Core-Shell Beads as Microreactors for Phylogrouping of E. coli Strains

 ,
, 
 ,
,  ,
,  ,
, 
Abstract
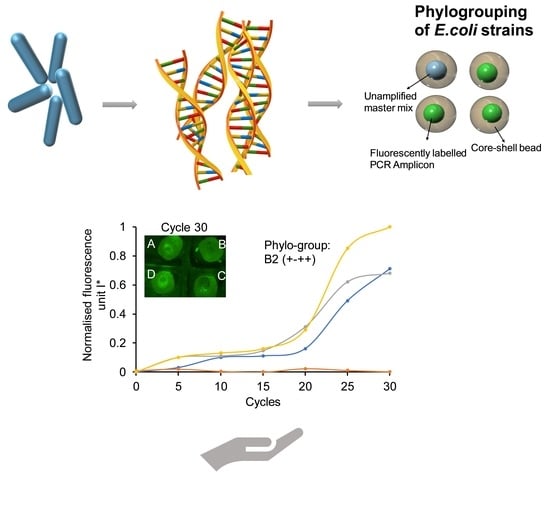
1. Introduction
2. Materials and Methods
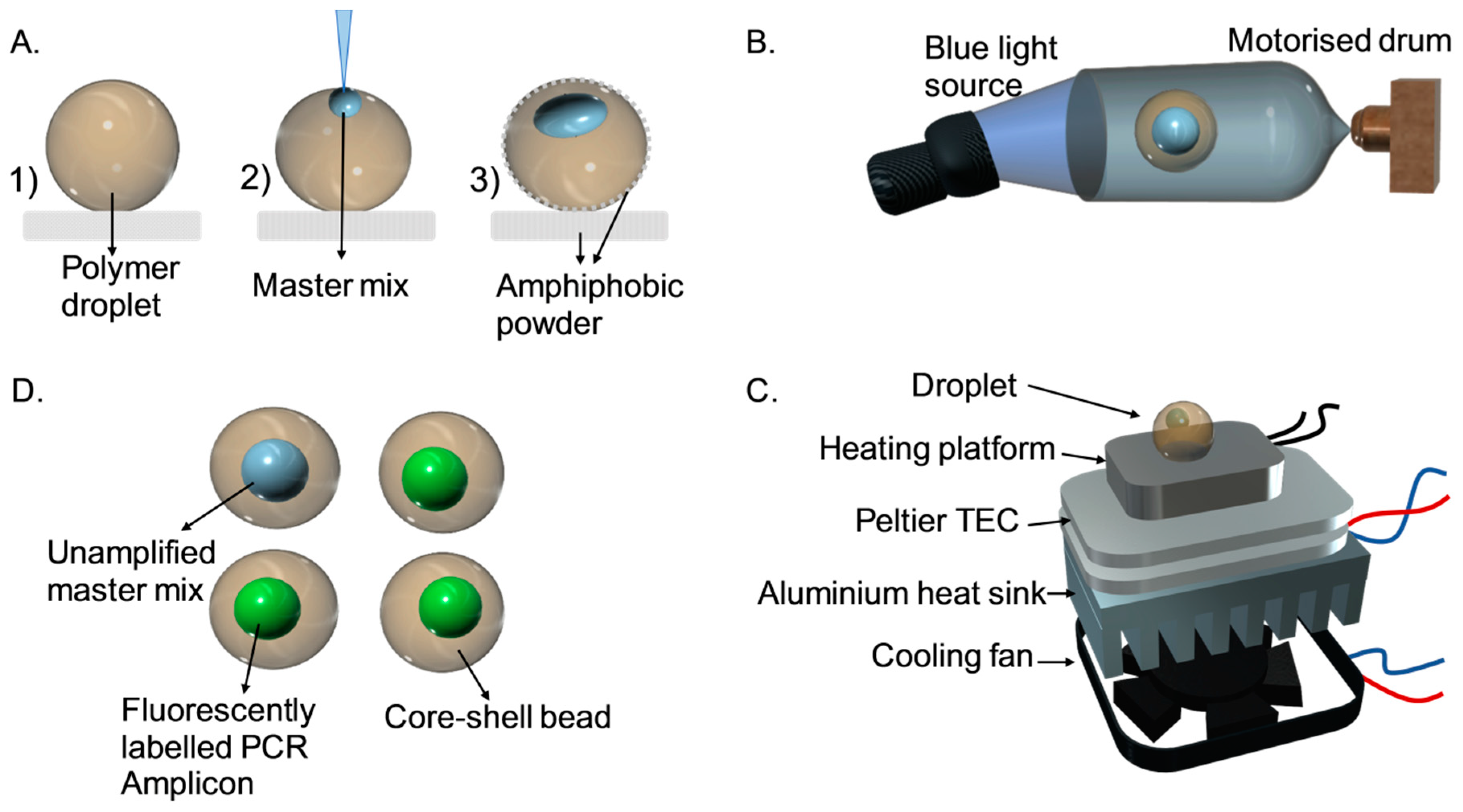
2.1. Fabrication of the Core-Shell Bead
2.2. Thermal Cycler
2.3. DNA Amplification
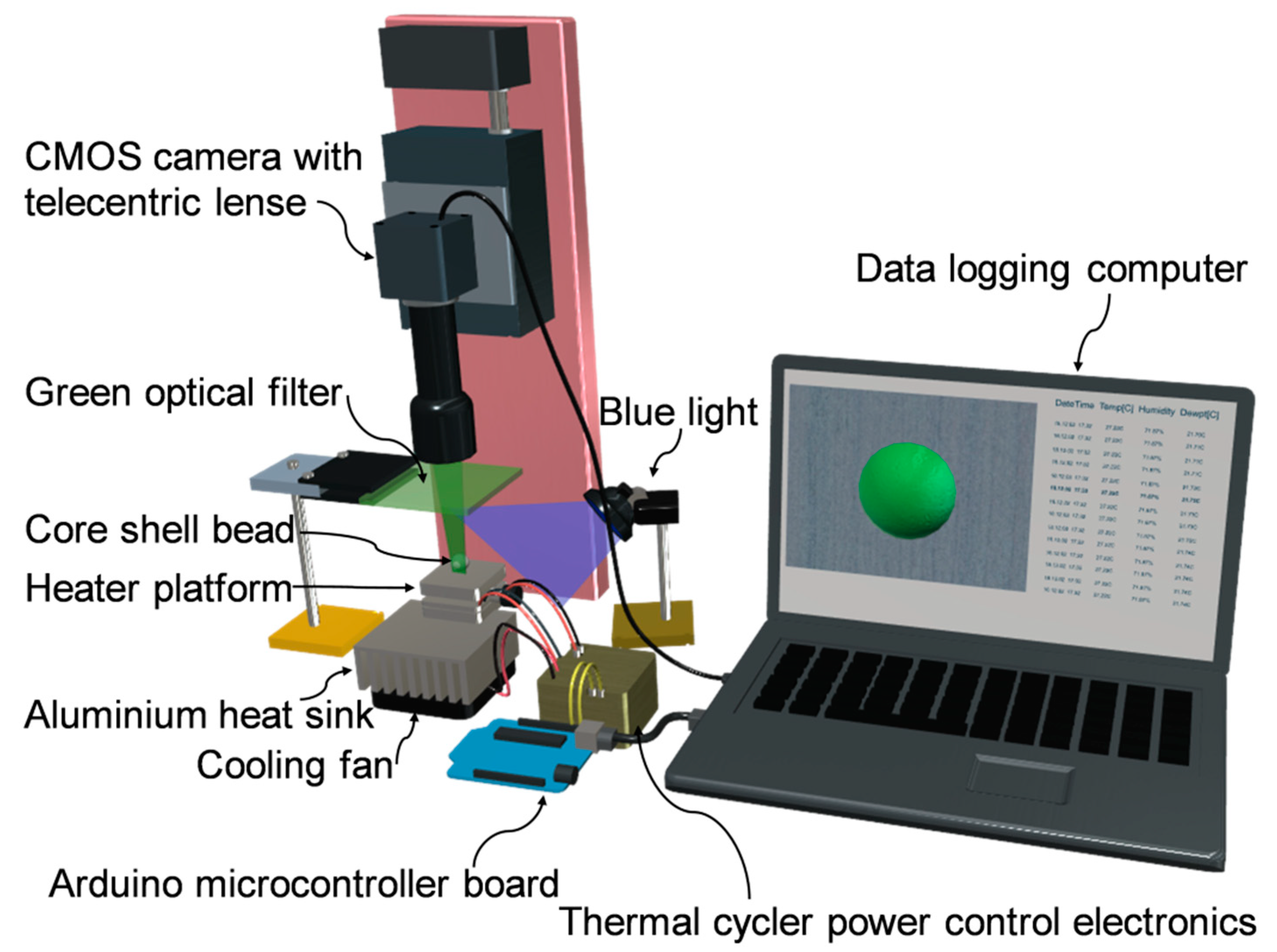
2.4. Optical Detection
3. Results and Discussion
3.1. Assay Principle
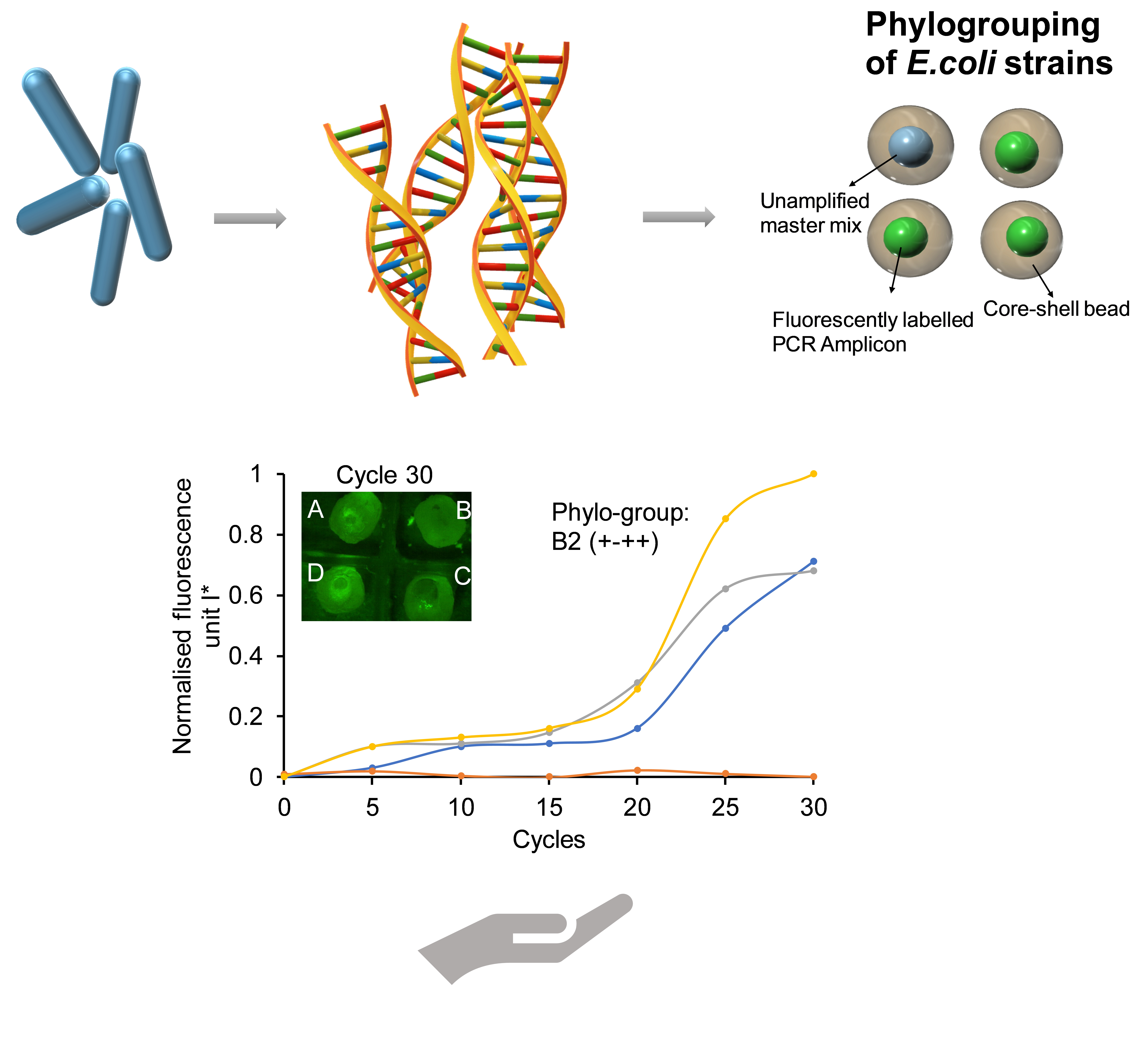
3.2. Phylogrouping Using Core Shell Beads
3.3. Validation of Phylogrouping by Gel Electrophoresis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chen, S.L.; Wu, M.; Henderson, J.P.; Hooton, T.M.; Hibbing, M.E.; Hultgren, S.J.; Gordon, J.I. Genomic diversity and fitness of E. coli strains recovered from the intestinal and urinary tracts of women with recurrent urinary tract infection. Sci. Transl. Med. 2013, 5, 184ra160. [Google Scholar] [CrossRef] [PubMed]
- Phillips-Houlbracq, M.; Ricard, J.-D.; Foucrier, A.; Yoder-Himes, D.; Gaudry, S.; Bex, J.; Messika, J.; Margetis, D.; Chatel, J.; Dobrindt, U. Pathophysiology of Escherichia coli pneumonia: Respective contribution of pathogenicity islands to virulence. Int. J. Med. Microbiol. 2018, 308, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Herzer, P.J.; Inouye, S.; Inouye, M.; Whittam, T.S. Phylogenetic distribution of branched RNA-linked multicopy single-stranded DNA among natural isolates of Escherichia coli. J. Bacteriol. 1990, 172, 6175–6181. [Google Scholar] [CrossRef] [PubMed]
- Clermont, O.; Bonacorsi, S.; Bingen, E. Rapid and simple determination of the Escherichia coli phylogenetic group. Appl. Environ. Microbiol. 2000, 66, 4555–4558. [Google Scholar] [CrossRef]
- Bingen, E.; Picard, B.; Brahimi, N.; Mathy, S.; Desjardins, P.; Elion, J.; Denamur, E. Phylogenetic analysis of Escherichia coli strains causing neonatal meningitis suggests horizontal gene transfer from a predominant pool of highly virulent B2 group strains. J. Infect. Dis. 1998, 177, 642–650. [Google Scholar] [CrossRef]
