Microfluidic Array Chip for Parallel Detection of Waterborne Bacteria





Abstract
1. Introduction
2. Materials and Methods
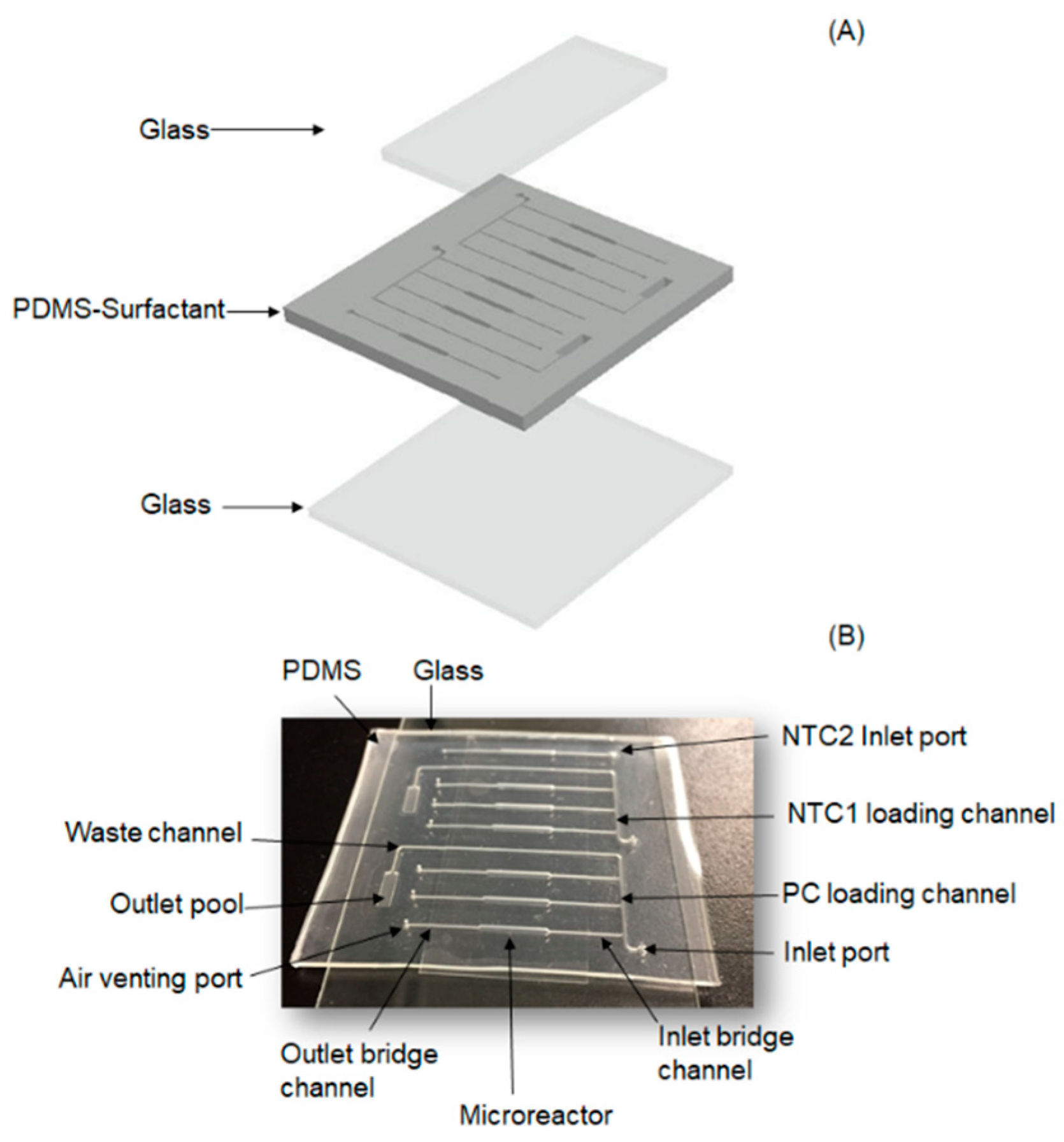
2.1. Chip Design
2.2. Chip Fabrication
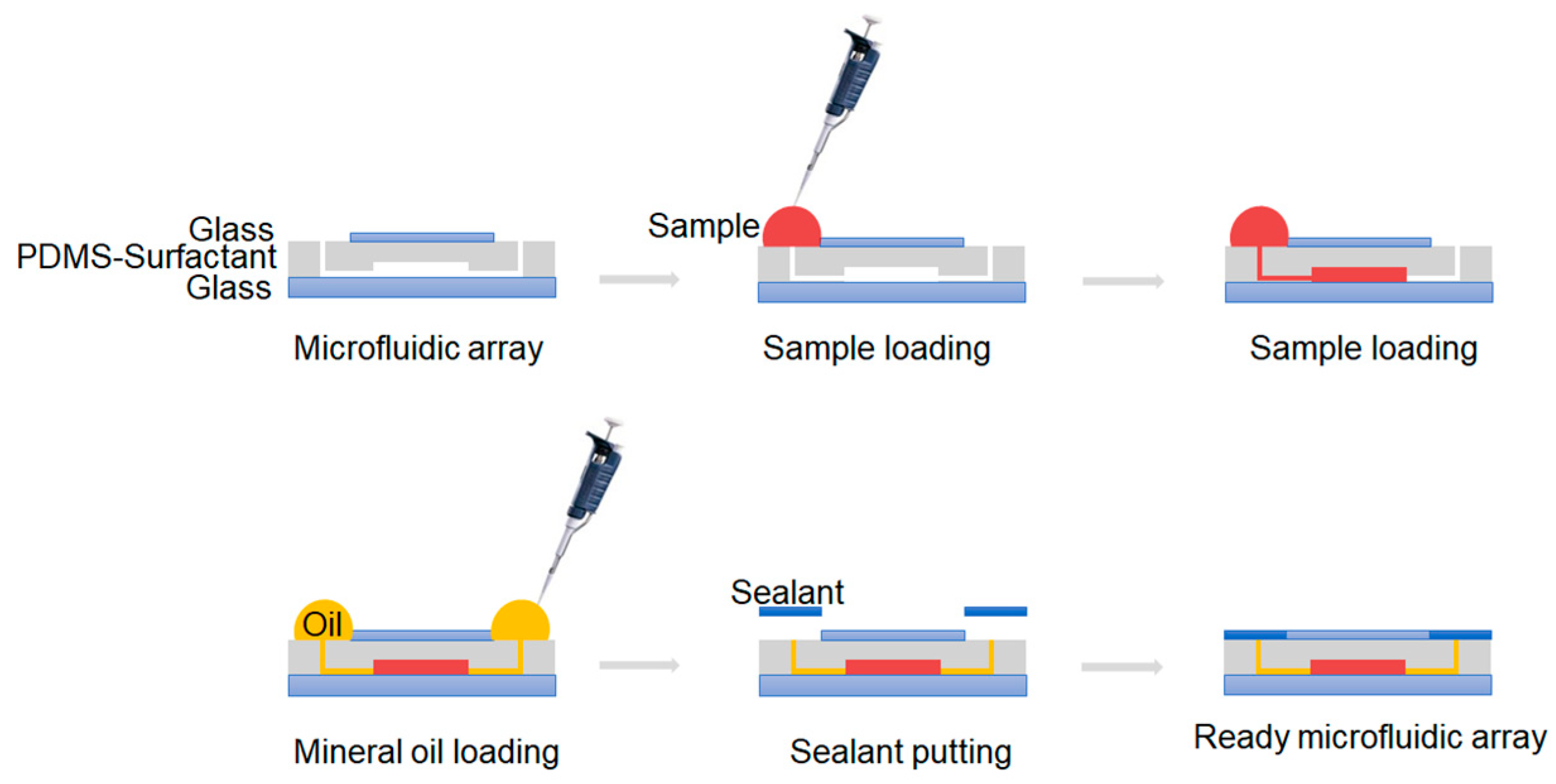
2.3. Chip Operation
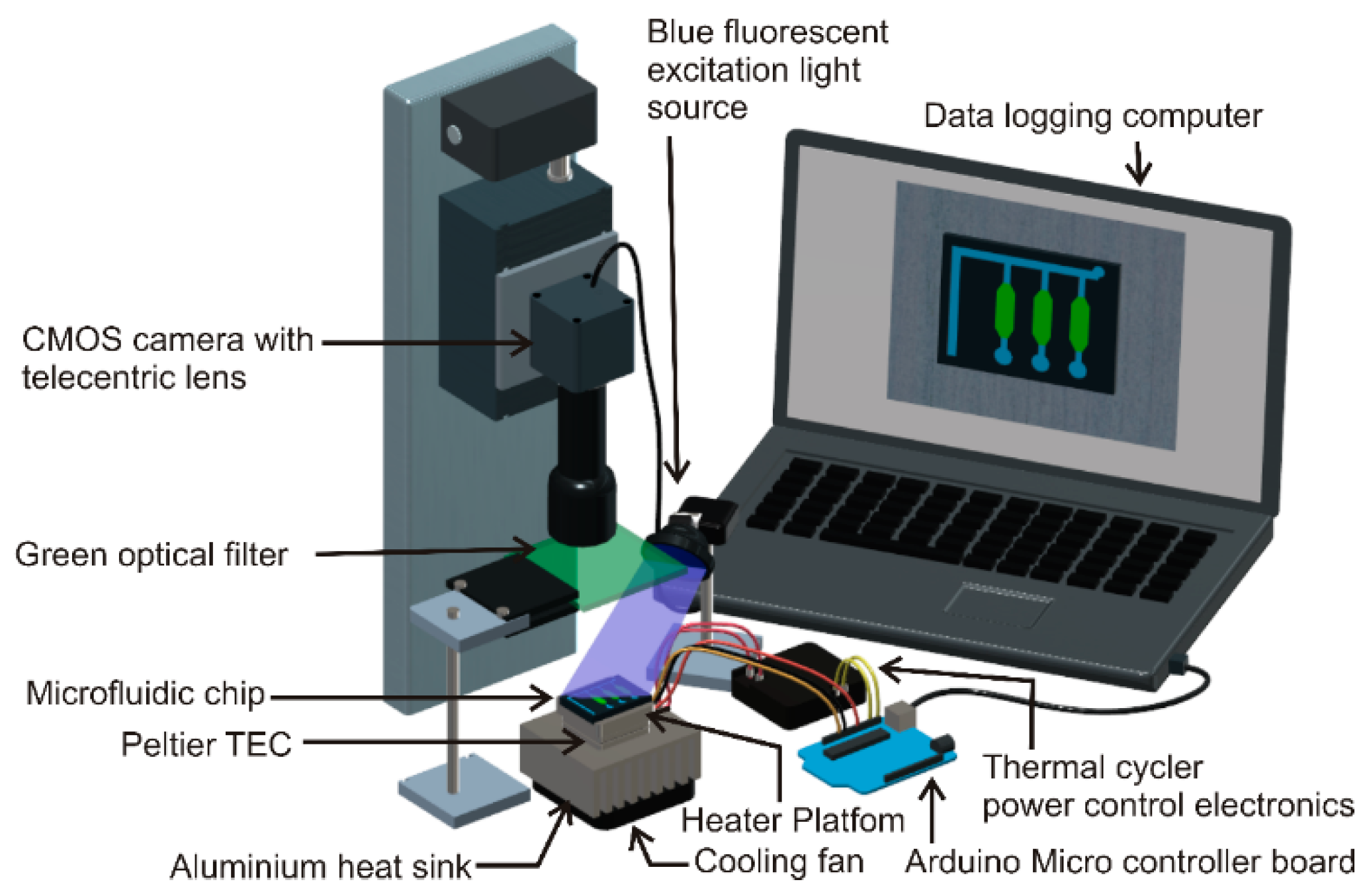
2.4. Real-Time Polymerase Chain Reaction (PCR) Instrument
2.5. Bacterial Samples and PCR Experiment
3. Results
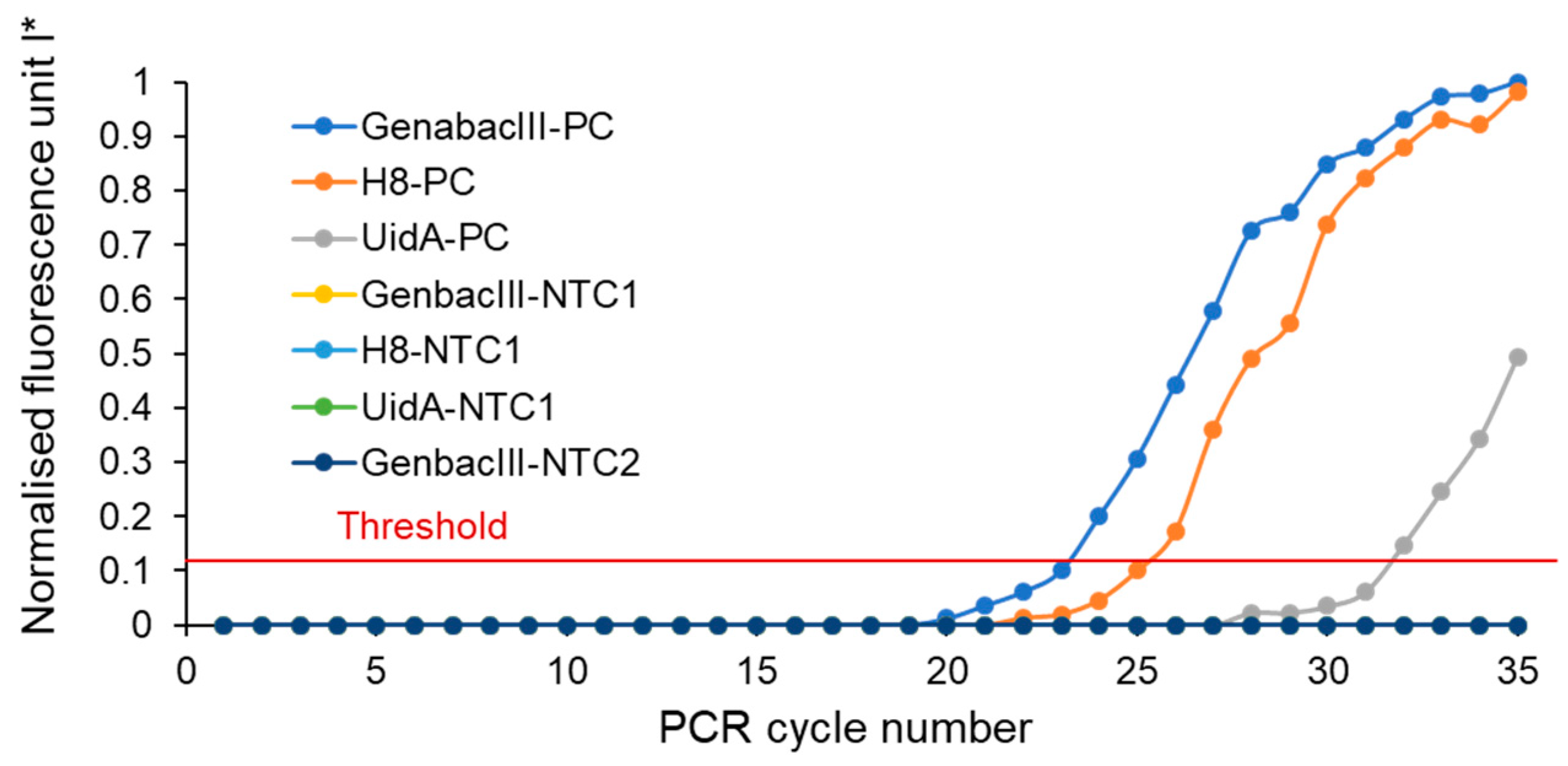
3.1. Simultaneous Detection of Microbial Faecal Source Tracking (MST) Markers Using Microfluidic Device
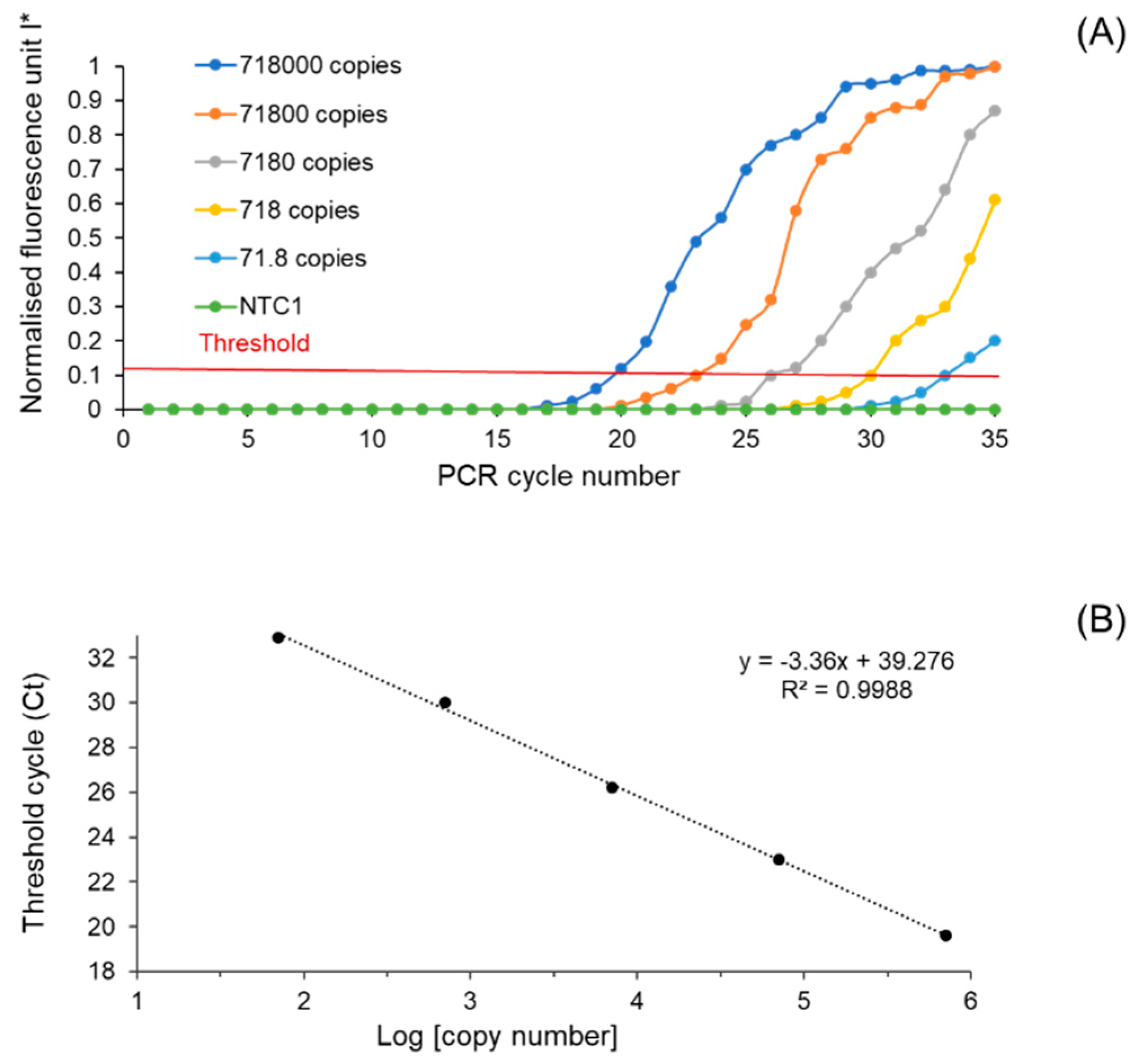
3.2. Performance Characteristics of the Developed Microfluidic Device
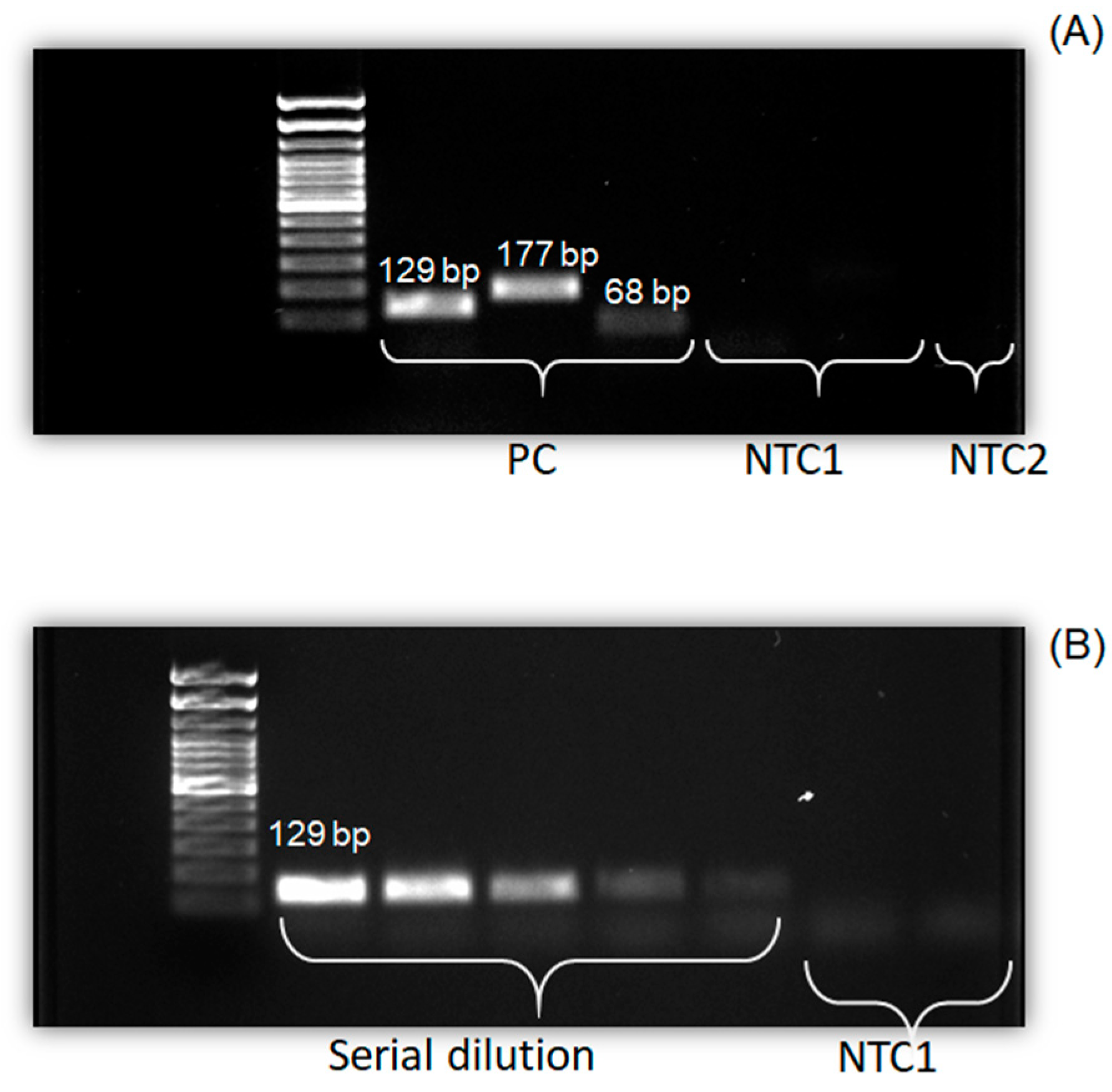
3.3. Validation of Real-Time PCR Using Gel Electrophoresis
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Napier, M.D.; Haugland, R.; Poole, C.; Dufour, A.P.; Stewart, J.R.; Weber, D.J.; Varma, M.; Lavender, J.S.; Wade, T.J. Exposure to human-associated fecal indicators and self-reported illness among swimmers at recreational beaches: A cohort study. Environ. Health 2017, 16, 103. [Google Scholar] [CrossRef] [PubMed]
- Farnleitner, A.H.; Ryzinska-Paier, G.; Reischer, G.H.; Burtscher, M.M.; Knetsch, S.; Kirschner, A.K.; Dirnböck, T.; Kuschnig, G.; Mach, R.L.; Sommer, R. Escherichia coli and enterococci are sensitive and reliable indicators for human, livestock and wildlife faecal pollution in alpine mountainous water resources. J. Appl. Microbiol. 2010, 109, 1599–1608. [Google Scholar] [CrossRef] [PubMed]
