OOCHIP: Compartmentalized Microfluidic Perfusion System with Porous Barriers for Enhanced Cell–Cell Crosstalk in Organ-on-a-Chip



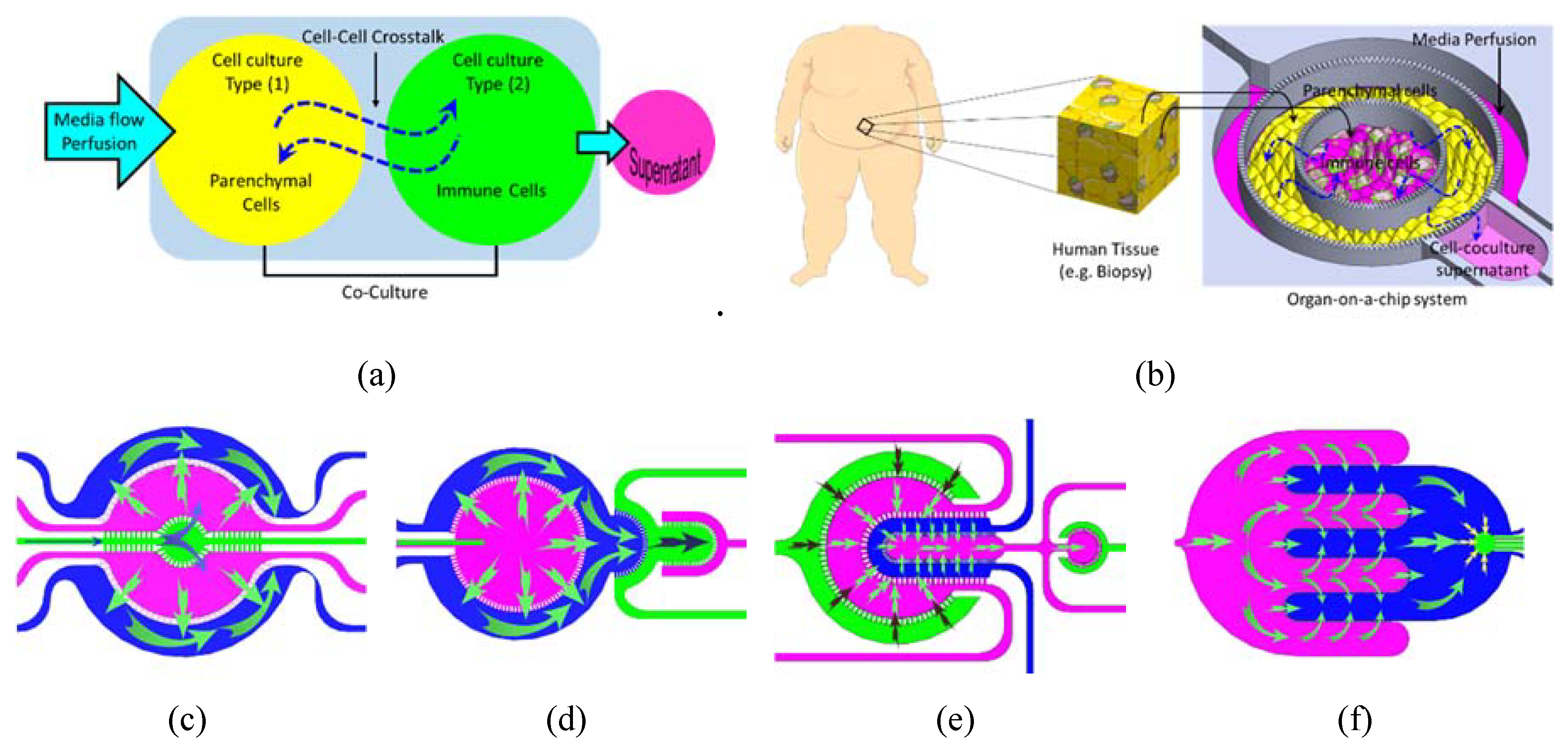{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
- (1)
- Maintaining a reasonable and physiologically relevant tissue/tissue and tissue/liquid ratio between the different organs. This is achievable due to the miniaturized nature of the microfluidic structure and the continuous perfusion of cell nutrients and stimuli without compromising the reagent cost. For example, when cells are cultured in a conventional culture flask with a surface area of 75 cm2, 10 mL of culture media is added onto the cell culture which results in a media/cell ratio (volume/volume) of 67 times. On the other hand, when the same cells are cultured in a microfluidic chip with a surface area of 1–2 cm2, the volume of media covering the cells is within the range of 20–30 μL which results in a ratio of media/cell of 6 only.
