A Novel Electroporation System for Living Cell Staining and Membrane Dynamics Interrogation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Electroporation
1.2. Focal Adhesion
1.3. Cell Motility
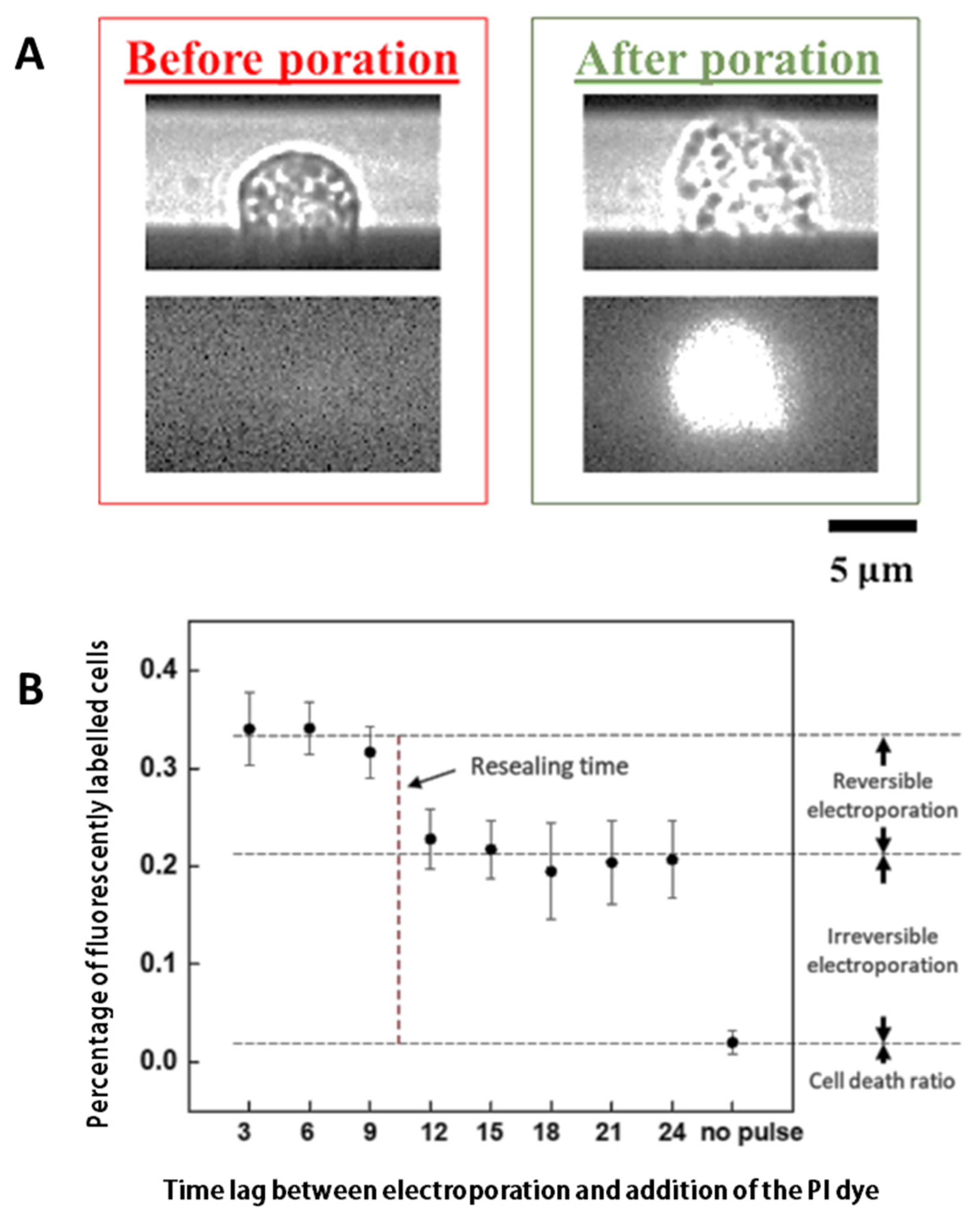
1.4. Membrane Resealing
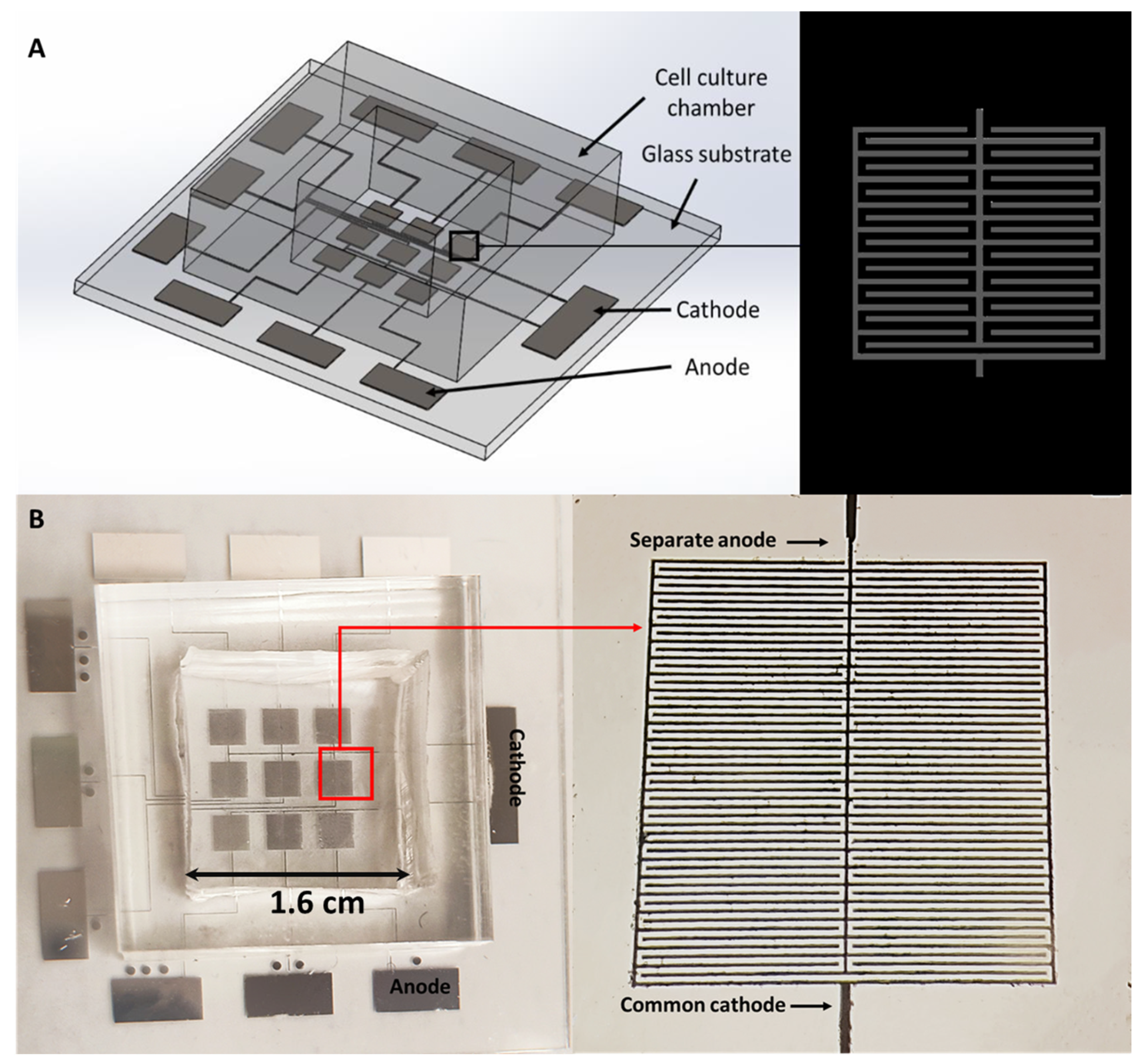
1.5. Electroporation Chip
2. Material and Method
2.1. Fabrication of the Microelectrodes on a Glass Substrate
2.2. Program-Controlled Pulse Generator
2.3. Cell Culture
2.4. Plasmid Extraction
3. Results and Discussion
3.1. Characterizing the Resealing of Membrane Pores Induced by Electroporation
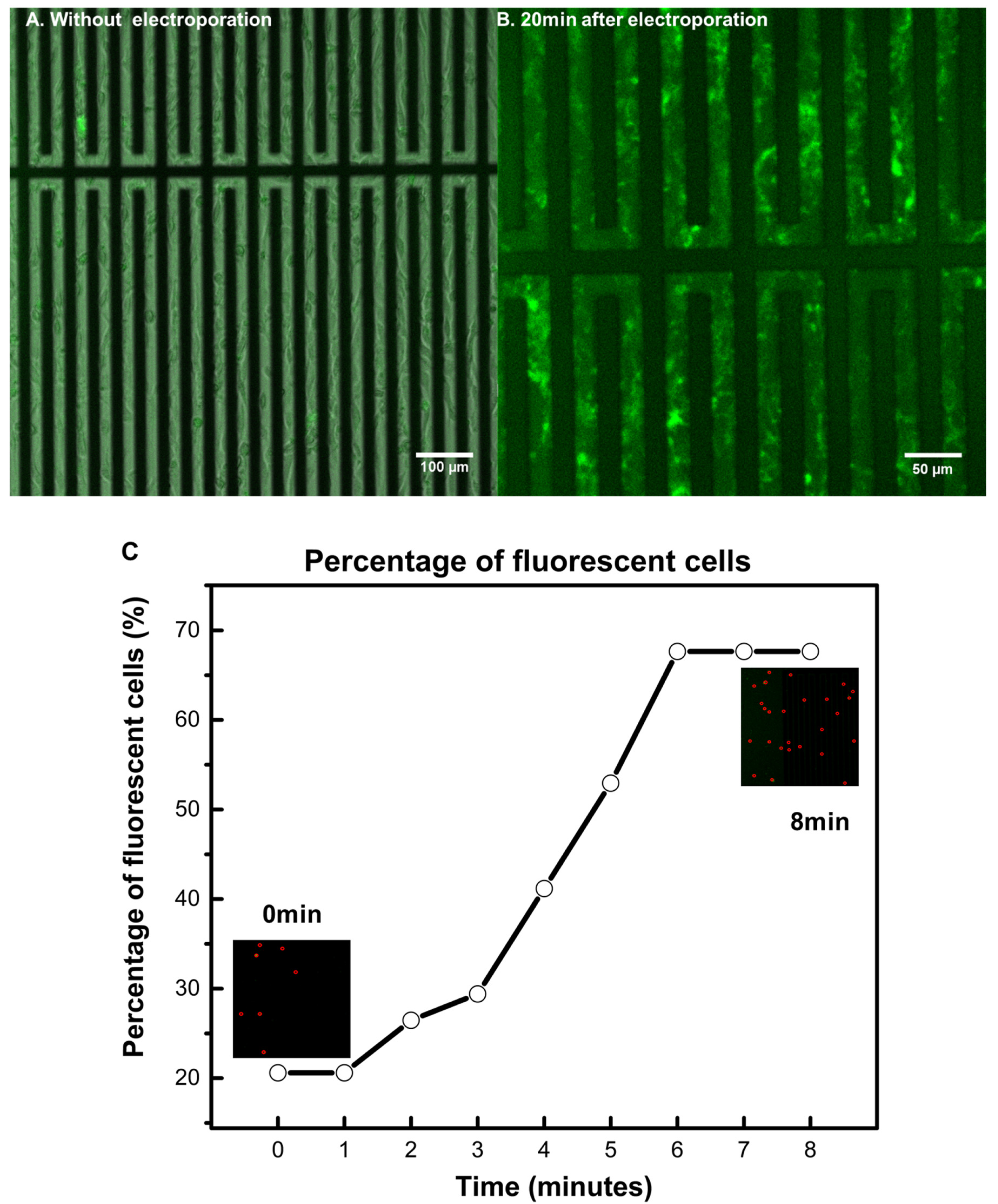
3.2. Achieving Live Cell F-Actin Staining
3.3. Achieving Fast Electrotransfection
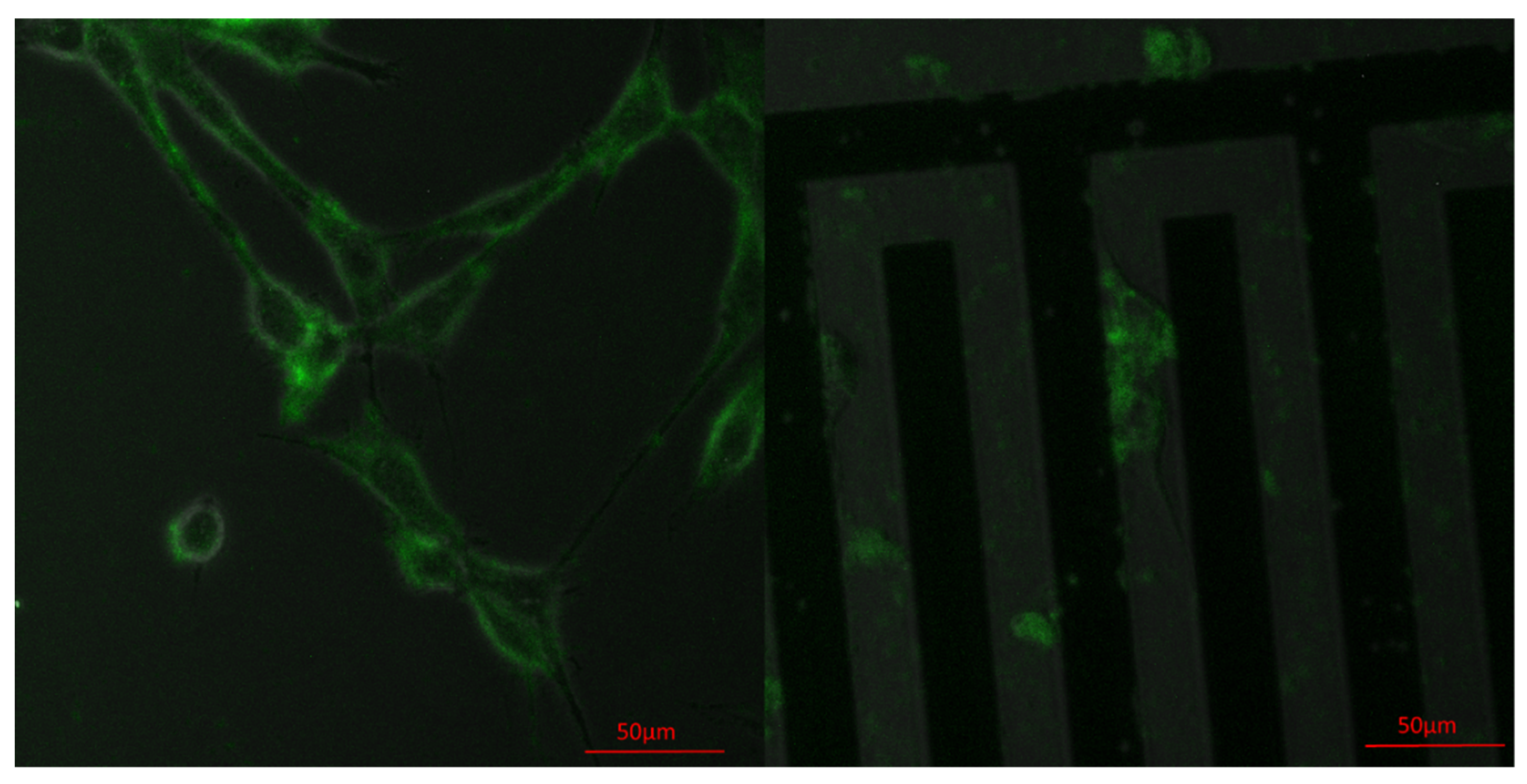
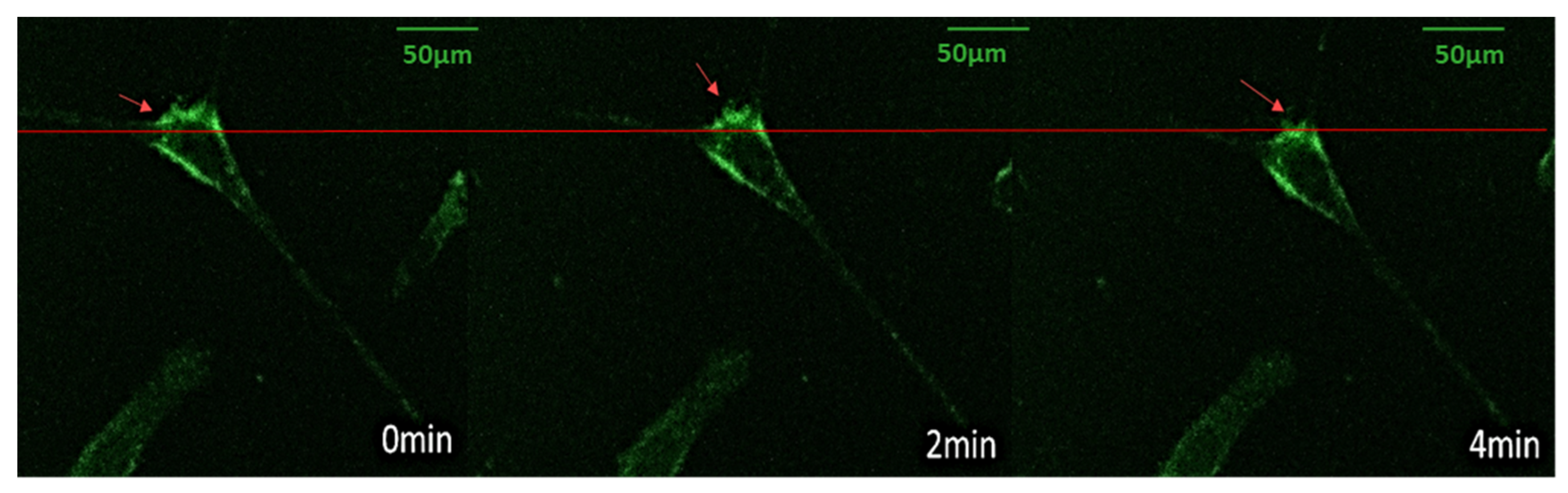
3.4. Vinculin Dynamics during Cell Migration
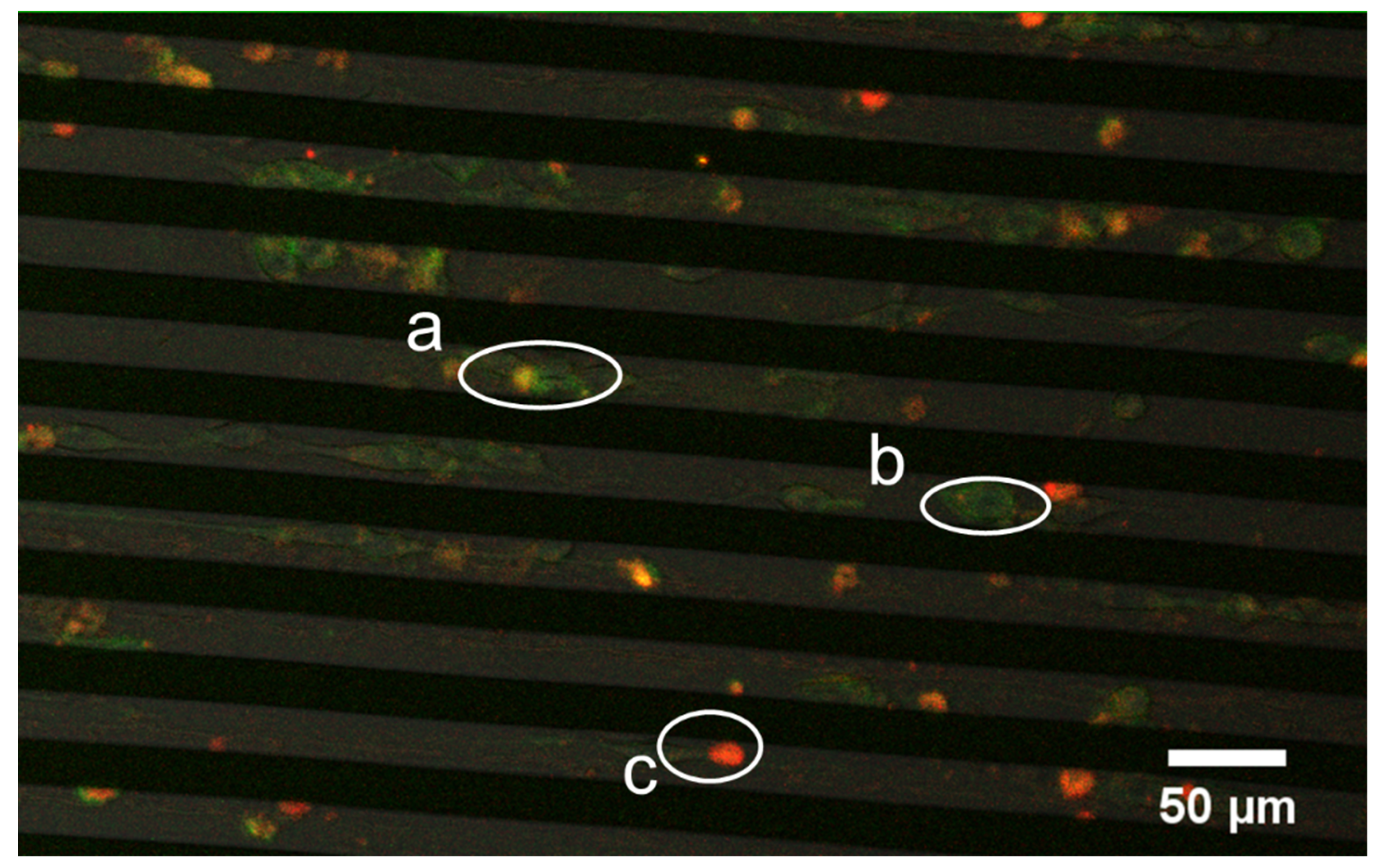
3.5. Successive Labelling of Vinculin and F-Actin
