential growth, including cascaded catalyzed hairpin assembly [
11], branched or hyperbranched hybridization chain reaction [
12,
13], and dendritic amplification [
14], but perhaps the most promising is the recent demonstration of a dumbbell DNA amplification scheme [
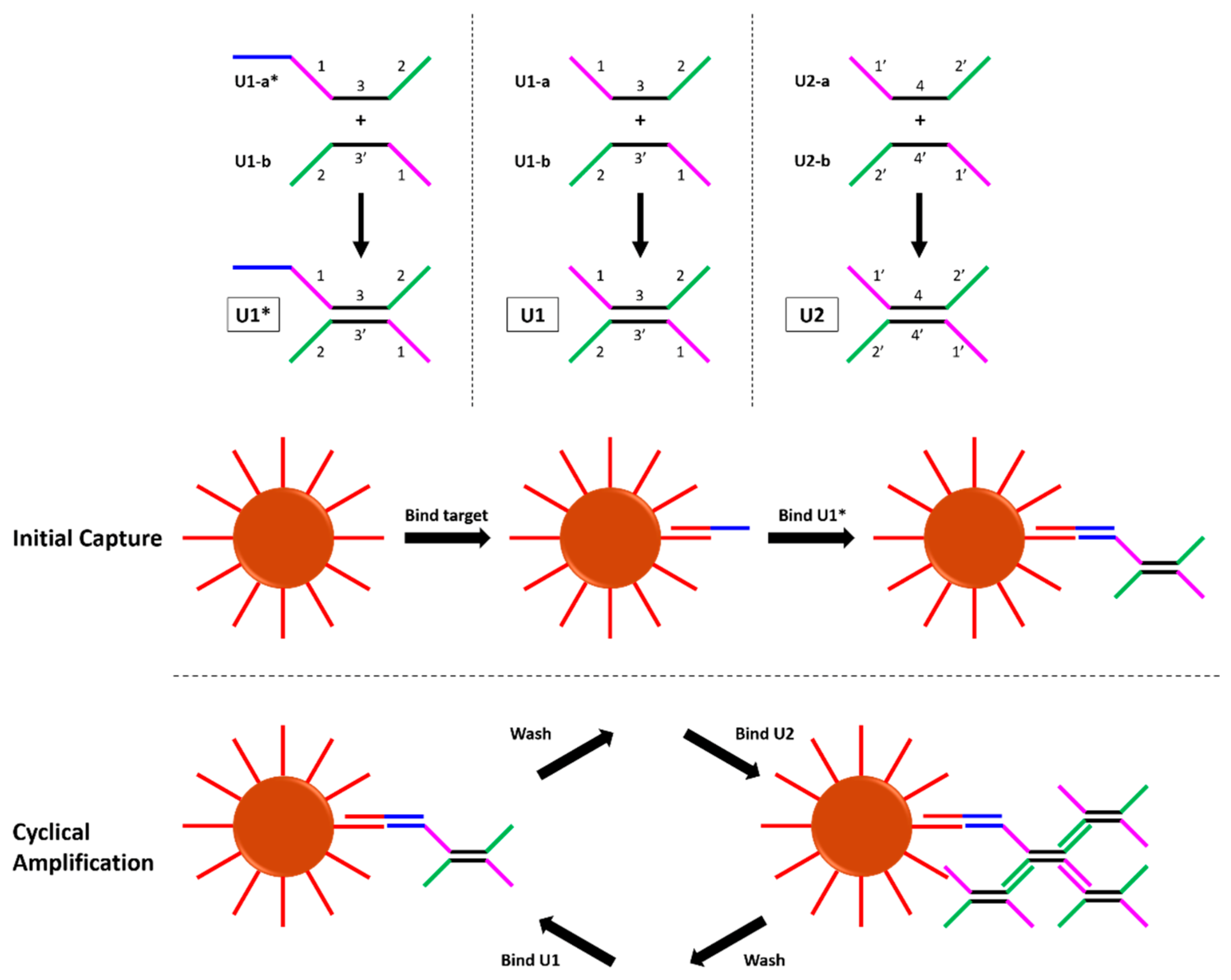
15]. Each DNA dumbbell, shown in
Figure 1, has four binding domains which bind the opposite dumbbell. The original authors performed the amplification assay by capturing target DNA on magnetic beads and sequentially incubating the beads with U1, then U2, then U1, etc. Over the course of 35 dumbbell incubations, a limit of detection of 5 copies/reaction was reported; this far surpasses limits of detection typical of other non-enzymatic NATs.