- Picard, B.; Garcia, J.S.; Gouriou, S.; Duriez, P.; Brahimi, N.; Bingen, E.; Elion, J.; Denamur, E. The link between phylogeny and virulence in Escherichia coli extraintestinal infection. Infect. Immun. 1999, 67, 546–553. [Google Scholar] [CrossRef]
- Duriez, P.; Clermont, O.; Bonacorsi, S.; Bingen, E.; Chaventré, A.; Elion, J.; Picard, B.; Denamur, E. Commensal Escherichia coli isolates are phylogenetically distributed among geographically distinct human populations. Microbiology 2001, 147, 1671–1676. [Google Scholar] [CrossRef]
- Gordon, D.M.; Cowling, A. The distribution and genetic structure of Escherichia coli in Australian vertebrates: Host and geographic effects. Microbiology 2003, 149, 3575–3586. [Google Scholar] [CrossRef]
- Escobar-Páramo, P.; Le Menac’H, A.; Le Gall, T.; Amorin, C.; Gouriou, S.; Picard, B.; Skurnik, D.; Denamur, E. Identification of forces shaping the commensal Escherichia coli genetic structure by comparing animal and human isolates. Environ. Microbiol. 2006, 8, 1975–1984. [Google Scholar] [CrossRef]
- Lautenbach, E.; Bilker, W.B.; Tolomeo, P.; Maslow, J.N. Impact of diversity of colonizing strains on strategies for sampling Escherichia coli from fecal specimens. J. Clin. Microbiol. 2008, 46, 3094–3096. [Google Scholar] [CrossRef]
- Moreno, E.; Andreu, A.; Pigrau, C.; Kuskowski, M.A.; Johnson, J.R.; Prats, G. Relationship between Escherichia coli strains causing acute cystitis in women and the fecal E. coli population of the host. J. Clin. Microbiol. 2008, 46, 2529–2534. [Google Scholar] [CrossRef] [PubMed]
- Moreno, E.; Johnson, J.R.; Pérez, T.; Prats, G.; Kuskowski, M.A.; Andreu, A. Structure and urovirulence characteristics of the fecal Escherichia coli population among healthy women. Microbe. Infect. 2009, 11, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Nowrouzian, F.L.; Wold, A.E.; Adlerberth, I. Escherichia coli strains belonging to phylogenetic group B2 have superior capacity to persist in the intestinal microflora of infants. J. Infect. Dis. 2005, 191, 1078–1083. [Google Scholar] [CrossRef]