- Hagedorn, C.; Blanch, A.R.; Harwood, V.J. Microbial Source Tracking: Methods, Applications, and Case Studies; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2011. [Google Scholar]
- Harwood, V.J.; Staley, C.; Badgley, B.D.; Borges, K.; Korajkic, A. Microbial source tracking markers for detection of fecal contamination in environmental waters: Relationships between pathogens and human health outcomes. FEMS Microbiol. Rev. 2014, 38, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Gorgannezhad, L.; Umer, M.; Islam, M.N.; Nguyen, N.-T.; Shiddiky, M.J. Circulating tumor DNA and liquid biopsy: Opportunities, challenges, and recent advances in detection technologies. Lab Chip 2018, 18, 1174–1196. [Google Scholar] [CrossRef] [PubMed]
- Gorgannezhad, L.; Umer, M.; Kamal Masud, M.; Hossain, M.S.A.; Tanaka, S.; Yamauchi, Y.; Salomon, C.; Kline, R.; Nguyen, N.T.; Shiddiky, M.J. Detection of FGFR2: FAM76A Fusion Gene in Circulating Tumor RNA Based on Catalytic Signal Amplification of Graphene Oxide-loaded Magnetic Nanoparticles. Electroanalysis 2018, 30, 2293–2301. [Google Scholar] [CrossRef]
