Simultaneous Determination of Four DNA bases at Graphene Oxide/Multi-Walled Carbon Nanotube Nanocomposite-Modified Electrode



Abstract
1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. Instrumentation
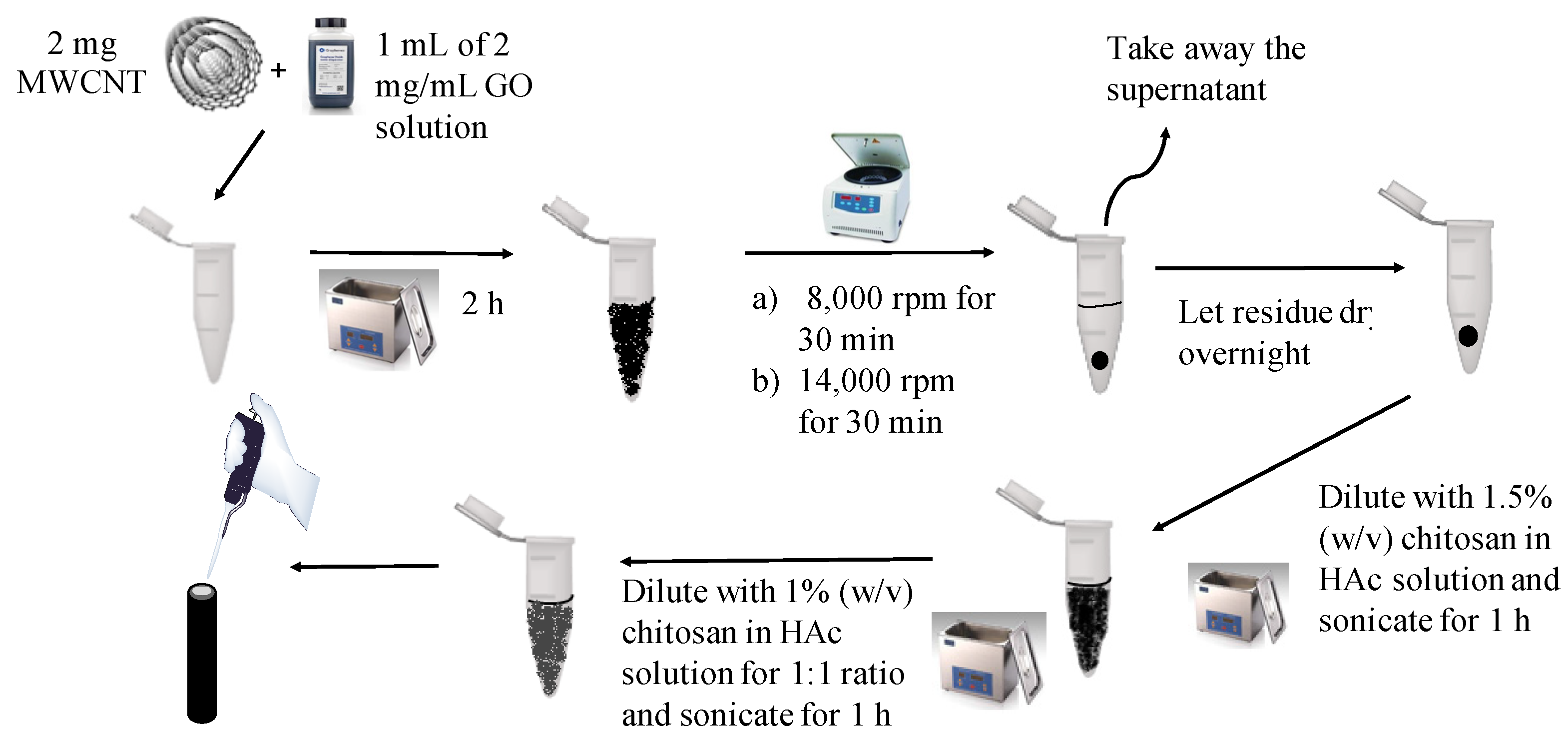
2.3. Synthesis of GO-MWCNT Hybrid in CHT
2.4. Modification of GCE Surface with GO-MWCNT Hybrid Nanocomposite in CHT
2.5. Spiked Human Serum Sample Preparation (10x dilution)
2.6. Spiked Human Saliva Sample and Artificial Saliva Sample Preparation (5x dilution)
3. Results and Discussion
3.1. Characterization of GO-MWCNT Hybrid Nanocomposite in CHT
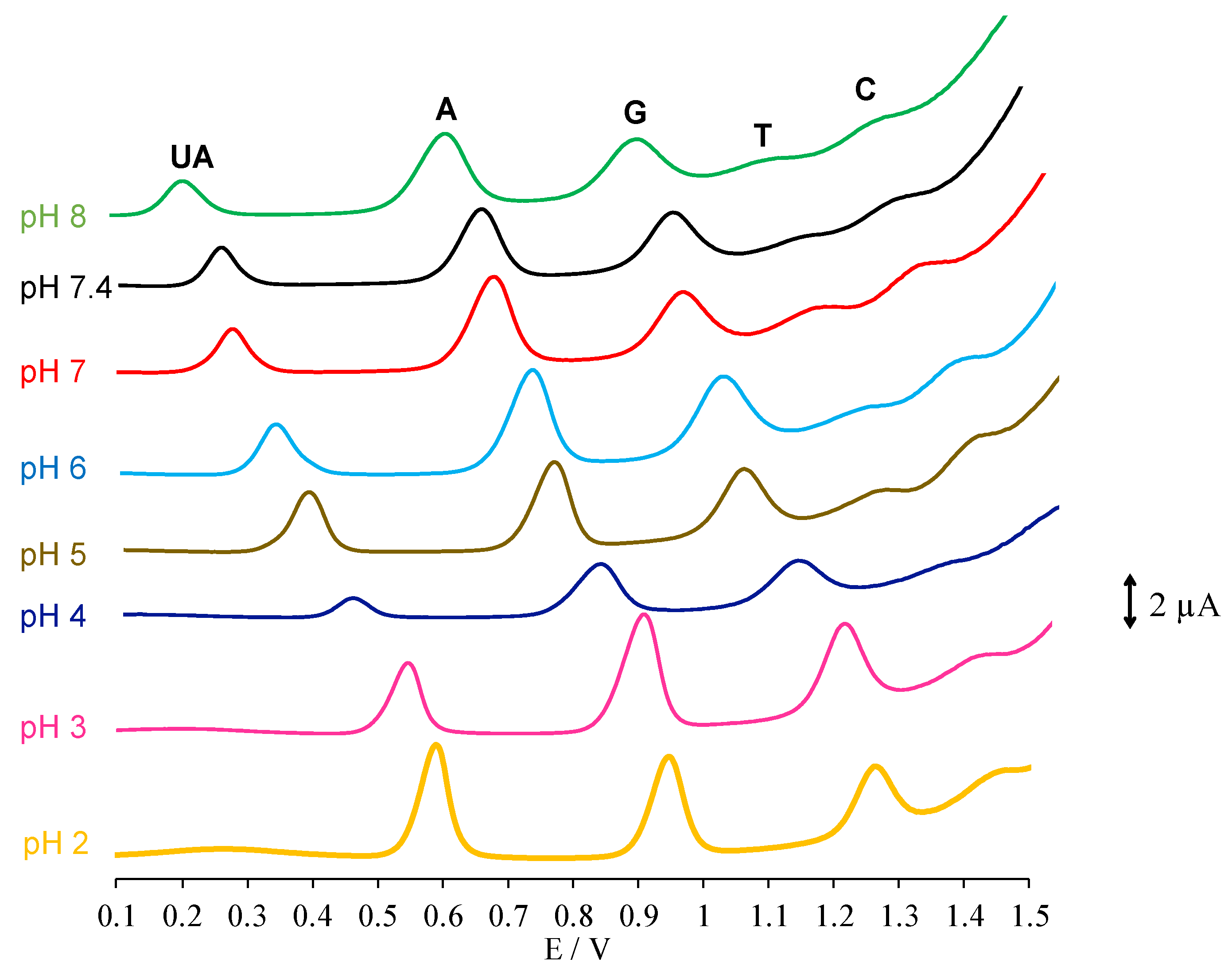
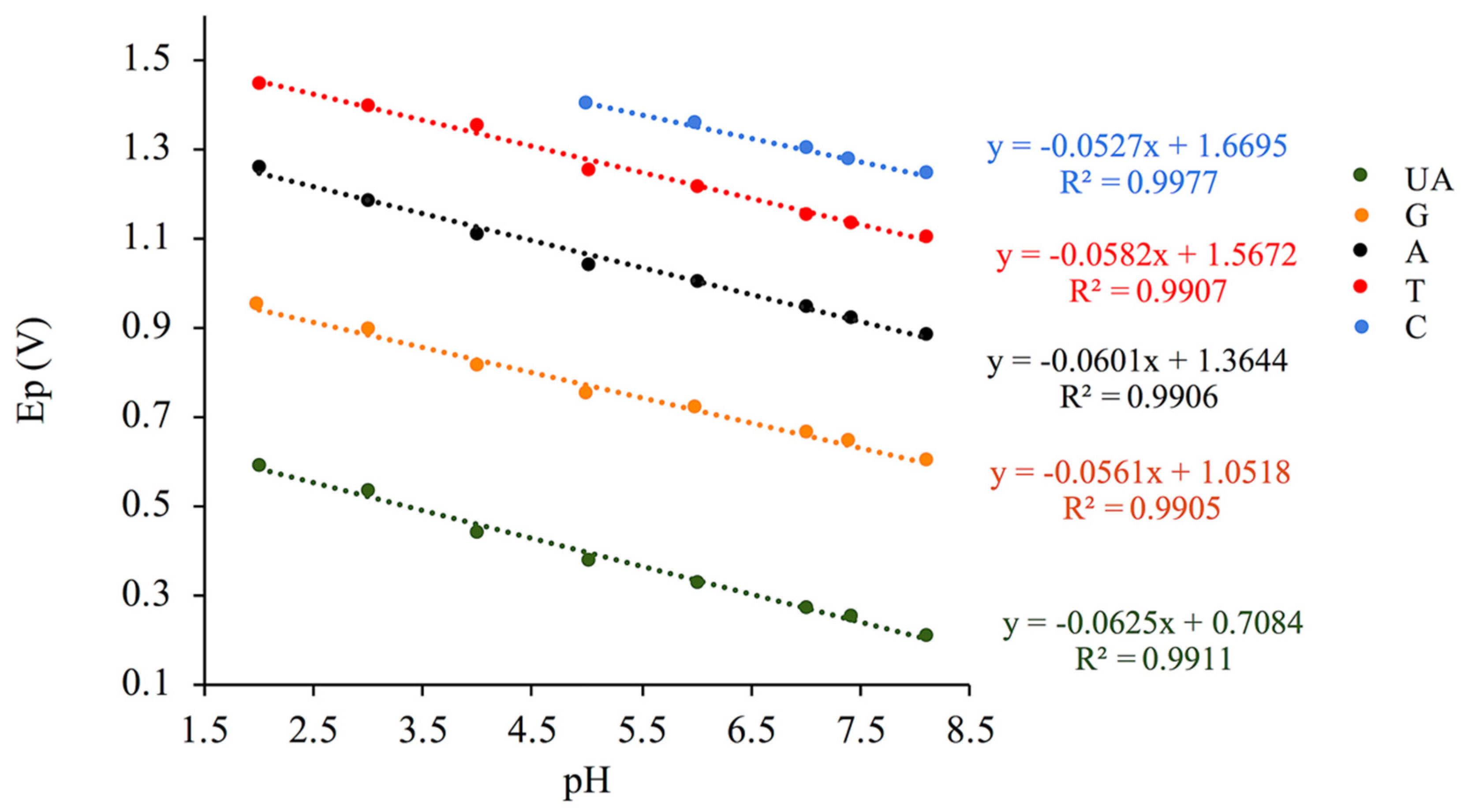
3.2. pH Effect
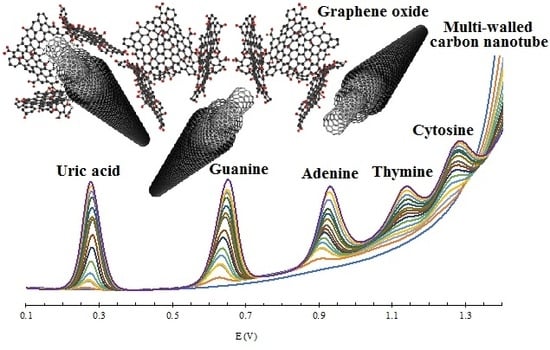
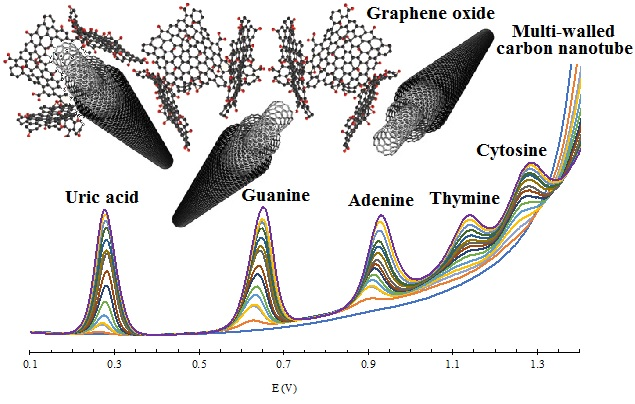
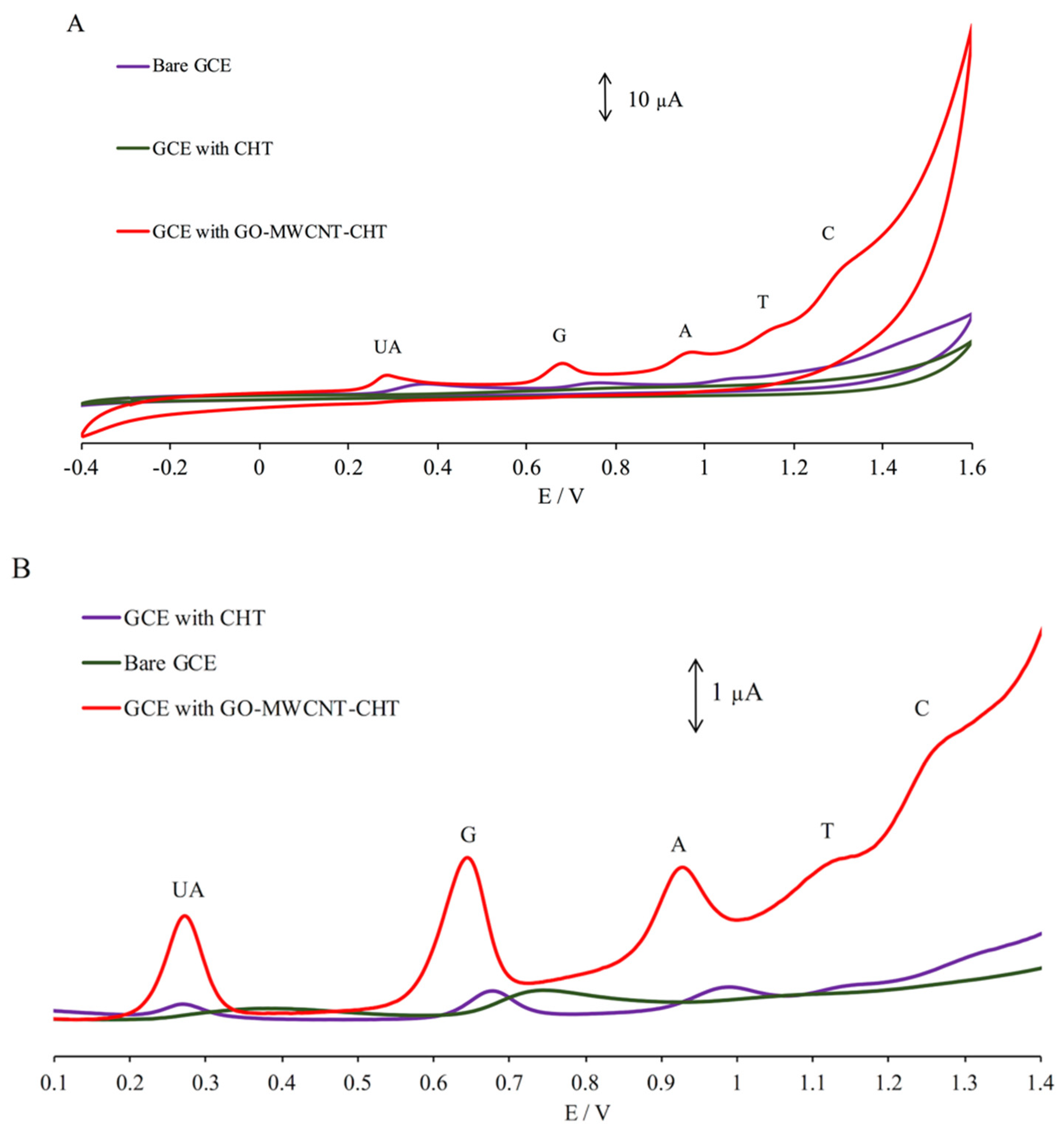
3.3. Electrochemical Behavior of UA and Nucleobases
3.4. Simultaneous Electrochemical Detection of Nucleobases in Presence of UA
3.5. Interference, Stability, and Reproducibility
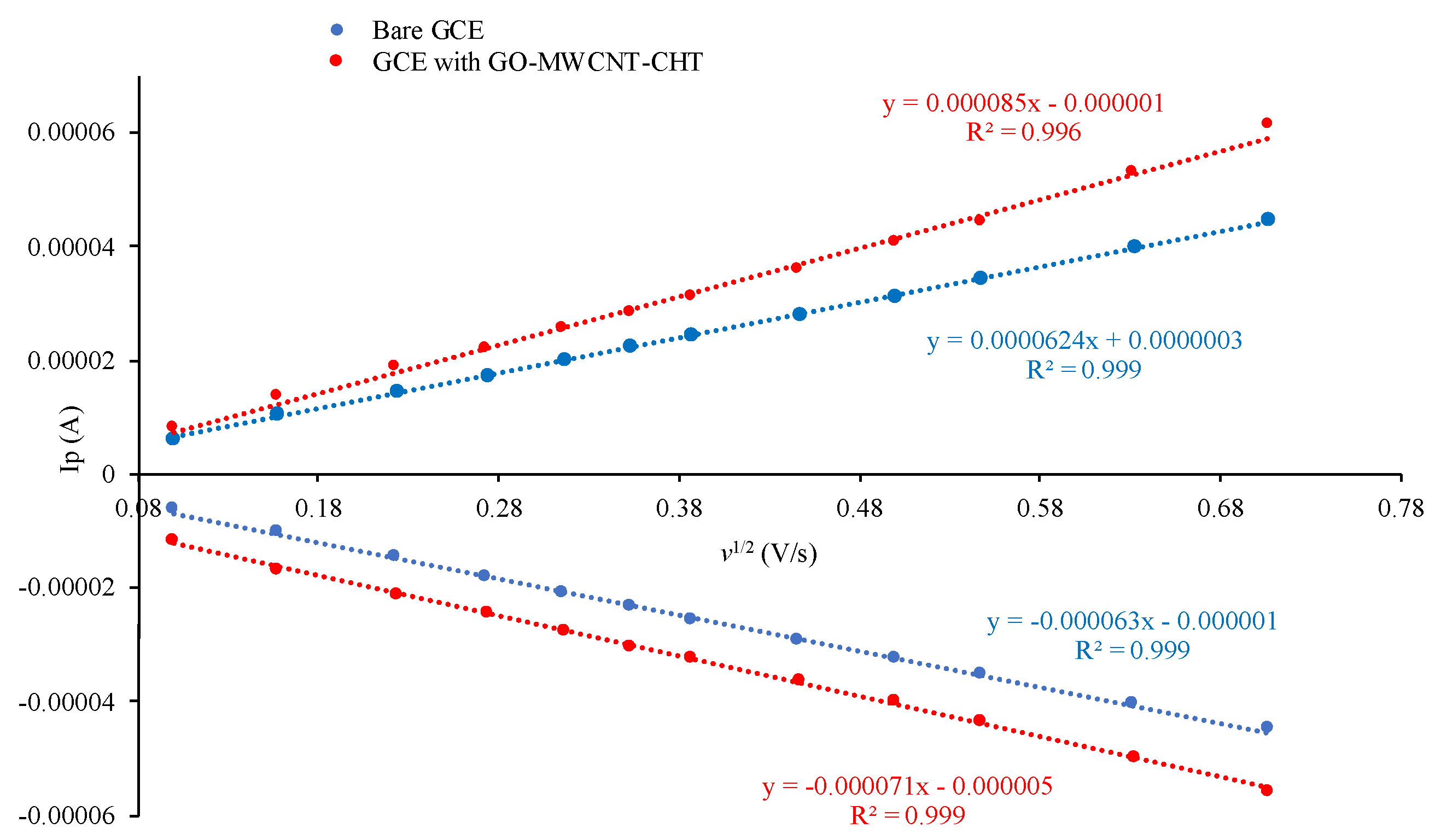
3.6. Scan Rate Dependence
3.7. Chronoamperometry
3.8. Real Samples
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ivandini, T.A.; Honda, K.; Rao, T.N.; Fujishima, A.; Einaga, Y. Simultaneous detection of purine and pyrimidine at highly boron-doped diamond electrodes by using liquid chromatography. Talanta 2007, 71, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Carolina Torres, A.; Barsan, M.M.; Brett, C.M.A. Simple electrochemical sensor for caffeine based on carbon and Nafion-modified carbon electrodes. Food Chem. 2014, 149, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Han, X.; Zhang, L.; Ye, J. Determination of purine and pyrimidine bases in DNA by micellar electrokinetic capillary chromatography with electrochemical detection. J. Chromatogr. A 2002, 954, 267–276. [Google Scholar] [CrossRef]