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Rubinsky, B.; Onik, G.; Mikus, P. Irreversible electroporation: A new ablation modality—Clinical implications. Technol. Cancer Res. Treat. 2007, 6, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Dermol-Černe, J.; Batista Napotnik, T.; Reberšek, M.; Miklavčič, D. Short microsecond pulses achieve homogeneous electroporation of elongated biological cells irrespective of their orientation in electric field. Sci. Rep. 2020, 10, 9149. [Google Scholar] [CrossRef] [PubMed]
- Yarmush, M.L.; Golberg, A.; Sersa, G.; Kotnik, T.; Miklavcic, D. Electroporation-Based Technologies for Medicine: Principles, Applications, and Challenges. Ann. Rev. Biomed. Eng. 2014, 16, 295–320. [Google Scholar] [CrossRef] [PubMed]
- Heller, R.; Heller, L.C. Gene electrotransfer clinical trials. Adv. Genet. 2015, 89, 235–262. [Google Scholar] [PubMed]
- Kotnik, T.; Frey, W.; Sack, M.; Meglic, S.H.; Peterka, M.; Miklavcic, D. Electroporation-based applications in biotechnology. Trends Biotechnol. 2015, 33, 480–488. [Google Scholar] [CrossRef]
- Hui, T.H.; Zhou, Z.L.; Fong, H.W.; Ngan, R.K.C.; Lee, T.Y.; Au, J.S.K.; Ngan, A.H.W.; Yip, T.T.C.; Lin, Y. Characterizing the malignancy and drug resistance of cancer cells from their membrane resealing response. Sci. Rep. 2016, 6, 26692. [Google Scholar] [CrossRef]
- Yan, Z.; Hui, T.H.; Fong, H.W.; Shao, X.; Cho, W.C.; Ngan, K.C.; Yip, T.C.; Lin, Y. An electroporation platform for Erlotinib resistance screening in living non-small cell lung cancer (NSCLC) cells. Biomed. Phys. Eng. Express 2018, 4, 6. [Google Scholar] [CrossRef]
- Li, Z.; Li, Z.; Li, S.; Wang, K.; Ma, F.; Tang, B. Potential application development of Sr/HCOOH metal organic framework in osteoarthritis. Microporous Mesoporous Mater. 2020, 294, 109835. [Google Scholar] [CrossRef]
- Guo, X.; Zhu, R. Controllable In-Situ cell electroporation with cell positioning and impedance monitoring using micro electrode array. Sci. Rep. 2016, 6, 31392. [Google Scholar] [CrossRef]
- Cheng, Z.; Kuru, E.; Sachdeva, A.; Vendrell, M. Fluorescent amino acids as versatile building blocks for chemical biology. Nat. Rev. Chem. 2020, 4, 275–290. [Google Scholar] [CrossRef]