A serious drawback of this work which was not discussed is the amount of time, labor, and reagents required. Due to the single-stranded complementary binding domains, combining the two dumbbells in solution will cause uncontrolled binding to one another, rather than controlled accumulation on the surface of the magnetic beads. Therefore, the dumbbells must be added to the beads one at a time, with wash steps in between each incubation. To perform 35 successive dumbbell incubations, as well as the initial target capture step, approximately 18 h were required. Over the course of those 18 h, several pipetting steps were required every 30 min, each one a new opportunity for human error. These challenges are exacerbated by the lack of an internal negative control; a second parallel reaction was required to measure non-specific amplification, doubling the total amount of labor, time, and reagents required. These two obstacles, the undesirably high number of hands-on steps and the lack of an internal negative control, make the dumbbell DNA assay impractical for diagnostic applications. If the difficulties of performing this assay can be alleviated, it would have all the qualities of a powerful NAT appropriate for use in low-resource settings.
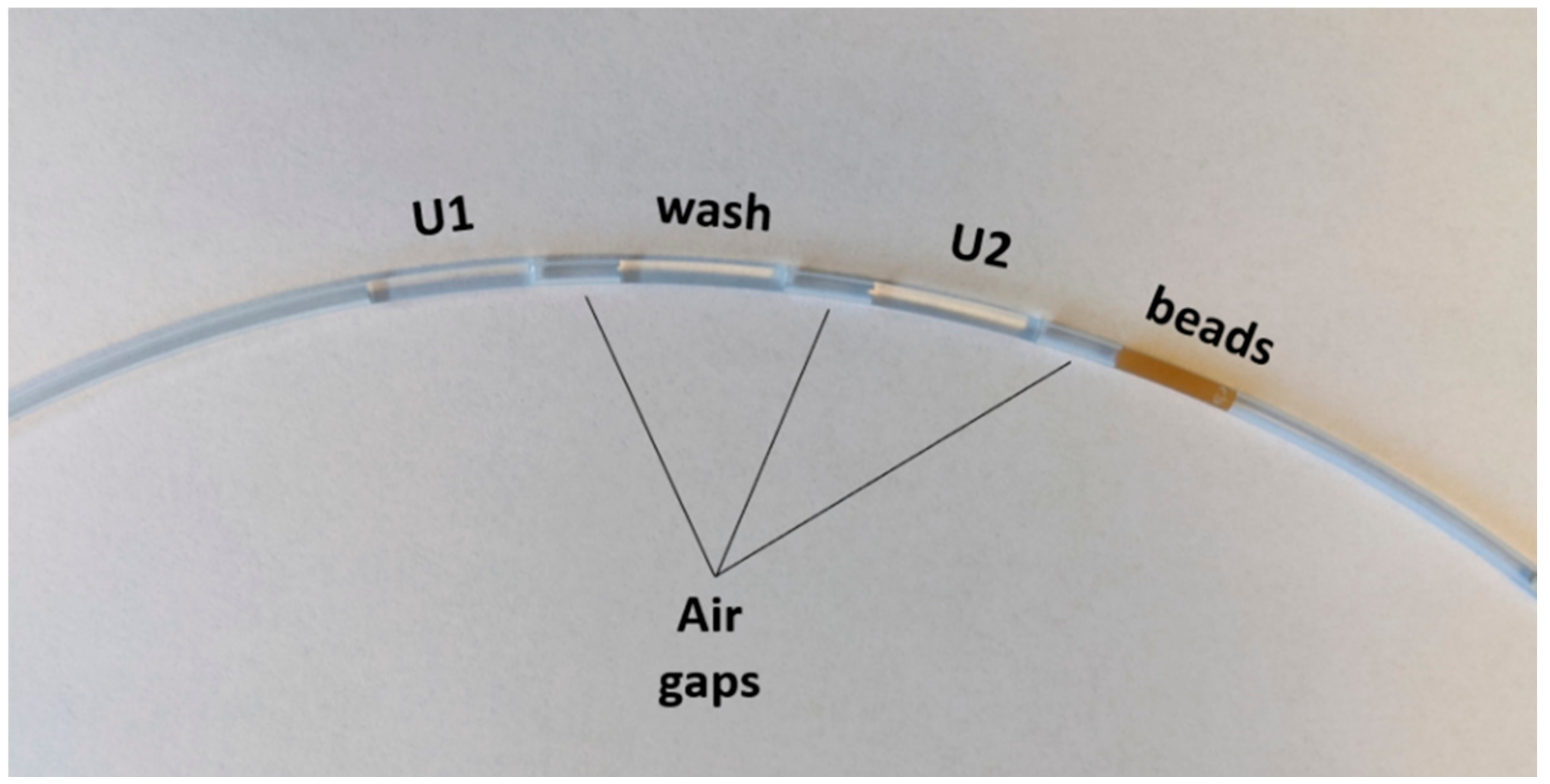
Previous works have pioneered the use of pre-arrayed reaction cassettes as a simplified and effective method of magnetic sample processing [
16,
17,
18]. In this approach, different solutions are loaded into microfluidic tubing and separated by air gaps. The surface tension of these solutions maintains the integrity of the air gaps, and sealing the ends of the tubing immobilizes the contents. Magnetic beads are then transported through the different solutions via movement of an external magnet. Inspired by these methods, we designed an automated reaction processor to perform the dumbbell amplification assay without the need for repeated human pipetting steps. After the initial target capture step, the magnetic beads are loaded into a reaction cassette along with each dumbbell, and automatic movement of the tubing relative to a stationary magnetic field performs the steps previously performed manually. Fluorescent labels on the dumbbells create a real-time optical readout that can be measured throughout the reaction.