- Gorgannezhad, L.; Stratton, H.; Nguyen, N.-T. Microfluidic-Based Nucleic Acid Amplification Systems in Microbiology. Micromachines 2019, 10, 408. [Google Scholar] [CrossRef]
- Ramírez-Castillo, F.; Loera-Muro, A.; Jacques, M.; Garneau, P.; Avelar-González, F.; Harel, J.; Guerrero-Barrera, A. Waterborne pathogens: Detection methods and challenges. Pathogens 2015, 4, 307–334. [Google Scholar] [CrossRef]
- Kong, R.; Lee, S.; Law, T.; Law, S.; Wu, R. Rapid detection of six types of bacterial pathogens in marine waters by multiplex PCR. Water Res. 2002, 36, 2802–2812. [Google Scholar] [CrossRef]
- Law, J.W.-F.; Ab Mutalib, N.-S.; Chan, K.-G.; Lee, L.-H. Rapid methods for the detection of foodborne bacterial pathogens: Principles, applications, advantages and limitations. Front. Microbiol. 2015, 5, 770. [Google Scholar] [CrossRef]
- Lee, D.-Y.; Shannon, K.; Beaudette, L.A. Detection of bacterial pathogens in municipal wastewater using an oligonucleotide microarray and real-time quantitative PCR. J. Microbiol. Methods 2006, 65, 453–467. [Google Scholar] [CrossRef]
- Yauk, C.L.; Berndt, M.L.; Williams, A.; Douglas, G.R. Comprehensive comparison of six microarray technologies. Nucleic Acids Res. 2004, 32, e124. [Google Scholar] [CrossRef] [PubMed]