- Wu, R.G.; Yang, C.S.; Cheing, C.C.; Tseng, F.G. Nanocapillary electrophoretic electrochemical chip: Towards analysis of biochemicals released by single cells. Interface Focus 2011, 1, 744–753. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Fu, A.; Yossifon, G. Active particles as mobile microelectrodes for selective bacteria electroporation and transport. Sci. Adv. 2020, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.D. Chapter One—The Dynamic Actin Cytoskeleton in Smooth Muscle. In Advances in Pharmacology; Khalil, R.A., Ed.; Academic Press: Cambridge, MA, USA, 2018; Volume 81, pp. 1–38. [Google Scholar]
- Haase, K.; Al-Rekabi, Z.; Pelling, A.E. Chapter Five—Mechanical Cues Direct Focal Adhesion Dynamics. In Progress in Molecular Biology and Translational Science; Engler, A.J., Kumar, S., Eds.; Academic Press: Cambridge, MA, USA, 2014; Volume 126, pp. 103–134. [Google Scholar]
- Plotnikov, V.; Pasapera, A.M.; Sabass, B.; Waterman, C.M. Force Fluctuations within Focal Adhesions Mediate ECM-Rigidity Sensing to Guide Directed Cell Migration. Cell 2012, 115, 1513–1527. [Google Scholar] [CrossRef] [PubMed]