To eliminate the need for a second control reaction, left-handed dumbbells with their own distinct fluorescent labels are included as a built-in negative control. Left-handed DNA (L-DNA) is the chiral enantiomer of right-handed DNA (D-DNA). Although only D-DNA is found in nature, advancements in chemical synthesis techniques have enabled the commercial production of synthetic L-DNA. L-DNA is identical in chemical composition, and exhibits identical solubility, thermodynamic properties, and binding behavior as its right-handed counterpart [
19,
20,
21]. Having previously demonstrated the utility of L-DNA as a built-in control for non-enzymatic DNA circuits [
10], we used them here as a measure of non-specific amplification in the dumbbell amplification assay.
Finally, as a proof of principle of the potential diagnostic applications for this automated, non-enzymatic DNA amplification reaction, we demonstrated the detection of
Schistosoma mansoni DNA.
S. mansoni is the most widespread member of the family of parasitic flatworms which cause schistosomiasis, a leading neglected tropical disease responsible for the loss of 4.5 million disability-adjusted life years (DALYs) [
22,
23,
24]. The target sequence is part of a 121 bp tandem repeat sequence which comprises approximately 12% of the
S. mansoni genome, and has previously been targeted with PCR assays [
25,
26,
27]. The PCR limit of detection was reported to be 1.28 pg/mL [
27]; given the highly-repeated nature of the target sequence in the genome, this corresponds to approximately 790,000 copies/mL of the target sequence.
2. Materials and Methods
2.1. Oligonucleotides
All D-DNA sequences were purchased from Integrated DNA Technologies (Skokie, IL, USA), and all L-DNA sequences were purchased from Biomers (Ulm, Germany). Each dumbbell component was modified with a 5′ FAM for D-DNA or a 5′ Texas Red (TXR) for L-DNA. All fluorescently-labeled oligonucleotides were HPLC purified. A complete list of DNA sequences is given in
Table 1, with distinct binding domains separated by underscores. Oligonucleotides were suspended in tris/EDTA (TE) buffer at a concentration of 100 µM and stored at −20 °C for long-term storage. For short-term storage, subsequent aliquots were created by diluting to 4 µM in 2X saline sodium citrate (SSC) buffer, and stored at 4 °C.
The dumbbell sequences (first 7 in
Table 1) were adapted from a previous work by Xu et al. [
15]. Sequences original to this work (last 5 in
Table 1) are those designed to detect
S. mansoni DNA, as well as ‘U1-a* tag’ and ‘U1* removal’, which were designed for a toehold-mediated strand displacement system to separate the dumbbells from the magnetic beads.
