Single-Molecule Mechanics in Ligand Concentration Gradient

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Preparation
2.2. Single-Molecule Manipulation and Imaging
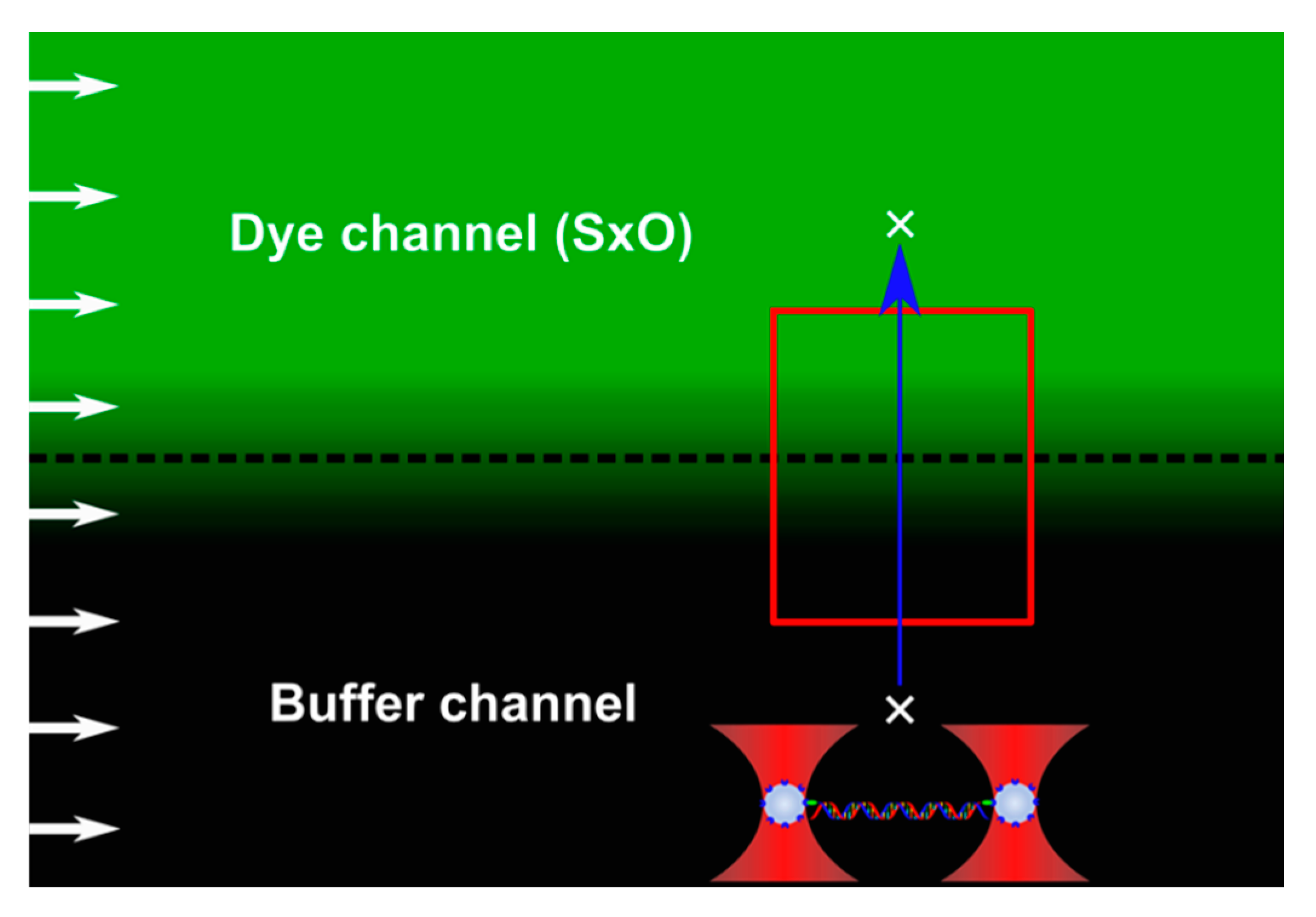
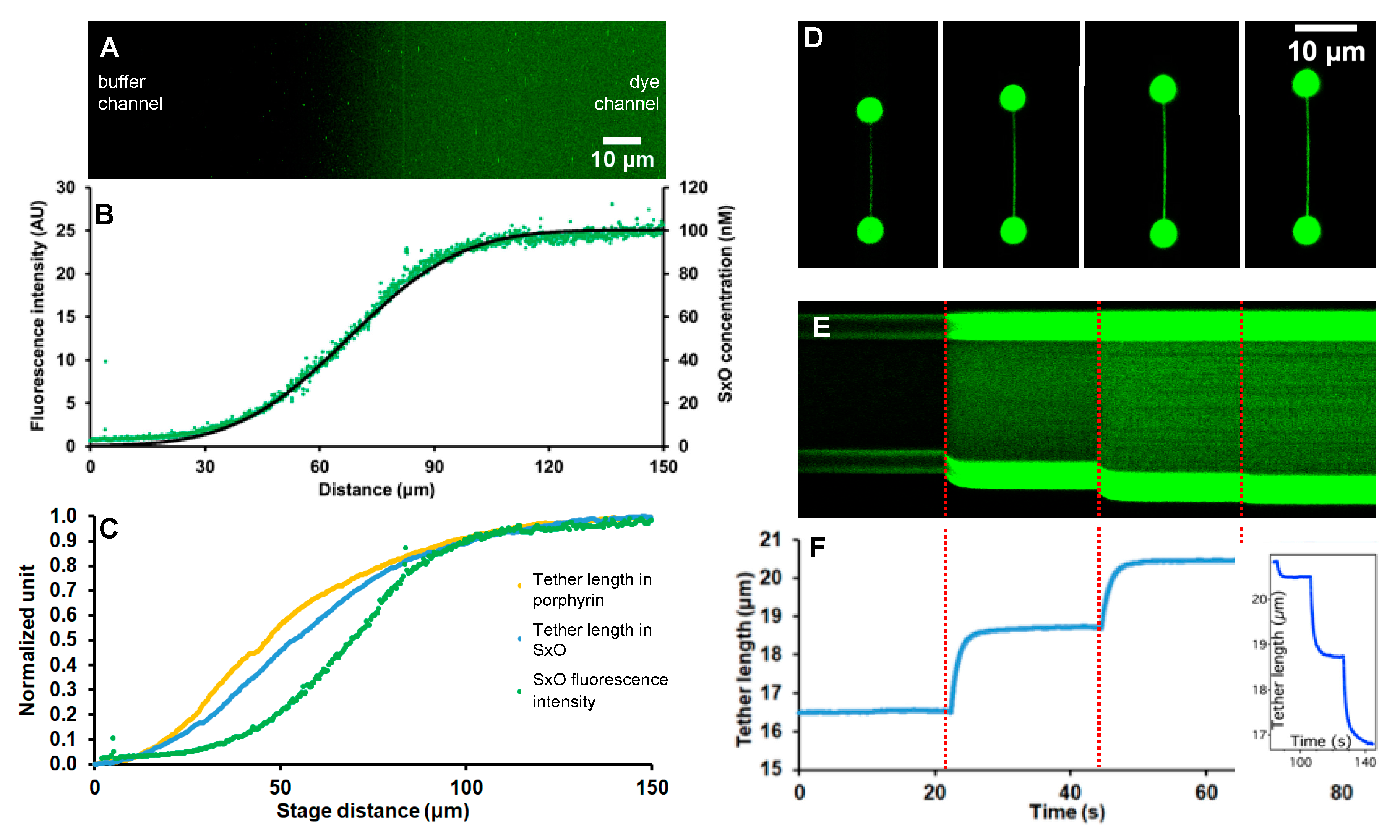
2.3. Calibration of Concentration Gradient with Fluorescence
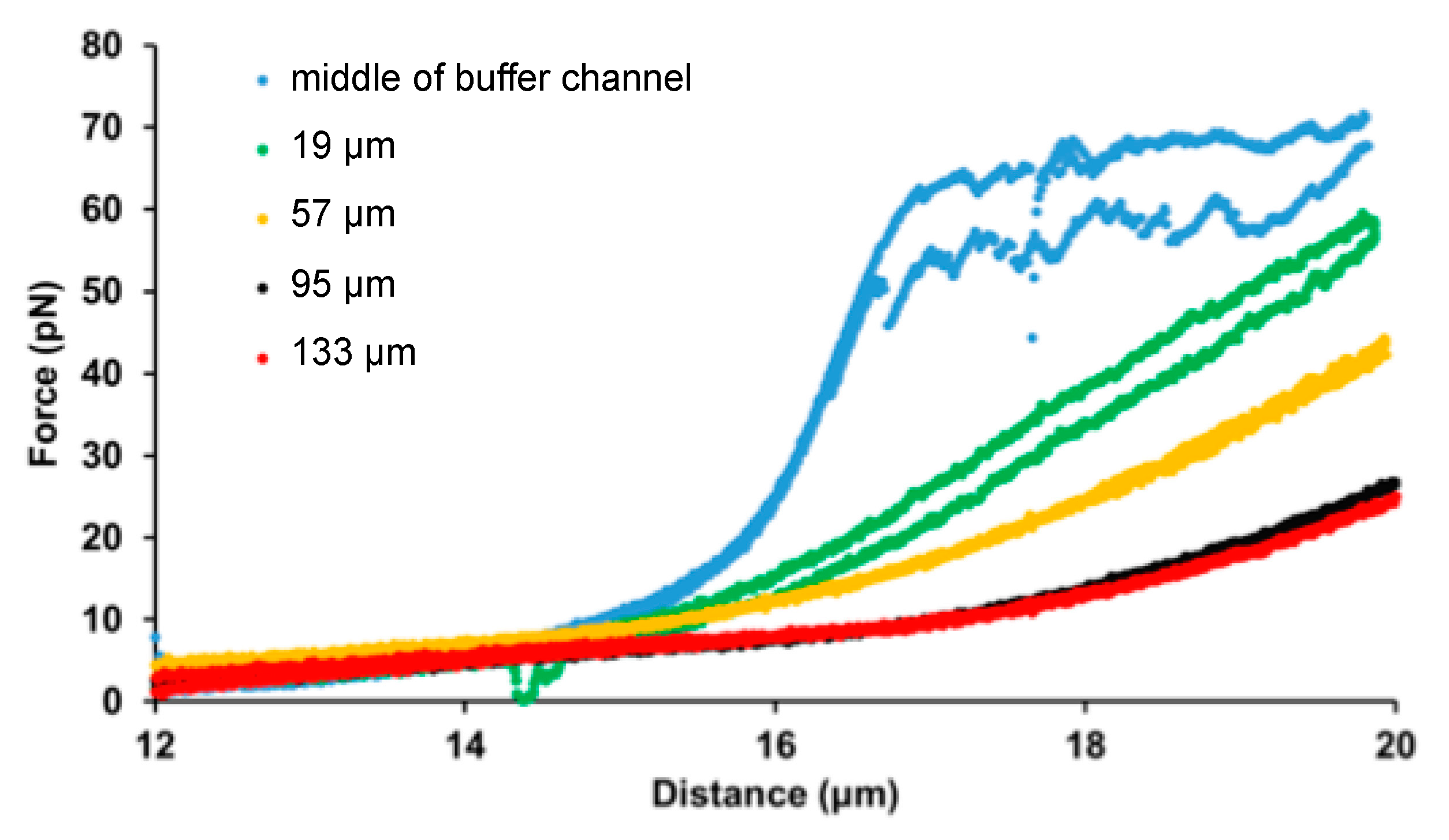
2.4. Molecular Scanning of the Ligand Concentration Gradient
2.5. Theory
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Block, S.M. Making light work with optical tweezers. Nature 1992, 360, 493–495. [Google Scholar] [CrossRef] [PubMed]
- Bustamante, C.; Bryant, Z.; Smith, S.B. Ten years of tension: Single-molecule DNA mechanics. Nature 2003, 421, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Su, Q.P.; Ju, L.A. Biophysical nanotools for single-molecule dynamics. Biophys. Rev. 2018, 10, 1349–1357. [Google Scholar] [CrossRef] [PubMed]
- Bustamante, C.; Macosko, J.C.; Wuite, G.J.L. Grabbing the cat by the tail: Manipulating molecules one by one. Nat. Rev. Mol. Cell Biol. 2000, 1, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Bustamante, C.; Smith, S.B.; Liphardt, J.; Smith, D. Single-molecule studies of DNA mechanics. Curr. Opin. Struct. Biol. 2000, 10, 279–285. [Google Scholar] [CrossRef]
- Forties, R.A.; Wang, M.D. Discovering the Power of Single Molecules. Cell 2014, 157, 4–7. [Google Scholar] [CrossRef][Green Version]
- Grier, D.G. A revolution in optical manipulation. Nature 2003, 424, 810–816. [Google Scholar] [CrossRef]
- Ishii, Y.; Ishijima, A.; Yanagida, T. Single molecule nanomanipulation of biomolecules. Trends Biotechnol. 2001, 19, 211–216. [Google Scholar] [CrossRef]
- Kim, H.; Ha, T. Single-molecule nanometry for biological physics. Rep. Prog. Phys. 2012, 76, 16601. [Google Scholar] [CrossRef]