- Vollmerhausen, T.L.; Ramos, N.L.; Gündoğdu, A.; Robinson, W.; Brauner, A.; Katouli, M. Population structure and uropathogenic virulence-associated genes of faecal Escherichia coli from healthy young and elderly adults. J. Med. Microbiol. 2011, 60, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.A.; Whitlock, J.E.; Harwood, V.J. Diversity and distribution of Escherichia coli genotypes and antibiotic resistance phenotypes in feces of humans, cattle, and horses. Appl. Environ. Microbiol. 2006, 72, 6914–6922. [Google Scholar] [CrossRef] [PubMed]
- Escobar-Páramo, P.; Grenet, K.; Le Menac’h, A.; Rode, L.; Salgado, E.; Amorin, C.; Gouriou, S.; Picard, B.; Rahimy, M.C.; Andremont, A. Large-scale population structure of human commensal Escherichia coli isolates. Appl. Environ. Microbiol. 2004, 70, 5698–5700. [Google Scholar] [CrossRef] [PubMed]
- Staji, H. Detection of enterohemorrhagic Escherichia coli related genes in E. coli strains belonging to B2 phylogroup isolated from urinary tract infections in combination with antimicrobial resistance phenotypes. JMB 2017, 6, 36–44. [Google Scholar]
- Sperner, B.; Schalch, B.; Eisgruber, H.; Stolle, A. Short protocol for pulsed field gel electrophoresis of a variety of Clostridia species. FEMS Immunol. Med. Microbiol. 1999, 24, 287–292. [Google Scholar] [CrossRef]
- Mouwen, D.; Weijtens, M.; Capita, R.; Alonso-Calleja, C.; Prieto, M. Discrimination of enterobacterial repetitive intergenic consensus PCR types of Campylobacter coli and Campylobacter jejuni by Fourier transform infrared spectroscopy. Appl. Environ. Microbiol. 2005, 71, 4318–4324. [Google Scholar] [CrossRef]