2.2. Design of the AutoPiLOT Reaction Processor
The automated Pi-powered looping oligonucleotide transporter (autoPiLOT) is an automated reaction processing device controlled by a Raspberry Pi B 3+ microcomputer. The Pi is connected via USB to two Arduino Unos, called the sensor Arduino and motor Arduino. Both Arduinos are connected to a 6 mm tactile button that, when pressed by the user, activates different segments of their onboard code. A circuit diagram is shown in
Supplementary Materials. The master code for the Raspberry Pi, as well as the onboard code for both Arduinos, are available upon request.
Two infrared reflective object sensors (Digi-Key, QRD1114) are connected to the sensor Arduino for detection of liquid/air interfaces. A stepper motor (Applied Motion Products, STR4 drive, HT23 motor) is connected to the motor Arduino, which turns gears controlling the movement of the microfluidic tubing. Fluorinated ethylene propylene (FEP) tubing—also commonly called PTFE tubing—with 1/8” outer diameter and 3/32” inner diameter (McMaster-Carr, 9369T24) was used to house the reaction contents. 5/8” × 5/8” × 1/4” neodymium magnets (K&J Magnetics Inc, BAA4) were used to control the magnetic beads. A Qiagen ESElog fluorometer was used for fluorescence measurements. Housing for the tubing, magnets, and fluorometer was printed using a MarkForged Mark 2 3D printer, and the 3D drawing is also available upon request.
The autoPiLOT is shown in
Figure 2. The housing was 3D printed to hold the magnets, IR sensors, fluorometer, and microfluidic tubing. Not pictured is the Raspberry Pi microcomputer and Arduinos which control the device. The low cost and portability of these components make this device easily transportable and independent of a laptop or other computer for operation. The microfluidic tubing is held between the gears, which are controlled by a stepper motor. Rotation of the gears therefore moves the microfluidic tubing which contains the various reaction fluids, as previously described for one-directional sample prep devices [
28,
29]. The reaction cassette was moved at a speed of 0.2 cm/s to transport the magnetic beads, and 7.5 cm/s to break the beads out of the magnetic field.
To account for variations in air gap size or fluid volume during the preparation of the reaction cassette, a pair of infrared sensor/receivers was interfaced with the system to detect the locations of the liquid/air interfaces in the tubing. Before performing the cyclical amplification reaction, a “reconnaissance run” is performed which detects changes in IR transmittance as the tubing moves from one end to the other. These changes in transmission indicate a change from liquid to air, and the locations and sizes of each fluid chamber are mapped using this information. Because it uses these measurements to guide the movement of the tubing, the autoPiLOT reaction is adaptable to variations in the pre-loaded reaction cassettes.
The movement of the magnetic beads between chambers is a result of the stationary magnetic field holding the beads as the tubing is moved by the gears. Tween 20 decreases the surface tension of the fluid chambers, which allows the beads to break through the interface. Several different concentrations of Tween were tested to see if they impacted the binding of the dumbbells to one another, and the results (shown in
Supplementary Materials) show that none inhibited binding. Since dumbbell binding appeared equally efficient for all tested buffer conditions, 0.05% Tween in 5X SSC buffer was used in accordance with the protocol for the original dumbbell assay [
15].
The movement of the magnetic beads through the four reaction chambers (shown in
Figure 3) is as follows: from their starting place in the beads chamber, they are carried into U2. Here, the beads are mixed and then removed from the magnetic field (via a fast movement speed) and then incubated for 30 min. The beads are then recollected in the magnetic field and carried into the wash chamber, where they are dispersed across the length of the chamber. The wash chamber is moved across the optical path of the fluorometer for fluorescence readings, and then the beads are transported into U1. Again, they are mixed and incubated to bind dumbbells. The beads are then moved in the reverse direction to U2, and the cycle begins again. Fluorescence measurements are collected in the wash chamber after dumbbell incubations number 1, 3, 5, etc. All reaction steps after the incubation with dumbbell U1* are therefore automated.