- Mehta, A.D.; Rief, M.; Spudich, J.A. Biomechanics, One molecule at a Time. J. Biol. Chem. 1999, 274, 14517–14520. [Google Scholar] [CrossRef]
- Mehta, A.D.; Rief, M.; Spudich, J.A.; Smith, D.A.; Simmons, R.M. Single-molecule biomechanics with optical methods. Science 1999, 283, 1689–1695. [Google Scholar] [CrossRef] [PubMed]
- Dickson, R.M.; Cubitt, A.B.; Tsien, R.Y.; Moerner, W.E. On/off blinking and switching behaviour of single molecules of green fluorescent protein. Nature 1997, 388, 355–358. [Google Scholar] [CrossRef] [PubMed]
- Kellermayer, M.S.Z.; Smith, S.B.; Granzier, H.L.; Bustamante, C. Folding-Unfolding Transitions in Single Titin Molecules Characterized with Laser Tweezers. Science 1997, 276, 1112–1116. [Google Scholar] [CrossRef] [PubMed]
- Rief, M.; Gautel, M.; Oesterhelt, F.; Fernandez, J.M.; Gaub, H.E. Reversible unfolding of individual Titin immunoglobulin domains by AFM. Science 1997, 276, 1109–1112. [Google Scholar] [CrossRef]
- Tskhovrebova, L.; Trinick, J.; Sleep, J.A.; Simmons, R.M. Elasticity and unfolding of single molecules of the giant muscle protein titin. Nature 1997, 387, 308–312. [Google Scholar] [CrossRef]
- Finer, J.T.; Simmons, R.M.; Spudich, J.A. Single myosin molecule mechanics: Piconewton forces and nanometre steps. Nature 1994, 368, 113–119. [Google Scholar] [CrossRef]
- Molloy, J.E.; Burns, J.E.; Kendrick-Jones, J.; Tregear, R.T.; White, D.C.S. Movement and force produced by a single myosin head. Nature 1995, 378, 209–212. [Google Scholar] [CrossRef]
- Svoboda, K.; Schmidt, C.F.; Schnapp, B.J.; Block, S.M. Direct observation of kinesin stepping by optical trapping interferometry. Nature 1993, 365, 721–727. [Google Scholar] [CrossRef]
- Ashkin, A. Acceleration and trapping of particles by radiation pressure. Phys. Rev. Lett. 1970, 24, 156–159. [Google Scholar] [CrossRef]