- Bing, D.H.; Boles, C.; Rehman, F.N.; Audeh, M.; Belmarsh, M.; Kelley, B.; Adams, C.P. Bridge amplification: A solid phase PCR system for the amplification and detection of allelic differences in single copy genes. In Proceedings of the Genetic Identity Conference Proceedings, Seventh International Symposium on Human Identification, Scottsdale, AZ, USA, 18 September 1996; pp. 18–20. [Google Scholar]
- Korostynska, O.; Mason, A.; Al-Shamma’a, A. Monitoring pollutants in wastewater: Traditional lab based versus modern real-time approaches. In Smart Sensors for Real-Time Water Quality Monitoring; Springer: Berlin/Heidelberg, Germany, 2013; pp. 1–24. [Google Scholar]
- Nagai, H.; Murakami, Y.; Morita, Y.; Yokoyama, K.; Tamiya, E. Development of a microchamber array for picoliter PCR. Anal. Chem. 2001, 73, 1043–1047. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, Y.; Kerman, K.; Kobayashi, M.; Yamamura, S.; Morita, Y.; Tamiya, E. Microchamber array based DNA quantification and specific sequence detection from a single copy via PCR in nanoliter volumes. Biosens. Bioelectron. 2005, 20, 1482–1490. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hansen, C.; Quake, S.R. Solving the “world-to-chip” interface problem with a microfluidic matrix. Anal. Chem. 2003, 75, 4718–4723. [Google Scholar] [CrossRef] [PubMed]
- Lagally, E.T.; Simpson, P.C.; Mathies, R.A. Monolithic integrated microfluidic DNA amplification and capillary electrophoresis analysis system. Sens. Actuators B Chem. 2000, 63, 138–146. [Google Scholar] [CrossRef]