- (2)
- Controlling spatiotemporal fluidic flow into and from the cell culture compartments to emulate the local blood flow and mass transport of nutrients into and out of the tissue.
- (3)
- Maintaining physiologically relevant shear stresses onto the surface of tissue.
- (4)
- Allowing micro-scale spatial heterogeneity of cells and tissue.
- (5)
- Facilitating efficient cell–cell and tissue-tissue interaction between heterogeneous cell cultures.
- (6)
- Enabling the spatial organization of different types of cells/tissue in 2D architectures with a physiologically relevant order, which may allow to track the key physiological events in disease progression and treatment.
- (7)
- Allowing the recruitment of circulating immune cells with a high spatiotemporal resolution of cell–cell interaction.
2. Materials and Methods
2.1. Design and Simulation
- Chip 2 (C2): Two circular concentered compartments interface with a downstream semi-circular compartment (Figure 2b).
- Chip 3 (C3): Four U-shaped concentered compartments with the inner compartment connected through a channel to another circular compartment (Figure 2c).
- Chip 4 (C4): Two interdigitated compartments interfaced with a small circular compartment located inside the upstream compartment (Figure 2d).
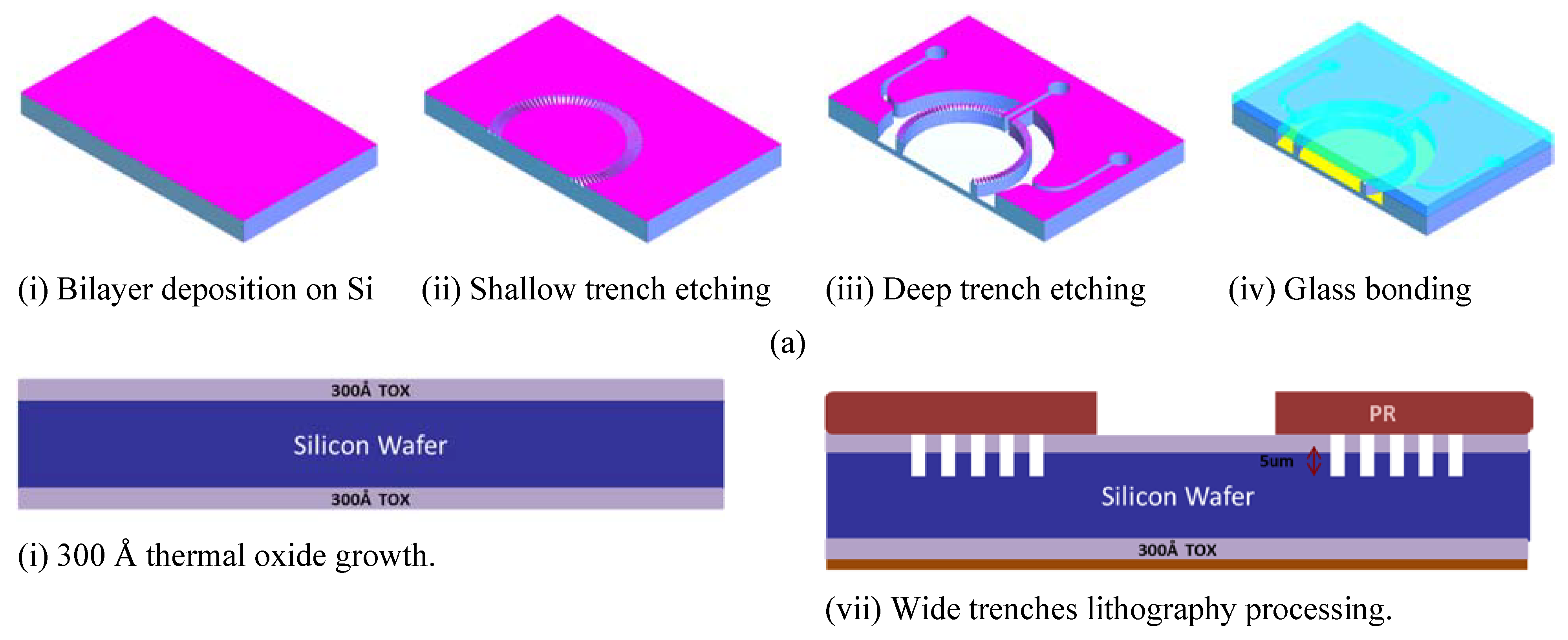
2.2. Chips Fabrication
2.3. Perfusion Setup
2.4. Finite Element Analysis
2.5. Characterization of Inter-Compartment Permeability
2.6. Cell Culture
2.7. Lipid Droplets Staining and Quantification
2.8. Immune-Metabolic Profile/Cytokine Secretion-Glucose Uptake
3. Results and Discussion
3.1. Finite Element Analysis
3.2. Microfluidic Testing: Inter-Compartment Permeability
3.3. System Testing with Parenchymal-Immune Cell Co-Culture
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Meyvantsson, I.; Beebe, D.J. Cell culture models in microfluidic systems. Annu. Rev. Anal. Chem. 2008, 1, 423–449. [Google Scholar] [CrossRef] [PubMed]
- Huh, D.; Hamilton, G.A.; Ingber, D.E. From 3D cell culture to organs-on-chips. Trends Cell. Biol. 2011, 21, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Bogdanowicz, D.R.; Lu, H.H. Multifunction Co-culture Model for Evaluating Cell–Cell Interactions. Methods Mol. Biol. 2014, 1202, 29–36. [Google Scholar] [PubMed]
- Proffen, B.L.; Haslauer, C.M.; Harris, C.E.; Murray, M.M. Mesenchymal stem cells from the retropatellar fat pad and peripheral blood stimulate ACL fibroblast migration, proliferation, and collagen gene expression. Connect. Tissue Res. 2013, 54, 14–21. [Google Scholar] [CrossRef]
- Wang, I.N.E.; Shan, J.; Choi, R.; Oh, S.; Kepler, C.K.; Chen, F.H.; Lu, H.H. Role of Osteoblast-Fibroblast Interactions in the Formation of the Ligament-to-Bone Interface. J. Orthop. Res. 2007, 25, 1609–1620. [Google Scholar] [CrossRef]
- Zhang, Y.; Chai, C.; Jiang, X.S.; Teoh, S.H.; Leong, K.W. Co-culture of umbilical cord blood CD34+ cells with human mesenchymal stem cells. Tissue Eng. 2006, 12, 2161–2170. [Google Scholar] [CrossRef]