2.3. Gel Electrophoresis Studies
A series of experiments were conducted using agarose gel electrophoresis to investigate whether the predicted DNA hybridization events were taking place. Gels contained 3% ultra-pure agarose (Thermo Fisher) in 0.5X tris/borate/EDTA (TBE) buffer, and were stained with 1X GelRed nucleic acid stain (GoldBio, originally 10,000X in water). An ultra-low range DNA ladder (ThermoFisher) was included in each gel at a concentration of 2 ng/µL. Gels were run at 60V for approximately 1 h, then imaged using a Bio-Rad Gel Doc EZ Imager.
2.4. Dumbbell Formation
To form the double-stranded dumbbell structures, the two oligonucleotides (for example, U1-a and U1-b form dumbbell U1) were combined at equal molar ratios to the desired final concentration and then heated with the following thermal profile: 95 °C for 5 min, followed by 50 °C for 10 min, and finally by 37 °C for 10 min. This process was performed in a Qiagen Rotor-Gene PCR thermal cycler. The dumbbells were then stored at room temperature until use. For experiments using both D-DNA and L-DNA dumbbells, both D-DNA and L-DNA were combined in the same tube. For example, D-DNA U1-a and U1-b were combined with L-DNA U1-a and U1-b in the same tube for a final solution of right- and left-handed U1.
2.5. Magnetic Bead Functionalization
Dynabeads MyOne Streptavidin T1 magnetic beads (ThermoFisher) were functionalized with a biotinylated capture probe designed to bind the DNA target of interest. First, the beads were washed three times using hybridization buffer (5X SSC buffer + 0.05% Tween 20). Each wash consists of magnetically separating the beads from solution and, while separated, removing the supernatant and adding fresh buffer. After washing, the beads were resuspended in the biotinylated capture probe at a ratio of 2.5 nanomoles DNA per mg beads, briefly vortexed, and incubated on a rotisserie for 20 min. The beads were then washed three times with hybridization buffer and resuspended in blocking buffer (1X PBS + 16 µM free biotin + 0.05% Tween 20) and incubated on the rotisserie for 30 min. Experiments investigating other blocking methods used blocking buffers in which the free biotin was replaced with 2% bovine serum albumin (BSA) by volume, 200 µg/mL salmon sperm DNA, or removed altogether. The beads were then washed three times with hybridization buffer and stored in stocking buffer (1X PBS + 0.05% Tween 20) at a final concentration of 1 mg/mL. D-Biotin and BSA were purchased from Thermo Fisher, and Tween 20 was purchased from Sigma Aldrich.
2.6. Dumbbell Amplification Reactions
An amount of 50 µg of functionalized magnetic beads was used in each reaction. The beads were washed three times in hybridization buffer and resuspended in 100 µL of sample solution. The solution was vortexed to disperse the beads throughout and incubated on a rotating rotisserie. Sequence-specific nucleic acid capture via functionalized magnetic beads has previously been shown to be a slow process, with low efficiency for incubation times shorter than 30 min [
17]. Therefore, 30 min was chosen as the incubation time. Next, 10 µL of U1* at 1 µM was added, and the beads incubated for another 30 min on the rotisserie. The beads were then washed three times in hybridization buffer, and resuspended in 75 µL hybridization buffer. At this point, the functionalized beads have bound target, which has in turn bound the first dumbbell U1*. Incubations in dumbbells U1 and U2 were then performed automatically using the autoPiLOT reaction processor.
The microfluidic FEP tubing was loaded with the following fluid chambers, each separated by a small air gap approximately 1 cm in length: 100 µL U1 at 250 nM, 100 µL hybridization buffer, 100 µL U2 at 250 nM, 100 µL magnetic bead solution. To load a fluid chamber into the tubing, the liquid was pipetted directly into the tube with the pipette tip flush against the opening. The tubing was then gently tipped to move the fluid chamber further along, clearing room for the next solution to be inserted. After the four fluid chambers were loaded in the tubing, both ends were sealed with Cha-seal tube sealing compound. The loaded tube (shown in
Figure 3) was run on the autoPiLOT to move the magnetic beads back and forth between the two dumbbell solutions, reading fluorescence on the beads in the wash chamber after every other dumbbell incubation. The total reaction time was a function of the number of dumbbell incubations; 15 incubations on the autoPiLOT took approximately 8 h (15 incubations × 0.5 h/incubation, plus the time to move the beads back and forth between chambers).