- Case, L.B.; Baird, M.A.; Shtengel, G.; Campbell, S.L.; Hess, H.F.; Davidson, M.W.; Waterman, C.M. Molecular mechanism of vinculin activation and nanoscale spatial organization in focal adhesions. Nat. Cell Biol. 2015, 17, 880–892. [Google Scholar] [CrossRef]
- Goldmann, W.H. Role of vinculin in cellular mechanotransduction. Cell Biol. Int. 2016, 40, 241–256. [Google Scholar] [CrossRef]
- Padhi, A.; Singh, K.; Franco-Barraza, J.; Marston, D.J.; Cukierman, E.; Hahn, K.M.; Kapania, R.K.; Nain, A.S. Force-exerting perpendicular lateral protrusions in fibroblastic cell contraction. Commun. Biol. 2020, 3, 390. [Google Scholar] [CrossRef]
- Pollard, T.D.; Borisy, G.G. Cellular motility driven by assembly and disassembly of actin filaments. Cell 2003, 112, 453–465. [Google Scholar] [CrossRef]
- Yang, L.; Gong, Z.; Lin, Y.; Chinthapenta, V.; Li, Q.; Webster, T.J.; Sheldon, B.W. Disordered Topography Mediates Filopodial Extension and Morphology of Cells on Stiff Materials. Adv. Funct. Mater. 2017, 27, 1702689. [Google Scholar] [CrossRef]