- Lavanya, N.; Nizeyimana Claude, J.; Sekar, C. Electrochemical determination of purine and pyrimidine bases using copper doped cerium oxide nanoparticles. J. Colloid Interface Sci. 2018, 530, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.L.; Khor, S.M. Graphite-based nanocomposite electrochemical sensor for multiplex detection of adenine, guanine, thymine, and cytosine: A biomedical prospect for studying DNA damage. Anal. Chem. 2017, 89, 10004–10012. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, X.; Luo, L.; Ding, Y.; Liu, B.; Xu, D. Simultaneous determination of purine and pyrimidine bases in DNA using poly(3,4-ethylenedioxythiophene)/graphene composite film. J. Electroanal. Chem. 2014, 735, 51–56. [Google Scholar] [CrossRef]
- Oliveira-Brett, A.M.; Piedade, J.A.P.; Silva, L.A.; Diculescu, V.C. Voltammetric determination of all DNA nucleotides. Anal. Biochem. 2004, 332, 321–329. [Google Scholar] [CrossRef]
- Xiao, F.; Zhao, F.; Li, J.; Liu, L.; Zeng, B. Characterization of hydrophobic ionic liquid-carbon nanotubes-gold nanoparticles composite film coated electrode and the simultaneous voltammetric determination of guanine and adenine. Electrochim. Acta 2008, 53, 7781–7788. [Google Scholar] [CrossRef]
- Yang, F.Q.; Guan, J.; Li, S.P. Fast simultaneous determination of 14 nucleosides and nucleobases in cultured Cordyceps using ultra-performance liquid chromatography. Talanta 2007, 73, 269–273. [Google Scholar] [CrossRef]
- Geng, X.; Bao, J.; Huang, T.; Wang, X.; Hou, C.; Hou, J.; Samalo, M.; Yang, M.; Huo, D. Electrochemical sensor for the simultaneous detection of guanine and adenine based on a PPyox/MWNTs-MoS2 modified electrode. J. Electrochem. Soc. 2019, 166, B498–B504. [Google Scholar] [CrossRef]
- Jankowska-Śliwińska, J.; Dawgul, M.; Kruk, J.; Pijanowska, D.G. Comparison of electrochemical determination of purines and pyrimidines by means of carbon, graphite and gold paste electrodes. Int. J. Electrochem. Sci. 2017, 12, 2329–2343. [Google Scholar] [CrossRef]
- Huang, Q.; Kaiser, K.; Benner, R. A simple high performance liquid chromatography method for the measurement of nucleobases and the RNA and DNA content of cellular material. Limnol. Oceanogr. Methods 2012, 10, 608–616. [Google Scholar] [CrossRef]