- Ramalingam, N.; San, T.C.; Kai, T.J.; Mak, M.Y.M.; Gong, H.-Q. Microfluidic devices harboring unsealed reactors for real-time isothermal helicase-dependent amplification. Microfluid. Nanofluidics 2009, 7, 325. [Google Scholar] [CrossRef]
- Fu, Y.; Zhou, H.; Jia, C.; Jing, F.; Jin, Q.; Zhao, J.; Li, G. A microfluidic chip based on surfactant-doped polydimethylsiloxane (PDMS) in a sandwich configuration for low-cost and robust digital PCR. Sens. Actuators B Chem. 2017, 245, 414–422. [Google Scholar] [CrossRef]
- Freeman, D.; Gray, M.; Aranyosi, A. Manufacturing a PDMS microfluidic device via a Silicon Wafer Master; Harvard-MIT Division of Health Sciences and Technology: Cambridge, MA, USA, 2007. [Google Scholar]
- Liu, H.-B.; Gong, H.-Q.; Ramalingam, N.; Jiang, Y.; Dai, C.-C.; Hui, K.M. Micro air bubble formation and its control during polymerase chain reaction (PCR) in polydimethylsiloxane (PDMS) microreactors. J. Micromech. Microeng. 2007, 17, 2055. [Google Scholar] [CrossRef]
- Sperandio, V.; Nguyen, Y. Enterohemorrhagic E. coli (EHEC) pathogenesis. Front. Cell. Infect. Microbiol. 2012, 2, 90. [Google Scholar]
- Zitomersky, N.L.; Atkinson, B.J.; Franklin, S.W.; Mitchell, P.D.; Snapper, S.B.; Comstock, L.E.; Bousvaros, A. Characterization of adherent bacteroidales from intestinal biopsies of children and young adults with inflammatory bowel disease. PLoS ONE 2013, 8, e63686. [Google Scholar] [CrossRef]