- Stuelten, C.H.; Parent, C.A.; Montell, D.J. Cell motility in cancer invasion and metastasis: Insights from simple model organisms. Nat. Rev. Cancer 2018, 18, 296–312. [Google Scholar] [CrossRef]
- Cheng, F.; Eriksson, J.E. Intermediate Filaments and the Regulation of Cell Motility during Regeneration and Wound Healing. Cold Spring Harb. Perspect. Biol. 2017, 9, 14. [Google Scholar] [CrossRef]
- Lin, Y.; Shenoy, V.B.; Hu, B.; Bai, L. A microscopic formulation for the actin-driven motion of listeria in curved paths. Biophys. J. 2010, 99, 1043–1052. [Google Scholar] [CrossRef]
- Aranjuez, G.; Burtscher, A.; Sawant, K.; Majumder, P.; McDonald, J.A. Dynamic myosin activation promotes collective morphology and migration by locally balancing oppositional forces from surrounding tissue. Mol. Biol. Cell 2016, 27, 1898–1910. [Google Scholar] [CrossRef] [PubMed]
- Pandya, P.; Orgaz, J.L.; Sanz-Moreno, V. Modes of invasion during tumour dissemination. Mol. Oncol. 2017, 11, 5–27. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y. A model of cell motility leading to biphasic dependence of transport speed on adhesive strength. J. Mech. Phys. Solids 2010, 58, 502–514. [Google Scholar] [CrossRef]
- Blazek, A.D.; Paleo, B.J.; Weisleder, N. Plasma Membrane Repair: A Central Process for Maintaining Cellular Homeostasis. Physiology 2015, 30, 438–448. [Google Scholar] [CrossRef] [PubMed]
- Kotnik, T.; Rems, L.; Tarek, M.; Miklavčič, D. Membrane Electroporation and Electropermeabilization: Mechanisms and Models. Annu. Rev. Biophys. 2019, 48, 63–91. [Google Scholar] [CrossRef]
- Li, X.L.; Li, G.H.; Fu, J.; Fu, Y.W.; Zhang, L.; Chen, W.; Arakaki, C.; Zhang, J.P.; Wen, W.; Zhao, M.; et al. Highly efficient genome editing via CRISPR-Cas9 in human pluripotent stem cells is achieved by transient BCL-XL overexpression. Nucleic Acids Res. 2018, 46, 10195–10215. [Google Scholar] [CrossRef]