- Ranjbar, R.; Tabatabaee, A.; Behzadi, P.; Kheiri, R. Enterobacterial repetitive intergenic consensus polymerase chain reaction (ERIC-PCR) genotyping of Escherichia coli strains isolated from different animal stool specimens. Iran. J. Pathol. 2017, 12, 25. [Google Scholar] [CrossRef]
- Kumar, N.S.; Gurusubramanian, G. Random amplified polymorphic DNA (RAPD) markers and its applications. Sci. Vis. 2011, 11, 116–124. [Google Scholar]
- Lin, W.-J.; Tung, C.-Y.; Yen, M.-Y.; Chan, Y.-J.; Lin, C.-H.; Hsueh, P.-R. A novel target pathogen identification and tracking system using capillary electrophoresis-random amplified polymorphic DNA. Sci. Rep. 2018, 8, 15365. [Google Scholar] [CrossRef] [PubMed]
- Huber, C.A.; Foster, N.F.; Riley, T.V.; Paterson, D.L. Challenges for standardization of Clostridium difficile typing methods. J. Clin. Microbiol. 2013, 51, 2810–2814. [Google Scholar] [CrossRef] [PubMed]
- Roohollah, K.; Naser, H.; Mehrouz, D. Efficacy evaluation of four different culture and PCR-based methods of Escherichia coli detection in water samples. Adv. Environ. Biol. 2013, 7, 2689–2694. [Google Scholar]
- Diego, J.G.-B.; Fernández-Soto, P.; Crego-Vicente, B.; Alonso-Castrillejo, S.; Febrer-Sendra, B.; Gómez-Sán chez, A.; Vicente, B.; López-Abán, J.; Muro, A. Progress in loop-mediated isothermal amplification assay for detection of Schistosoma mansoni DNA: Towards a ready-to-use test. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Fawley, W.N.; Knetsch, C.; MacCannell, D.R.; Harmanus, C.; Du, T.; Mulvey, M.R.; Paulick, A.; Anderson, L.; Kuijper, E.; Wilcox, M.H. Development and validation of an internationally-standardized, high-resolution capillary gel-based electrophoresis PCR-ribotyping protocol for Clostridium difficile. PLoS ONE 2015, 10, e0118150. [Google Scholar] [CrossRef]
- Mackay, I.M.; Arden, K.E.; Nitsche, A. Real-time PCR in virology. Nucleic Acids Res. 2002, 30, 1292–1305. [Google Scholar] [CrossRef]