- Ashkin, A. Forces of a single-beam gradient laser trap on a dielectric sphere in the ray optics regime. Biophys. J. 1992, 61, 569–582. [Google Scholar] [CrossRef]
- Svoboda, K.; Block, S.M. Biological applications of optical forces. Annu. Rev. Biophys. Biomol. Struct. 1994, 23, 247–285. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.B.; Cui, Y.; Bustamante, C. Overstretching B-DNA: The elastic response of individual double-stranded and single-stranded DNA molecules. Science 1996, 271, 795–799. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.D.; Yin, H.; Landick, R.; Gelles, J.; Block, S.M. Stretching DNA with optical tweezers. Biophys. J. 1997, 72, 1335–1346. [Google Scholar] [CrossRef]
- Liphardt, J.; Onoa, B.; Smith, S.B.; Tinoco, I.J.; Bustamante, C. Reversible unfolding of single RNA molecules by mechanical force. Science 2001, 292, 733–737. [Google Scholar] [CrossRef] [PubMed]
- Boer, G.; Johann, R.; Rohner, J.; Merenda, F.; Delacrétaz, G.; Renaud, P.; Salathé, R.-P. Combining multiple optical trapping with microflow manipulation for the rapid bioanalytics on microparticles in a chip. Rev. Sci. Instrum. 2007, 78, 116101. [Google Scholar] [CrossRef]
- Gross, P.; Farge, G.; Peterman, E.J.G.; Wuite, G.J.L. Combining Optical Tweezers, Single-Molecule Fluorescence Microscopy, and Microfluidics for Studies of DNA–Protein Interactions. Methods Enzymol. 2010, 475, 427–453. [Google Scholar] [CrossRef]
- Abrahamse, H.; Hamblin, M.R. New photosensitizers for photodynamic therapy. Biochem. J. 2016, 473, 347–364. [Google Scholar] [CrossRef]
- Almaqwashi, A.A.; Zhou, W.; Naufer, M.N.; Riddell, I.A.; Yilmaz, Ö.H.; Lippard, S.J.; Williams, M.C. DNA intercalation facilitates efficient DNA-targeted covalent binding of phenanthriplatin. J. Am. Chem. Soc. 2019, 141, 1537–1545. [Google Scholar] [CrossRef]