4. Discussion
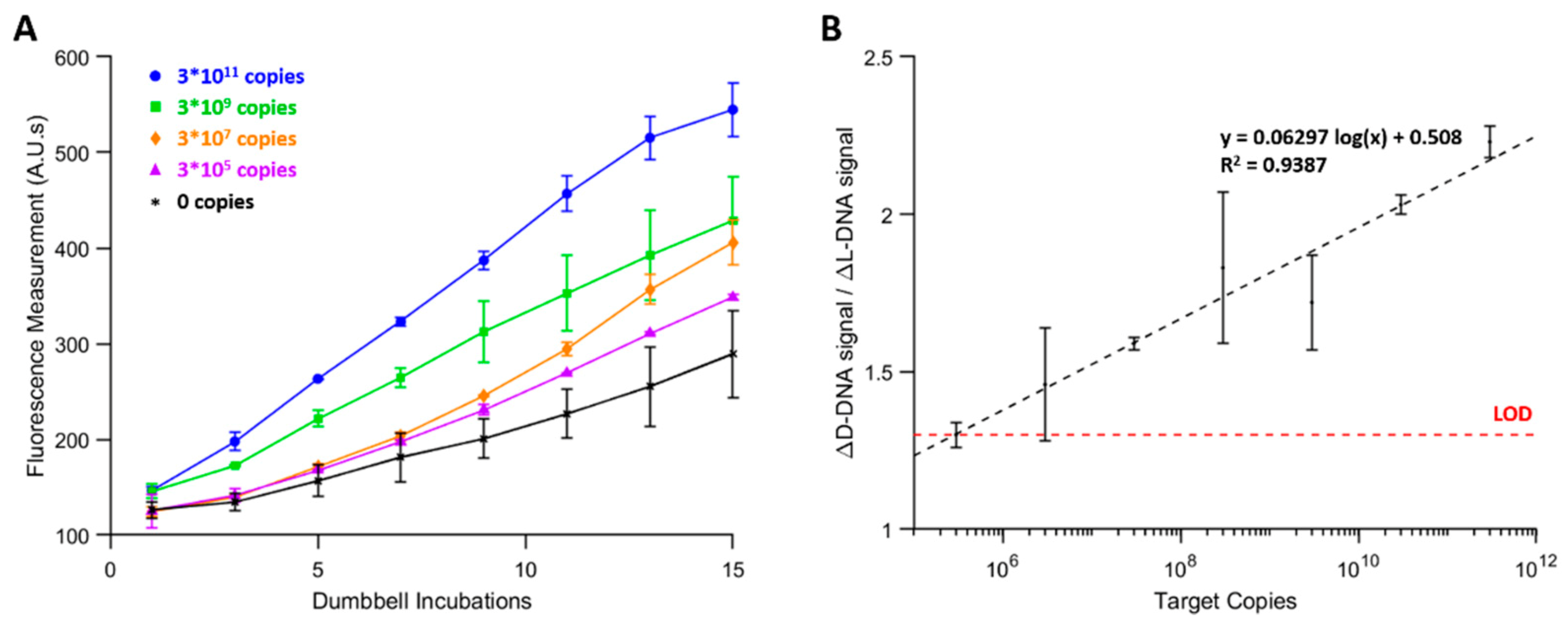
Real-time fluorescence measurements were shown to reflect the amount of DNA which had accumulated on the magnetic beads. Signal amplification was approximately linear, and the slope increased with increasing amounts of target DNA in the sample, as shown in
Figure 7. The autoPiLOT assay exhibited a limit of detection of 5 fM, and showed promising results when applied to the detection of
S. mansoni DNA, demonstrating the potential for a sensitive, low-cost, point-of-care non-enzymatic NAT.
The observed linear amplification is not in agreement with the previously-reported exponential dumbbell DNA amplification. This discrepancy is likely due to a combination of factors. When the beads are transported into a new chamber in the autoPiLOT, they are briefly mixed back and forth to disperse them across the length of the chamber. Despite this, the beads quickly settle to the bottom of the tubing where they remain for most of the 30 min. Dumbbell binding efficiency would likely improve if the beads were homogenously dispersed throughout the chamber; the effective exposed surface area of the beads would be much larger. Another potential cause of decreased amplification is steric hinderance from the surrounding dumbbells. The more dumbbells are attached to the bead surface, the more chaotic and intertwined the growing network of dumbbells becomes. This would result in amplification which becomes less efficient the more dumbbells are bound; this behavior is suggested in the amplification curves shown in
Figure 8. At lower fluorescence values (in the 100–250 A.U. range), the amplification has some concavity indicative of exponential amplification. This is most visible for the 3 × 10
5 and 3 × 10
7 copy number curves. As the reaction progresses and more dumbbells attach to the beads, the curve becomes linear, and even begins to taper off at high enough values. Future work should focus on optimization of the autoPiLOT magnetic bead control strategies, in an effort to more efficiently bind dumbbells and achieve exponential amplification.