- Gorgannezhad, L.; Sreejith, K.R.; Zhang, J.; Kijanka, G.; Christie, M.; Stratton, H.; Nguyen, N.-T. Microfluidic array chip for parallel detection of waterborne bacteria. Micromachines 2019, 10, 883. [Google Scholar] [CrossRef]
- Nguyen, N.-T.; Hejazian, M.; Ooi, C.H.; Kashaninejad, N. Recent advances and future perspectives on microfluidic liquid handling. Micromachines 2017, 8, 186. [Google Scholar] [CrossRef]
- Gorgannezhad, L.; Stratton, H.; Nguyen, N.-T. Microfluidic-based nucleic acid amplification systems in microbiology. Micromachines 2019, 10, 408. [Google Scholar] [CrossRef]
- Gorgannezhad, L.; Umer, M.; Islam, M.N.; Nguyen, N.-T.; Shiddiky, M.J. Circulating tumor DNA and liquid biopsy: Opportunities, challenges, and recent advances in detection technologies. Lab Chip 2018, 18, 1174–1196. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Huang, Q.; Xie, L.; Xiang, G.; Wang, L.; Xu, H.; Ma, L.; Luo, X.; Xin, J.; Zhou, X. A rapid, low-cost, and microfluidic chip-based system for parallel identification of multiple pathogens related to clinical pneumonia. Sci. Rep. 2017, 7, 6441. [Google Scholar] [CrossRef] [PubMed]
- Gorgannezhad, L.; Umer, M.; Kamal Masud, M.; Hossain, M.S.A.; Tanaka, S.; Yamauchi, Y.; Salomon, C.; Kline, R.; Nguyen, N.T.; Shiddiky, M.J. Detection of FGFR2: FAM76A fusion gene in circulating tumor RNA based on catalytic signal amplification of graphene oxide-loaded magnetic nanoparticles. Electroanalysis 2018, 30, 2293–2301. [Google Scholar] [CrossRef]
- Song, C.; Nguyen, N.-T.; Tan, S.-H.; Asundi, A.K. Modelling and optimization of micro optofluidic lenses. Lab Chip 2009, 9, 1178–1184. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Z.; Huang, X.; Nguyen, N.-T.; Abgrall, P. Thermocapillary actuation of droplet in a planar microchannel. Microfluid. Nanofluidics 2008, 5, 205–214. [Google Scholar] [CrossRef]