- Bahira, M.; McCauley, M.J.; Almaqwashi, A.A.; Lincoln, P.; Westerlund, F.; Rouzina, I.; Williams, M.C. A ruthenium dimer complex with a flexible linker slowly threads between DNA bases in two distinct steps. Nucleic Acids Res. 2015, 43, 8856–8867. [Google Scholar] [CrossRef]
- Crisafuli, F.A.P.; Cesconetto, E.C.; Ramos, E.B.; Rocha, M.S. DNA–cisplatin interaction studied with single molecule stretching experiments. Integr. Biol. 2012, 4, 568–574. [Google Scholar] [CrossRef]
- Sischka, A.; Toensing, K.; Eckel, R.; Wilking, S.D.; Sewald, N.; Ros, R.; Anselmetti, D. Molecular Mechanisms and Kinetics between DNA and DNA Binding Ligands. Biophys. J. 2005, 88, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Vladescu, I.D.; McCauley, M.J.; Nuñez, M.E.; Rouzina, I.; Williams, M.C. Quantifying force-dependent and zero-force DNA intercalation by single-molecule stretching. Nat. Methods 2007, 4, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Hembury, G.A.; Borovkov, V.V.; Inoue, Y. Chirality-sensing supramolecular systems. Chem. Rev. 2008, 108, 1–73. [Google Scholar] [CrossRef] [PubMed]
- Kakiuchi, T.; Ito, F.; Nagamura, T. Time-resolved studies of energy transfer from meso-tetrakis(n-methylpyridinium-4-yl)-porphyrin to 3,3′-diethyl-2,2′-thiatricarbocyanine iodide along deoxyribonucleic acid chain. J. Phys. Chem. B 2008, 112, 3931–3937. [Google Scholar] [CrossRef] [PubMed]
- Mutsamwira, S.; Ainscough, E.W.; Partridge, A.C.; Derrick, P.J.; Filichev, V.V. DNA duplex as a scaffold for a ground state complex formation between a zinc cationic porphyrin and phenylethynylpyren-1-yl. J. Photochem. Photobiol. A Chem. 2014, 288, 76–81. [Google Scholar] [CrossRef]