- Schober, A.; Fernekorn, U.; Singh, S.; Schlingloff, G.; Gebinog, M.; Hampl, J.; Williamson, A. Mimicking the biological world: Methods for the 3D structuring of artificial cellular environments. Eng. Life Sci. 2013, 13, 352–367. [Google Scholar] [CrossRef]
- Esch, M.B.; Ueno, H.; Applegate, D.R.; Shuler, M.L. Modular, pumpless body-on-a-chip platform for the co-culture of GI tract epithelium and 3D primary liver tissue. Lab Chip 2016, 16, 2719–2729. [Google Scholar] [CrossRef]
- Skardal, A.; Murphy, S.V.; Devarasetty, M.; Mead, I.; Kang, H.W.; Seol, Y.J.; Zhang, Y.S.; Shin, S.R.; Zhao, L.; Aleman, J.; et al. Multi-tissue interactions in an integrated three-tissue organ-on-a-chip platform. Sci. Rep. 2017, 7, 8837. [Google Scholar] [CrossRef]
- Zhang, Y.S.; Aleman, J.; Shin, S.R.; Kilic, T.; Kim, D.; Shaegh, S.A.M.; Massa, S.; Riahi, R.; Chae, S.; Hu, N.; et al. Multisensor-integrated organs-on-chips platform for automated and continual in situ monitoring of organoid behaviors. Proc. Natl. Acad. Sci. USA 2017, 114, E2293–E2302. [Google Scholar] [CrossRef]
- Oleaga, C.; Bernabini, C.; Smith, A.S.; Srinivasan, B.; Jackson, M.; McLamb, W.; Platt, V.; Bridges, R.; Cai, Y.; Santhanam, N.; et al. Multi-Organ toxicity demonstration in a functional human in vitro system composed of four organs. Sci. Rep. 2016, 6, 20030. [Google Scholar] [CrossRef] [PubMed]
- Tsamandouras, N.; Chen, W.L.K.; Edington, C.D.; Stokes, C.L.; Griffith, L.G.; Cirit, M. Integrated Gut and Liver Microphysiological Systems for Quantitative In Vitro Pharmacokinetic Studies. AAPS J. 2017, 19, 1499–1512. [Google Scholar] [CrossRef] [PubMed]
- Wikswo, J.P.; Curtis, E.L.; Eagleton, Z.E.; Evans, B.C.; Kole, A.; Hofmeister, L.H.; Matloff, W.J. Scaling and systems biology for integrating multiple organs-on-a-chip. Lab Chip 2013, 13, 3496–3511. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Cilfone, N.A.; Large, E.M.; Sarkar, U.; Wishnok, J.S.; Tannenbaum, S.R.; Hughes, D.J.; Lauffenburger, D.A.; Griffith, L.G.; Stokes, C.L.; et al. Quantitative Systems Pharmacology Approaches Applied to Microphysiological Systems (MPS): Data Interpretation and Multi-MPSIntegration. CPT Pharmacomet. Syst. Pharmacol. 2015, 4, 585–594. [Google Scholar] [CrossRef]
- Abaci, H.E.; Shuler, M.L. Human-on-a-chip design strategies and principles for physiologically based pharmacokinetics/pharmacodynamics modeling. Integr. Biol. (Camb.) 2015, 7, 383–391. [Google Scholar] [CrossRef]
- Maschmeyer, I.; Lorenz, A.K.; Schimek, K.; Hasenberg, T.; Ramme, A.P.; Hübner, J.; Lindner, M.; Drewell, C.; Bauer, S.; Thomas, A.; et al. A four-organ-chip for interconnected long-term co-culture of human intestine, liver, skin and kidney equivalents. Lab Chip 2015, 15, 2688–2699. [Google Scholar] [CrossRef]