- Ingham, H.; Rich, G.; Selkon, J.; Hale, J.; Roxby, C.; Betty, M.; Johnson, R.; Uldall, P. Treatment with metronidazole of three patients with serious infections due to Bacteroides fragilis. J. Antimicrob. Chemother. 1975, 1, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Rueden, C.T.; Hiner, M.C.; Eliceiri, K.W. The ImageJ ecosystem: An open platform for biomedical image analysis. Mol. Reprod. Dev. 2015, 82, 518–529. [Google Scholar] [CrossRef] [PubMed]
- Sreejith, K.R.; Gorgannezhad, L.; Jin, J.; Ooi, C.H.; Stratton, H.; Dao, D.; Nguyen, N.-T. Liquid marble as a biochemical reactor for polymerase chain reaction. Lab Chip 2019, 19, 3220–3227. [Google Scholar] [CrossRef] [PubMed]
- Veldwijk, M.R.; Topaly, J.; Laufs, S.; Hengge, U.R.; Wenz, F.; Zeller, W.J.; Fruehauf, S. Development and optimization of a real-time quantitative PCR-based method for the titration of AAV-2 vector stocks. Mol. Ther. 2002, 6, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, N.; Rui, Z.; Liu, H.-B.; Dai, C.-C.; Kaushik, R.; Ratnaharika, B.; Gong, H.-Q. Real-time PCR-based microfluidic array chip for simultaneous detection of multiple waterborne pathogens. Sens. Actuators B Chem. 2010, 145, 543–552. [Google Scholar] [CrossRef]