- Pathak, P.; Yao, W.; Hook, K.D.; Vik, R.; Winnerdy, F.R.; Brown, J.Q.; Gibb, B.C.; Pursell, Z.F.; Phan, A.T.; Jayawickramarajah, J. Bright G-Quadruplex nanostructures functionalized with porphyrin lanterns. J. Am. Chem. Soc. 2019, 141, 12582–12591. [Google Scholar] [CrossRef]
- Stulz, E. Nanoarchitectonics with porphyrin functionalized DNA. Acc. Chem. Res. 2017, 50, 823–831. [Google Scholar] [CrossRef]
- Biebricher, A.S.; Heller, I.; Roijmans, R.F.H.; Hoekstra, T.P.; Peterman, E.J.G.; Wuite, G.J.L. The impact of DNA intercalators on DNA and DNA-processing enzymes elucidated through force-dependent binding kinetics. Nat. Commun. 2015, 6, 7304. [Google Scholar] [CrossRef]
- Almaqwashi, A.A.; Paramanathan, T.; Rouzina, I.; Williams, M.C. Mechanisms of small molecule–DNA interactions probed by single-molecule force spectroscopy. Nucleic Acids Res. 2016, 44, 3971–3988. [Google Scholar] [CrossRef]
- Fiel, R.J. Porphyrin—Nucleic acid interactions: A Review. J. Biomol. Struct. Dyn. 1989, 6, 1259–1274. [Google Scholar] [CrossRef]
- Sehlstedt, U.; Kim, S.K.; Carter, P.; Goodisman, J.; Vollano, J.F.; Norden, B.; Dabrowiak, J.C. Interaction of cationic porphyrins with DNA. Biochemistry 1994, 33, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.-J.; Cui, Y.; Chen, C.-S.; Wei, W.H.; Chang, R.-S.; Shu, W.-Y.; Hsu, I.C. Freezing shortens the lifetime of DNA molecules under tension. J. Biol. Phys. 2017, 43, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.L.; Erbay, C.; Sadr, R.; Han, A. Dynamic flow characteristics and design principles of laminar flow microbial fuel cells. Micromachines 2018, 9, 479. [Google Scholar] [CrossRef] [PubMed]
- Bokstein, B.S.; Mendelev, M.I.; Srolovitz, D.J. Thermodynamics and Kinetics in Materials Science: A Short Course; Oxford University Press: New York, NY, USA, 2005; ISBN 9780198528036. [Google Scholar]