- Zong, S.Y.; Han, H.; Wang, B.; Li, N.; Dong, T.T.X.; Zhang, T.; Tsim, K.W.K.; McPhee, D.J. Fast simultaneous determination of 13 nucleosides and nucleobases in cordyceps sinensis by UHPLC-ESI-MS/MS. Molecules 2015, 20, 21816–21825. [Google Scholar] [CrossRef] [PubMed]
- Brohi, R.O.Z.; Khuhawar, M.Y.; Channa, A.; Laghari, A.J.; Abbasi, K. Gas chromatographic determination of purines and pyrimidines from dna using ethyl chloroformate as derivatizing reagent. Pak. J. Anal. Environ. Chem. 2016, 17, 50–57. [Google Scholar] [CrossRef]
- Li, T.; Li, B.; Dong, S. Aptamer-based label-free method for hemin recognition and DNA assay by capillary electrophoresis with chemiluminescence detection. Anal. Bioanal. Chem. 2007, 389, 887–893. [Google Scholar] [CrossRef]
- Heisler, I.; Keller, J.; Tauber, R.; Sutherland, M.; Fuchs, H. A colorimetric assay for the quantitation of free adenine applied to determine the enzymatic activity of ribosome-inactivating proteins. Anal. Biochem. 2002, 302, 114–122. [Google Scholar] [CrossRef]
- Zhu, Z.; Zhan, L.; Hou, C.; Wang, Z. Nanostructured metal-enhanced raman spectroscopy for DNA base detection. IEEE Photonics J. 2012, 4, 1333–1339. [Google Scholar]
- Xu, Q.; Liu, X.; Li, H.-B.; Yin, L.N.; Hu, X. Electrochemical determination of purine and pyrimidine DNA bases based on the recognition properties of azocalix [4] arene. Biosens. Bioelectron. 2013, 42, 355–361. [Google Scholar]
- Hasoň, S.; Fojt, L.; Šebest, M.F.P. Improved electrochemical detection of purine nucleobases at mechanically roughened edge-plane pyrolytic graphite electrode. Electroanalysis 2009, 21, 666–670. [Google Scholar] [CrossRef]
- Zhang, J.; Han, D.; Wang, S.; Zhang, X.; Yang, R.; Ji, Y.; Yu, X. Electrochemical detection of adenine and guanine using a three-dimensional WS2 nanosheet/graphite microfiber hybrid electrode. Electrochem. Commun. 2019, 99, 75–80. [Google Scholar] [CrossRef]
- Jesny, S.; Girish Kumar, K. Electrocatalytic resolution of guanine, adenine and cytosine along with uric acid on poly (4-amino-3-hydroxy naphthalene-1-sulfonic acid) modified glassy carbon electrode. J. Electroanal. Chem. 2017, 801, 153–161. [Google Scholar] [CrossRef]