Future research should also work toward the implementation of a lower-cost fluorescence imaging system. The fluorometer used in the autoPiLOT is the only expensive component, with a price tag of several thousand dollars. Recently, smartphone-based fluorescence microscopes have grown in popularity and sophistication, bringing high quality digital cameras and multifunctionality to the point of care [
31,
32]. It is estimated that approximately 80% of the world population uses smartphones, making a smartphone optical readout highly applicable, even in low-resource settings [
33].
An alternative strategy to avoid use of a costly fluorometer would be a colorimetric readout. The original demonstration of the dumbbell amplification assay utilized a colorimetric readout, in which dumbbells were labeled with biotin, and avidin-labeled horseradish peroxidase (HRP) was added to the beads at the end of the end of the reaction [
15]. The drawback of this strategy is that it prohibits the use of two distinct readouts, one for D-DNA and one for L-DNA. The use of a dual-chirality design allowed for the monitoring of not only target signal, but also non-specific amplification in each reaction. Switching to a colorimetric readout simplifies the autoPiLOT components, but sacrifices the innovative dual-chirality functionality.
Reaction automation in the autoPiLOT removes the need for many hands-on pipetting steps, but does not by default decrease the total reaction time. Future research may also investigate shortening the overall autoPiLOT reaction time. We have shown (in
Supplementary Materials) that the incubation period in each dumbbell can be greatly reduced while still observing signal amplification. The exact effect on signal-to-noise ratio, however, remains unknown. Further testing of several incubation times to determine a balance between reaction time and sensitivity would be useful for diagnostic applications in which time is limited. Additionally, a more flexible design in which the reaction runs only as long as necessary could be developed; samples with higher target concentrations would amplify quicker, terminating the reaction quicker than samples with less target.
Finally, the performance of the autoPiLOT diagnostic platform should be evaluated using real urine samples. The preliminary data presented here suggest that the autoPiLOT is an effective strategy to detect
S. mansoni DNA, which has previously been detected in urine samples via PCR. The autoPiLOT demonstrated a limit of detection of 3,000,000/mL target copies, nearly as sensitive as previously described PCR assays. It is possible that reaction sensitivity will decrease when applied to DNA extracted from urine, since urine is known to contain inhibitors such as nucleases. In the case of nucleases, heating the urine to 75 °C for 10 min has been shown to deactivate nucleases in urine [
10]. Sample-prep steps such as heating are likely required to maximize extraction efficiency from urine. Losses due to extraction efficiency may be countered by increasing sample volume; large volumes are easily obtained. These characteristics suggest that the autoPiLOT has the potential to be a highly-sensitive, low-resource, non-enzymatic NAT, and the first of its kind.
5. Conclusions
The dumbbell DNA amplification scheme has previously been shown to be a highly-sensitive, non-enzymatic method of detecting target DNA. The largest obstacles to its use in diagnostic applications were the extremely high number of manual pipetting steps and hands-on time required, and the need for an additional control reaction. The autoPiLOT platform has overcome these obstacles through automation of the reaction in a self-contained, microfluidic reaction cassette. The components are primarily 3D-printed parts and low-cost electronics, making the autoPiLOT a low-resource compatible diagnostic platform.
The autoPiLOT assay also demonstrates the utility of left-handed L-DNA as an internal control. Whereas a traditional assay format would require the performance of two parallel reactions, one with and one without target, for interpretation of results, the dual-chirality design used here discretely measured both specific and non-specific signals in the same reaction. In addition to saving time and reagents, this opens the door to testing unpurified biological samples which may exhibit varying rates of non-specific amplification that cannot be accurately simulated with a parallel control reaction.


 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}