- Yap, Y.-F.; Tan, S.-H.; Nguyen, N.-T.; Murshed, S.S.; Wong, T.-N.; Yobas, L. Thermally mediated control of liquid microdroplets at a bifurcation. J. Phys. D Appl. Phys. 2009, 42, 065503. [Google Scholar] [CrossRef]
- Duncombe, T.A.; Tentori, A.M.; Herr, A.E. Microfluidics: Reframing biological enquiry. Nat. Rev. Mol. Cell Biol. 2015, 16, 554–567. [Google Scholar] [CrossRef]
- Bormashenko, E.; Balter, R.; Aurbach, D. Micropump based on liquid marbles. Appl. Phys. Lett. 2010, 97, 091908. [Google Scholar] [CrossRef]
- Mahadevan, L.; Pomeau, Y. Rolling droplets. Phys. Fluids. 1999, 11, 2449–2453. [Google Scholar] [CrossRef]
- Tian, J.; Arbatan, T.; Li, X.; Shen, W. Liquid marble for gas sensing. Chem. Commun. 2010, 46, 4734–4736. [Google Scholar] [CrossRef]
- Xue, Y.; Wang, H.; Zhao, Y.; Dai, L.; Feng, L.; Wang, X.; Lin, T. Magnetic liquid marbles: A “precise” miniature reactor. Adv. Mater. 2010, 22, 4814–4818. [Google Scholar] [CrossRef] [PubMed]
- Leite, Á.J.; Oliveira, N.M.; Song, W.; Mano, J.F. Bioactive hydrogel marbles. Sci. Rep. 2018, 8, 15215. [Google Scholar] [CrossRef] [PubMed]
- Sreejith, K.R.; Gorgannezhad, L.; Jin, J.; Ooi, C.H.; Stratton, H.; Dao, D.V.; Nguyen, N.-T. Liquid marbles as biochemical reactors for the polymerase chain reaction. Lab Chip 2019, 19, 3220–3227. [Google Scholar] [CrossRef] [PubMed]
- Sreejith, K.R.; Gorgannezhad, L.; Jin, J.; Ooi, C.H.; Takei, T.; Hayase, G.; Stratton, H.; Lamb, K.; Shiddiky, M.; Dao, D.V.; et al. Core-shell beads made by composite liquid marble technology as a versatile microreactor for polymerase chain reaction. Micromachines 2020, 11, 242. [Google Scholar] [CrossRef] [PubMed]
- Hayase, G.; Kanamori, K.; Hasegawa, G.; Maeno, A.; Kaji, H.; Nakanishi, K. A superamphiphobic macroporous silicone monolith with marshmallow-like flexibility. Angew. Chem. Int. Ed. 2013, 52, 10788–10791. [Google Scholar] [CrossRef] [PubMed]