- Fernández-Carballo, B.L.; McGuiness, I.; McBeth, C.; Kalashnikov, M.; Borrós, S.; Sharon, A.; Sauer-Budge, A.F. Low-cost, real-time, continuous flow PCR system for pathogen detection. Biomed. Microdevices 2016, 18, 34. [Google Scholar] [CrossRef]
- Trinh, K.T.L.; Lee, N.Y. Glass-polytetrafluoroethylene-glass based sandwich microdevice for continuous-flow polymerase chain reaction and its application for fast identification of foodborne pathogens. Talanta 2018, 176, 544–550. [Google Scholar] [CrossRef]
- Siefring, S.; Varma, M.; Atikovic, E.; Wymer, L.; Haugland, R. Improved real-time PCR assays for the detection of fecal indicator bacteria in surface waters with different instrument and reagent systems. J. Water Health 2008, 6, 225–237. [Google Scholar] [CrossRef]
- Ohad, S.; Vaizel-Ohayon, D.; Rom, M.; Guttman, J.; Berger, D.; Kravitz, V.; Pilo, S.; Huberman, Z.; Kashi, Y.; Rorman, E. Microbial source tracking in adjacent karst springs. Appl. Environ. Microbiol. 2015, 81, 5037–5047. [Google Scholar] [CrossRef]
- Gomi, R.; Matsuda, T.; Matsui, Y.; Yoneda, M. Fecal source tracking in water by next-generation sequencing technologies using host-specific Escherichia coli genetic markers. Environ. Sci. Technol. 2014, 48, 9616–9623. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Organism | Primer | Sequence (5ʹ-3ʹ) | PCR Product Size (bp) |