- Li, D.; Yang, X.L.; Xiao, B.L.; Geng, F.Y.; Hong, J.; Sheibani, N.; Moosavi-Movahedi, A.A. Detection of guanine and adenine using an aminated reduced graphene oxide functional membrane-modified glassy carbon electrode. Sensors 2017, 17, 1652. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, T.M.B.F.; Morais, S. New generation of electrochemical sensors based on multi-walled carbon nanotubes. Appl. Sci. 2018, 8, 1925. [Google Scholar] [CrossRef]
- Aboutalebi, S.H.; Chidembo, A.T.; Salari, M.; Konstantinov, K.; Wexler, D.; Liu, H.K.; Dou, S.X. Comparison of GO, GO/MWCNTs composite and MWCNTs as potential electrode materials for supercapacitors. Energy Environ. Sci. 2011, 4, 1855–1865. [Google Scholar] [CrossRef]
- Kong, H.X. Hybrids of carbon nanotubes and graphene/graphene oxide. Curr. Opin. Solid State Mater. Sci. 2013, 17, 31–37. [Google Scholar] [CrossRef]
- Mani, V.; Devadas, B.; Chen, S.M. Direct electrochemistry of glucose oxidase at electrochemically reduced graphene oxide-multiwalled carbon nanotubes hybrid material modified electrode for glucose biosensor. Biosens. Bioelectron. 2013, 41, 309–315. [Google Scholar] [CrossRef]
- Arena, A.; Scandurra, G.; Ciofi, C. Copper oxide chitosan nanocomposite: Characterization and application in non-enzymatic hydrogen peroxide sensing. Sensors 2017, 17, 2198. [Google Scholar] [CrossRef]
- Li, S.; Noroozifar, M.; Kerman, K. Nanocomposite of ferricyanide-doped chitosan with multi-walled carbon nanotubes for simultaneous senary detection of redox-active biomolecules. J. Electroanal. Chem. 2019, 849, 113376. [Google Scholar] [CrossRef]
- Suh, J.H.; Lee, Y.Y.; Lee, H.J.; Kang, M.; Hur, Y.; Lee, S.N.; Yang, D.H.; Han, S.B. Dispersive liquid-liquid microextraction based on solidification of floating organic droplets followed by high performance liquid chromatography for the determination of duloxetine in human plasma. J. Pharm. Biomed. Anal. 2013, 75, 214–219. [Google Scholar] [CrossRef]