- Nematollahi, M.H.; Torkzadeh-Mahanai, M.; Pardakhty, A.; Ebrahimi Meimand, H.A.; Asadikaram, G. Ternary complex of plasmid DNA with NLS-Mu-Mu protein and cationic niosome for biocompatible and efficient gene delivery: A comparative study with protamine and lipofectamine. Artif. Cells Nanomed. Biotechnol. 2018, 46, 1781–1791. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Y.; Yan, Z.; Xia, X.; Lin, Y. A Novel Electroporation System for Living Cell Staining and Membrane Dynamics Interrogation. Micromachines 2020, 11, 767. https://doi.org/10.3390/mi11080767
Zhang Y, Yan Z, Xia X, Lin Y. A Novel Electroporation System for Living Cell Staining and Membrane Dynamics Interrogation. Micromachines. 2020; 11(8):767. https://doi.org/10.3390/mi11080767
Chicago/Turabian StyleZhang, Yuanjun, Zishen Yan, Xingyu Xia, and Yuan Lin. 2020. "A Novel Electroporation System for Living Cell Staining and Membrane Dynamics Interrogation" Micromachines 11, no. 8: 767. https://doi.org/10.3390/mi11080767
APA StyleZhang, Y., Yan, Z., Xia, X., & Lin, Y. (2020). A Novel Electroporation System for Living Cell Staining and Membrane Dynamics Interrogation. Micromachines, 11(8), 767. https://doi.org/10.3390/mi11080767