|---|---|---|---|
| Bacteroidales | GenbacIII-F GenbacIII-R | GGGGTTCTGAGAGGAAGGT CCGTCATCCTTCACGCTACT | 129 |
| Escherichia coli (E. coli) | H8-F H8-R | ACAGTCAGCGAGATTCTTC GAACGTCAGCACCACCAA | 177 |
| E. coli | UidA-F UidA-R | CGGAAGCAACGCGTAAACTC GAGCGTCGCAGAACATTACATT | 68 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gorgannezhad, L.; Sreejith, K.R.; Zhang, J.; Kijanka, G.; Christie, M.; Stratton, H.; Nguyen, N.-T. Microfluidic Array Chip for Parallel Detection of Waterborne Bacteria. Micromachines 2019, 10, 883. https://doi.org/10.3390/mi10120883
Gorgannezhad L, Sreejith KR, Zhang J, Kijanka G, Christie M, Stratton H, Nguyen N-T. Microfluidic Array Chip for Parallel Detection of Waterborne Bacteria. Micromachines. 2019; 10(12):883. https://doi.org/10.3390/mi10120883
Chicago/Turabian StyleGorgannezhad, Lena, Kamalalayam Rajan Sreejith, Jun Zhang, Gregor Kijanka, Melody Christie, Helen Stratton, and Nam-Trung Nguyen. 2019. "Microfluidic Array Chip for Parallel Detection of Waterborne Bacteria" Micromachines 10, no. 12: 883. https://doi.org/10.3390/mi10120883
APA StyleGorgannezhad, L., Sreejith, K. R., Zhang, J., Kijanka, G., Christie, M., Stratton, H., & Nguyen, N.-T. (2019). Microfluidic Array Chip for Parallel Detection of Waterborne Bacteria. Micromachines, 10(12), 883. https://doi.org/10.3390/mi10120883