- Madsen, K.D.; Sander, C.; Baldursdottir, S.; Pedersen, A.M.L.; Jacobsen, J. Development of an ex vivo retention model simulating bioadhesion in the oral cavity using human saliva and physiologically relevant irrigation media. Int. J. Pharm. 2013, 448, 373–381. [Google Scholar] [CrossRef]
- Tohidinia, M.; Noroozifar, M. Investigation of carbon allotropes for simultaneous determination of ascorbic acid, epinephrine, uric acid, nitrite and xanthine. Int. J. Electrochem. Sci. 2018, 13, 2310–2328. [Google Scholar] [CrossRef]
- Anu Prathap, M.U.; Srivastava, R.; Satpati, B. Simultaneous detection of guanine, adenine, thymine, and cytosine at polyaniline/MnO2 modified electrode. Electrochim. Acta 2013, 114, 285–295. [Google Scholar] [CrossRef]
- Veera Manohara Reddy, Y.; Sravani, B.; Agarwal, S.; Gupta, V.K.; Madhavi, G. Electrochemical sensor for detection of uric acid in the presence of ascorbic acid and dopamine using the poly(DPA)/SiO2@Fe3O4 modified carbon paste electrode. J. Electroanal. Chem. 2018, 820, 168–175. [Google Scholar] [CrossRef]
- Sharma, V.K.; Jelen, F.; Trnkova, L. Functionalized solid electrodes for electrochemical biosensing of purine nucleobases and their analogues: A review. Sensors 2015, 15, 1564–1600. [Google Scholar] [CrossRef]
- Sun, W.; Xi, M.; Zhang, L.; Zhan, T.; Gao, H.; Jiao, K. Electrochemical behaviors of thymine on a new ionic liquid modified carbon electrode and its detection. Electrochim. Acta 2010, 56, 222–226. [Google Scholar] [CrossRef]
- Ganesh, H.V.S.; Noroozifar, M.; Kerman, K. Epigallocatechin gallate-modified graphite paste electrode for simultaneous detection of redox-active biomolecules. Sensors 2018, 18, 23. [Google Scholar] [CrossRef]
- Yin, H.; Zhou, Y.; Ma, Q.; Ai, S.; Ju, P.; Zhu, L.; Lu, L. Electrochemical oxidation behavior of guanine and adenine on graphene-Nafion composite film modified glassy carbon electrode and the simultaneous determination. Process Biochem. 2010, 45, 1707–1712. [Google Scholar] [CrossRef]