- Sørensen, K.T.; Kristensen, A. Label-free monitoring of diffusion in microfluidics. Micromachines 2017, 8, 329. [Google Scholar] [CrossRef]
- Eriksson, E.; Enger, J.; Nordlander, B.; Erjavec, N.; Ramser, K.; Goksör, M.; Hohmann, S.; Nyström, T.; Hanstorp, D. A microfluidic system in combination with optical tweezers for analyzing rapid and reversible cytological alterations in single cells upon environmental changes. Lab Chip 2007, 7, 71–76. [Google Scholar] [CrossRef]
- Eriksson, E.; Scrimgeour, J.; Granéli, A.; Ramser, K.; Wellander, R.; Enger, J.; Hanstorp, D.; Goksör, M. Optical manipulation and microfluidics for studies of single cell dynamics. J. Opt. A Pure Appl. Opt. 2007, 9, S113. [Google Scholar] [CrossRef]
- Eriksson, E.; Sott, K.; Lundqvist, F.; Sveningsson, M.; Scrimgeour, J.; Hanstorp, D.; Goksör, M.; Granéli, A. A microfluidic device for reversible environmental changes around single cells using optical tweezers for cell selection and positioning. Lab Chip 2010, 10, 617–625. [Google Scholar] [CrossRef]
- Yan, X.; Habbersett, R.C.; Cordek, J.M.; Nolan, J.P.; Yoshida, T.M.; Jett, J.H.; Marrone, B.L. Development of a mechanism-based, DNA staining protocol using SYTOX orange nucleic acid stain and DNA fragment sizing flow cytometry. Anal. Biochem. 2000, 286, 138–148. [Google Scholar] [CrossRef]
- Kellermayer, M.S.Z.; Smith, S.B.; Bustamante, C.; Granzier, H.L. Complete unfolding of the titin molecule under external force. J. Struct. Biol. 1998, 122, 197–205. [Google Scholar] [CrossRef]
- Mandal, S.S.; Merz, D.R.; Buchsteiner, M.; Dima, R.I.; Rief, M.; Žoldák, G. Nanomechanics of the substrate binding domain of Hsp70 determine its allosteric ATP-induced conformational change. Proc. Natl. Acad. Sci. USA 2017, 114, 6040–6045. [Google Scholar] [CrossRef]