- Wagner, I.; Materne, E.M.; Brincker, S.; Süssbier, U.; Frädrich, C.; Busek, M.; Sonntag, F.; Sakharov, D.A.; Trushkin, E.V.; Tonevitsky, A.G.; et al. A dynamic multi-organ-chip for long-term cultivation and substance testing proven by 3D human liver and skin tissue co-culture. Lab Chip 2013, 13, 3538–3547. [Google Scholar] [CrossRef]
- Kimura, H.; Sakai, Y.; Fujii, T. Organ/body-on-a-chip based on microfluidic technology for drug discovery. Drug Metab. Pharmacokinet. 2018, 33, 43–48. [Google Scholar] [CrossRef]
- Hung, P.J.; Lee, P.; Sabounchi, P.; Lin, R.; Lee, L.P. Continuous Perfusion Microfluidic Cell Culture Array for High-Throughput Cell-Based Assays. Biotech. Bioeng. 2005, 89, 1–8. [Google Scholar] [CrossRef]
- Toh, Y.C.; Zhang, C.; Zhang, J.; Khong, Y.M.; Chang, S.; Samper, V.D.; van Noort, D.; Hutmacher, D.W.; Yu, H. A novel 3D mammalian cell perfusion-culture system in microfluidic channels. Lab Chip 2007, 7, 302–309. [Google Scholar] [CrossRef]
- Toh, Y.C.; Raja, A.; Yu, H.; Van Noort, D. A 3D Microfluidic Model to Recapitulate Cancer Cell Migration and Invasion. Bioengineering 2018, 5, 29. [Google Scholar] [CrossRef] [PubMed]
- Stevens, K.R.; Ungrin, M.D.; Schwartz, R.E.; Ng, S.; Carvalho, B.; Christine, K.S.; Chaturvedi, R.R.; Li, C.Y.; Zandstra, P.W.; Chen, C.S.; et al. InVERT molding for scalable control of tissue microarchitecture. Nat. Commun. 2013, 4, 1847. [Google Scholar] [CrossRef] [PubMed]
- Vunjak-Novakovic, G.; Bhatia, S.N.C.S.; Hirschi, K. HeLiVa platform: Integrated heart–liver–vascular systems for drug testing in human health and disease. Stem. Cell Res. Ther. 2013, 4 (Suppl. 1), S8. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.M.; Trappmann, B.; Stapleton, S.C.; Toro, E.; Chen, C.S. Microfl uidics embedded within extracellular matrix to defi ne vascular architectures and pattern diff usive gradients. Lab Chip 2013, 13, 3246–3252. [Google Scholar] [CrossRef]
- Nguyen, D.H.T.; Stapleton, S.C.; Yang, M.T.; Cha, S.S.; Choi, C.K.; Galie, P.A.; Chen, C.S. Biomimetic model to reconstitute angiogenic sprouting morphogenesis in vitro. Proc. Natl. Acad. Sci. USA 2013, 110, 6712–6717. [Google Scholar] [CrossRef]
- Liu, Y.X.; Kongsuphol, P.; Saha, S.; Gourikutty, S.B.; Biswas, S.; Ramadan, Q. Adipose-on-a-chip: A dynamic micro-physiological in vitro model of the human adipose for immune-metabolic analysis in type II diabetes. Lab Chip 2019, 19, 241–253. [Google Scholar] [CrossRef]
- Kongsuphol, P.; Saha, S.; Liu, Y.X.; Biswas, S.; Ramadan, Q. In vitro microphysiological model of the inflamed human adipose tissue for immune-metabolic analysis in type II diabetes. Sci. Rep. 2019, 9, 4887. [Google Scholar] [CrossRef]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Invest. 2003, 112, 1821–1830. [Google Scholar] [CrossRef]
- Hruskova, Z.; Biswas, S.K. A new “immunological” role for adipocytes in obesity. Cell Metab. 2013, 17, 315–317. [Google Scholar] [CrossRef][Green Version]