- Yari, A.; Derki, S. New MWCNT-Fe3O4@PDA-Ag nanocomposite as a novel sensing element of an electrochemical sensor for determination of guanine and adenine contents of DNA. Sensors Actuators B Chem. 2016, 227, 456–466. [Google Scholar] [CrossRef]
- Thangaraj, R.; Senthil Kumar, A. Simultaneous detection of guanine and adenine in DNA and meat samples using graphitized mesoporous carbon modified electrode. J. Solid State Electrochem. 2013, 17, 583–590. [Google Scholar] [CrossRef]
- Da Cruz, F.S.; de Souza Paula, F.; Franco, D.L.; dos Santos, W.T.P.; Ferreira, L.F. Electrochemical detection of uric acid using graphite screen-printed electrodes modified with Prussian blue/poly(4-aminosalicylic acid)/Uricase. J. Electroanal. Chem. 2017, 806, 172–179. [Google Scholar] [CrossRef]
- Ping, J.; Wu, J.; Wang, Y.; Ying, Y. Simultaneous determination of ascorbic acid, dopamine and uric acid using high-performance screen-printed graphene electrode. Biosens. Bioelectron. 2012, 34, 70–76. [Google Scholar] [CrossRef]
- Stozhko, N.; Bukharinova, M.; Galperin, L.; Brainina, K. A Nanostructured sensor based on gold nanoparticles and Nafion for determination of uric acid. Biosensors 2018, 8, 21. [Google Scholar] [CrossRef] [PubMed]
- Brohi, R.O.Z.Z.; Khuhawar, M.Y.; Khuhawar, T.M.J. GC-FID determination of nucleobases guanine, adenine, cytosine, and thymine from DNA by precolumn derivatization with isobutyl chloroformate. J. Anal. Sci. Technol. 2016, 7, 10. [Google Scholar] [CrossRef]
- Duan, B.; Wang, L.; Dai, X.; Huang, L.; Yang, M.; Chen, S. Identification and quantitative analysis of nucleosides and nucleobases in aqueous extracts of Fritillaria cirrhosa D. DON. using HPLC-DAD and HPLC-ESI-MS. Anal. Lett. 2011, 44, 2491–2502. [Google Scholar] [CrossRef]
- Cheng, W.; Zhang, X.; Song, Q.; Lu, W.; Wu, T.; Zhang, Q.; Li, C. Determination and comparative analysis of 13 nucleosides and nucleobases in natural fruiting body of Ophiocordyceps sinensis and its substitutes. Mycology 2017, 8, 318–326. [Google Scholar] [CrossRef] [PubMed]