- Caldarini, M.; Sonar, P.; Valpapuram, I.; Tavella, D.; Volonté, C.; Pandini, V.; Vanoni, M.A.; Aliverti, A.; Broglia, R.A.; Tiana, G.; et al. The complex folding behavior of HIV-1-protease monomer revealed by optical-tweezer single-molecule experiments and molecular dynamics simulations. Biophys. Chem. 2014, 195, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Canavan, C.; Taylor, S.S.; Maillard, R.A. Integrated method to attach DNA handles and functionally select proteins to study folding and protein-ligand interactions with optical tweezers. Sci. Rep. 2017, 7, 10843. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Cui, Y.; Selvam, S.; Ghimire, C.; Mao, H. Dissecting cooperative communications in a protein with a high-throughput single-molecule scalpel. ChemPhysChem 2015, 16, 223–232. [Google Scholar] [CrossRef]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kretzer, B.; Kiss, B.; Tordai, H.; Csík, G.; Herényi, L.; Kellermayer, M. Single-Molecule Mechanics in Ligand Concentration Gradient. Micromachines 2020, 11, 212. https://doi.org/10.3390/mi11020212
Kretzer B, Kiss B, Tordai H, Csík G, Herényi L, Kellermayer M. Single-Molecule Mechanics in Ligand Concentration Gradient. Micromachines. 2020; 11(2):212. https://doi.org/10.3390/mi11020212
Chicago/Turabian StyleKretzer, Balázs, Bálint Kiss, Hedvig Tordai, Gabriella Csík, Levente Herényi, and Miklós Kellermayer. 2020. "Single-Molecule Mechanics in Ligand Concentration Gradient" Micromachines 11, no. 2: 212. https://doi.org/10.3390/mi11020212
APA StyleKretzer, B., Kiss, B., Tordai, H., Csík, G., Herényi, L., & Kellermayer, M. (2020). Single-Molecule Mechanics in Ligand Concentration Gradient. Micromachines, 11(2), 212. https://doi.org/10.3390/mi11020212