- Kim, K.M.; Choi, Y.J.; Hwang, J.; Kim, A.R.; Cho, H.J.; Hwang, E.S.; Park, J.Y.; Lee, S.; Hong, J. Shear Stress Induced by an Interstitial Level of Slow Flow Increases the Osteogenic Differentiation of Mesenchymal Stem Cells through TAZ Activation. PLoS ONE 2014, 9, e92427. [Google Scholar] [CrossRef]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Invest. 2003, 112, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Cancello, R.; Henegar, C.; Viguerie, N.; Taleb, S.; Poitou, C.; Rouault, C.; Coupaye, M.; Pelloux, V.; Hugol, D.; Bouillot, J.L.; et al. Reduction of macrophage infiltration and chemoattractant gene expression changes in white adipose tissue of morbidly obese subjects after surgery-induced weight loss. Diabetes 2005, 54, 2277–2286. [Google Scholar] [CrossRef]
- Clement, K.; Langin, D. Regulation of inflammation-related genes in human adipose tissue. J. Int. Med. 2007, 262, 422–430. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Lee, Y.H.; Rousseau, E.; Cam, M.; Tataranni, P.A.; Baier, L.J.; Bogardus, C.; Permana, P.A. Increased expression of inflammation-related genes in cultured preadipocytes/stromal vascular cells from obese compared with non-obese Pima Indians. Diabetologia 2005, 48, 1784–1788. [Google Scholar] [CrossRef] [PubMed]
- Thurgood, P.; Baratchi, S.; Szydzik, C.; Mitchell, A.; Khoshmanesh, K. Porous PDMS structures for the storage and release of aqueous solutions into fluidic environments. Lab Chip 2017, 17, 2517–2527. [Google Scholar] [CrossRef]
- Zhu, J.Y.; Suarez, S.A.; Thurgood, P.; Nguyen, N.; Mohammed, M.; Abdelwahab, H.; Needham, S.; Pirogova, E.; Ghorbani, K.; Baratchi, S.; et al. Reconfigurable, Self-Sufficient Convective Heat Exchanger for Temperature Control of Microfluidic Systems. Anal. Chem. 2019, 91, 15784–15790. [Google Scholar] [CrossRef]











© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramadan, Q.; Gourikutty, S.B.N.; Zhang, Q. OOCHIP: Compartmentalized Microfluidic Perfusion System with Porous Barriers for Enhanced Cell–Cell Crosstalk in Organ-on-a-Chip. Micromachines 2020, 11, 565. https://doi.org/10.3390/mi11060565
Ramadan Q, Gourikutty SBN, Zhang Q. OOCHIP: Compartmentalized Microfluidic Perfusion System with Porous Barriers for Enhanced Cell–Cell Crosstalk in Organ-on-a-Chip. Micromachines. 2020; 11(6):565. https://doi.org/10.3390/mi11060565
Chicago/Turabian StyleRamadan, Qasem, Sajay Bhuvanendran Nair Gourikutty, and Qingxin Zhang. 2020. "OOCHIP: Compartmentalized Microfluidic Perfusion System with Porous Barriers for Enhanced Cell–Cell Crosstalk in Organ-on-a-Chip" Micromachines 11, no. 6: 565. https://doi.org/10.3390/mi11060565
APA StyleRamadan, Q., Gourikutty, S. B. N., & Zhang, Q. (2020). OOCHIP: Compartmentalized Microfluidic Perfusion System with Porous Barriers for Enhanced Cell–Cell Crosstalk in Organ-on-a-Chip. Micromachines, 11(6), 565. https://doi.org/10.3390/mi11060565