- Ferrag, C.; Noroozifar, M.; Kerman, K. Thiol functionalized carbon ceramic electrode modified with multi-walled carbon nanotubes and gold nanoparticles for simultaneous determination of purine derivatives. Mater. Sci. Eng. C 2020, 110, 110568. [Google Scholar] [CrossRef]
- Rajabi, H.; Noroozifar, M.; Khorasani-Motlagh, M. Graphite paste electrode modified with Lewatit® FO36 nano-resin for simultaneous determination of ascorbic acid, acetaminophen and tryptophan. Anal. Methods 2016, 8, 1924–1934. [Google Scholar] [CrossRef]
- Velasco, J.G. Determination of standard rate constants for electrochemical irreversible processes from linear sweep voltammograms. Electroanalysis 1997, 9, 880–882. [Google Scholar] [CrossRef]
- Noroozifar, M.; Khorasani-Motlagh, M.; Tavakkoli, H. Preparation of tetraheptylammonium iodide-iodine graphite-multiwall carbon nanotube paste electrode: Electrocatalytic determination of ascorbic acid in pharmaceuticals and foods. Anal. Sci. 2011, 27, 929–935. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analytes | Linear Relationships | Slopes (mV/pH) | Regression Coefficients |
|---|---|---|---|
| UA | Ep (UA) = −0.0625 pH + 0.7084 | 62.5 | R² = 0.9911 |
| G | Ep (G) = −0.0561 pH + 1.0518 | 56.1 | R² = 0.9905 |
| A | Ep (A) = −0.0601 pH + 1.3644 | 60.1 | R² = 0.9906 |
| T | Ep (T) = −0.0582 pH + 1.5672 | 58.2 | R² = 0.9907 |
| C | Ep (C) = −0.0458 pH + 1.6268 | 52.7 | R² = 0.9913 |
| Electrode | Analytes | Detection Limit (µM) | Linear Range (µM) | Reference |
|---|---|---|---|---|
| Graphene-Nafion modified GCE | G | 0.58 | 2–120 | [37] |
| A | 0.75 | 5–170 | ||
| MWCNT-Fe3O4 coated with PDA-Ag modified GPE | G | 1.5 | 8–130 | [38] |
| A | 5.7 | 0.1–120 | ||
| GMC modified GCE | G | 0.76 | 25–150 | [39] |
| A | 0.63 | 25–200 | ||
| PANI-MnO2 modified GCE | G | 4.8 | 10–100 | [32] |
| A | 2.9 | 10–100 | ||
| T | 1.3 | 10–100 | ||
| C | 1.3 | 10–100 | ||
| MWCNT-graphite deposited with Au-rGO modified GPE | G | 3.3 | 3–170 | [5] |
| A | 3.7 | 3–190 | ||
| T | 7.9 | 7.5–800 | ||
| C | 9.0 | 9–900 | ||