- Clermont, O.; Christenson, J.K.; Denamur, E.; Gordon, D.M. The C lermont Escherichia coli phylo-typing method revisited: Improvement of specificity and detection of new phylo-groups. Environ. Microbiol. Rep. 2013, 5, 58–65. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene | Primer ID | Sequence (5-3′) | PCR Product Size (bp) |
|---|---|---|---|
| chuA | chuA.1b chuA.2 | ATGGTACCGGACGAACCAAC TGCCGCCAGTACCAAAGACA | 288 |
| yjaA | yjaA.1b yjaA.2b | CAAACGTGAAGTGTCAGGAG AATGCGTTCCTCAACCTGTG | 211 |
| TspE4C2 | TspE4C2.1b TspE4C2.2b | CACTATTCGTAAGGTCATCC AGTTTATCGCTGCGGGTCGC | 152 |
| arpA | AceK.f ArpA1.r | AACGCTATTCGCCAGCTTGC TCTCCCCATACCGTACGCTA | 400 |
| Experiment ID | Bead A (chuA) | Bead B (arpA) | Bead C (TspE4C2) | Bead D (yjaA) | Phylo-Group |
|---|---|---|---|---|---|
| I | + | − | + | + | B2 |
| II | − | − | + | + | Unknown |
| III | − | − | − | + | Clade I/II |
| IV | − | − | + | − | Unknown |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gorgannezhad, L.; Sreejith, K.R.; Christie, M.; Jin, J.; Ooi, C.H.; Katouli, M.; Stratton, H.; Nguyen, N.-T. Core-Shell Beads as Microreactors for Phylogrouping of E. coli Strains. Micromachines 2020, 11, 761. https://doi.org/10.3390/mi11080761
Gorgannezhad L, Sreejith KR, Christie M, Jin J, Ooi CH, Katouli M, Stratton H, Nguyen N-T. Core-Shell Beads as Microreactors for Phylogrouping of E. coli Strains. Micromachines. 2020; 11(8):761. https://doi.org/10.3390/mi11080761
Chicago/Turabian StyleGorgannezhad, Lena, Kamalalayam Rajan Sreejith, Melody Christie, Jing Jin, Chin Hong Ooi, Mohammad Katouli, Helen Stratton, and Nam-Trung Nguyen. 2020. "Core-Shell Beads as Microreactors for Phylogrouping of E. coli Strains" Micromachines 11, no. 8: 761. https://doi.org/10.3390/mi11080761
APA StyleGorgannezhad, L., Sreejith, K. R., Christie, M., Jin, J., Ooi, C. H., Katouli, M., Stratton, H., & Nguyen, N.-T. (2020). Core-Shell Beads as Microreactors for Phylogrouping of E. coli Strains. Micromachines, 11(8), 761. https://doi.org/10.3390/mi11080761