| Prussian blue/ poly(4-aminosalicylic acid)/ uricase modified graphite screen-printed electrode | UA | 3.0 | 10–200 | [40] |
| Screen-printed graphene electrode | UA | 0.4 | 1.2–1.8 | [41] |
| Poly(DPA) on SiO2@Fe3O4 modified screen-printed graphene electrode | UA | 0.20 | 0.8–2500 | [33] |
| Au-Nafion-modified carbon screen-printed electrode | UA | 0.3 | 0.5–600 | [42] |
| GO-MWCNT hybrid with CHT modified GCE | UA | 0.12 | 1–151 | This work |
| G | 0.11 | 1–78 | ||
| A | 0.43 | 2–119.5 | ||
| T | 1.71 | 12.5–227.5 | ||
| C | 0.80 | 5–132.5 |
| Real Sample | Analytes | Spiked Concentration (μM) | Detected Concentration (μM) | % Recovery |
|---|---|---|---|---|
| Human serum | G | 5 | 5.0 | 100 |
| A | 10 | 10.4 | 104 | |
| T | 13.33 | 13.3 | 100 | |
| C | 83.3 | 80.8 | 97 | |
| Human saliva | G | 51.25 | 52.6 | 103 |
| A | 32.5 | 32.4 | 100 | |
| T | 62.5 | 59.1 | 95 | |
| C | 62.5 | 64.5 | 103 | |
| Artificial saliva | G | 50 | 48.1 | 96 |
| A | 50 | 48.2 | 96 | |
| T | 100 | 98.6 | 99 | |
| C | 100 | 105.0 | 105 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, S.; Ferrag, C.; Noroozifar, M.; Kerman, K. Simultaneous Determination of Four DNA bases at Graphene Oxide/Multi-Walled Carbon Nanotube Nanocomposite-Modified Electrode. Micromachines 2020, 11, 294. https://doi.org/10.3390/mi11030294
Wang S, Ferrag C, Noroozifar M, Kerman K. Simultaneous Determination of Four DNA bases at Graphene Oxide/Multi-Walled Carbon Nanotube Nanocomposite-Modified Electrode. Micromachines. 2020; 11(3):294. https://doi.org/10.3390/mi11030294
Chicago/Turabian StyleWang, Shuting, Celia Ferrag, Meissam Noroozifar, and Kagan Kerman. 2020. "Simultaneous Determination of Four DNA bases at Graphene Oxide/Multi-Walled Carbon Nanotube Nanocomposite-Modified Electrode" Micromachines 11, no. 3: 294. https://doi.org/10.3390/mi11030294
APA StyleWang, S., Ferrag, C., Noroozifar, M., & Kerman, K. (2020). Simultaneous Determination of Four DNA bases at Graphene Oxide/Multi-Walled Carbon Nanotube Nanocomposite-Modified Electrode. Micromachines, 11(3), 294. https://doi.org/10.3390/mi11030294