Secreted Phospholipases A2 from Animal Venoms in Pain and Analgesia

 and
and
Abstract
:1. Introduction
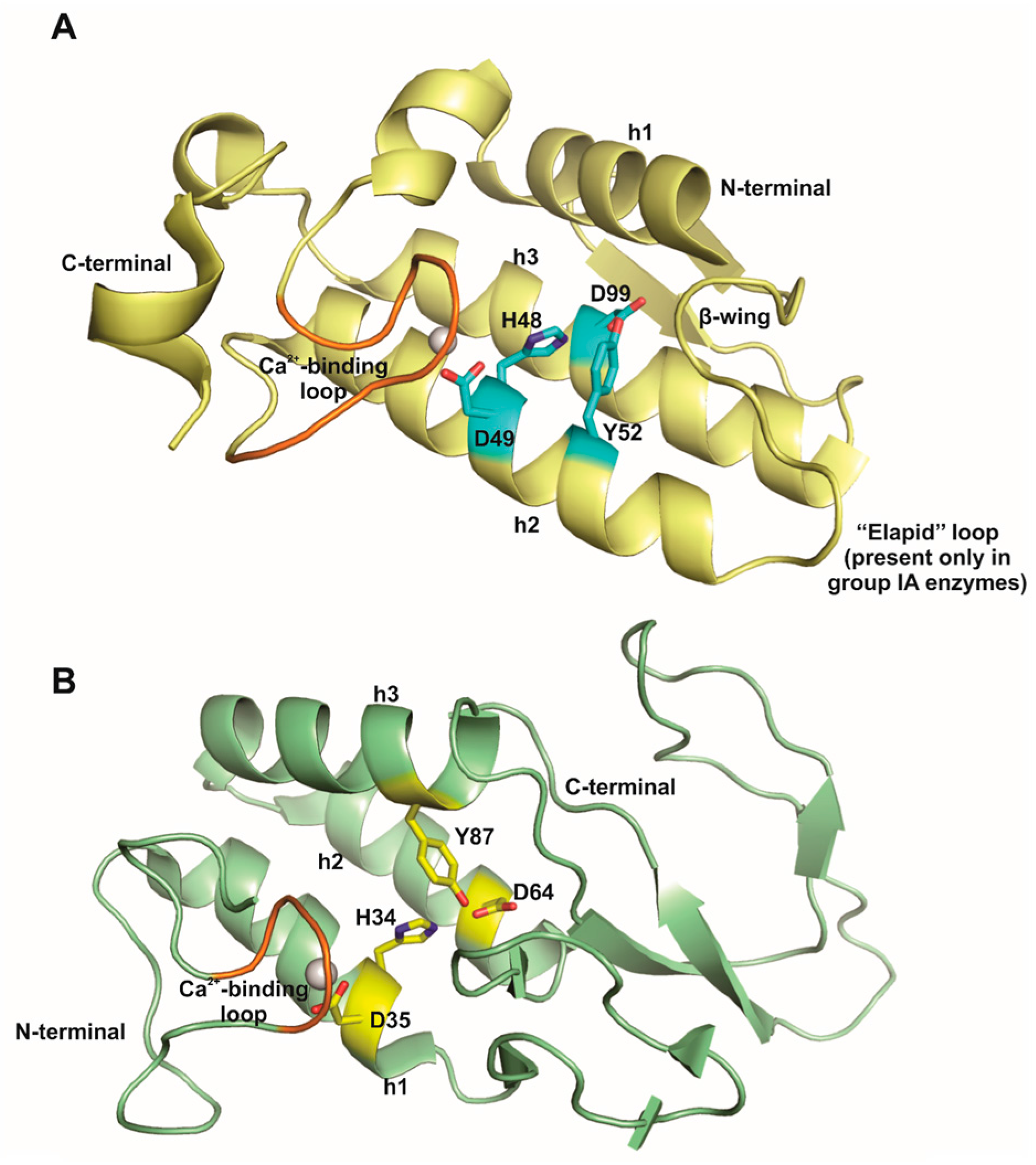
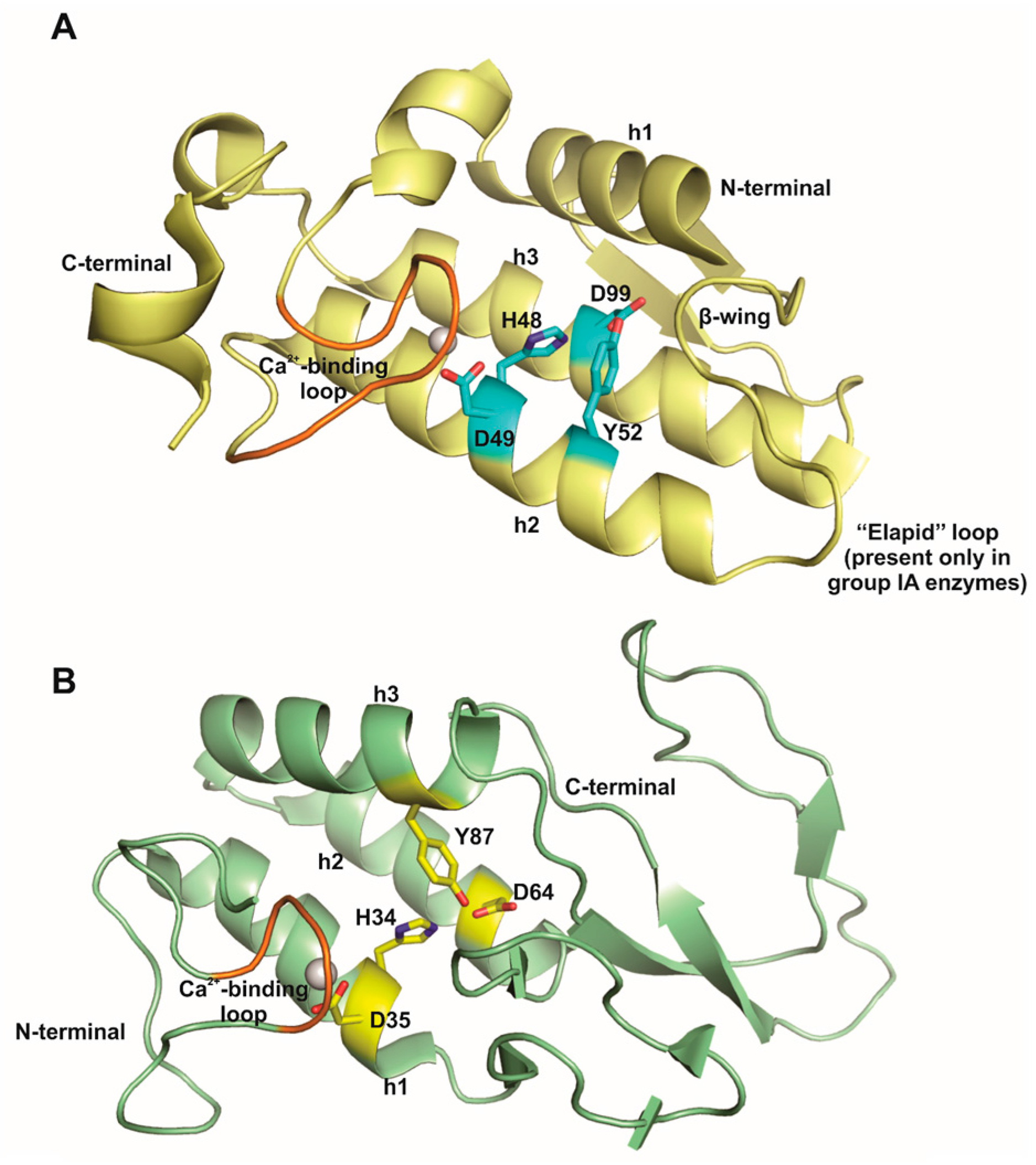
2. Animal Venom sPLA2s
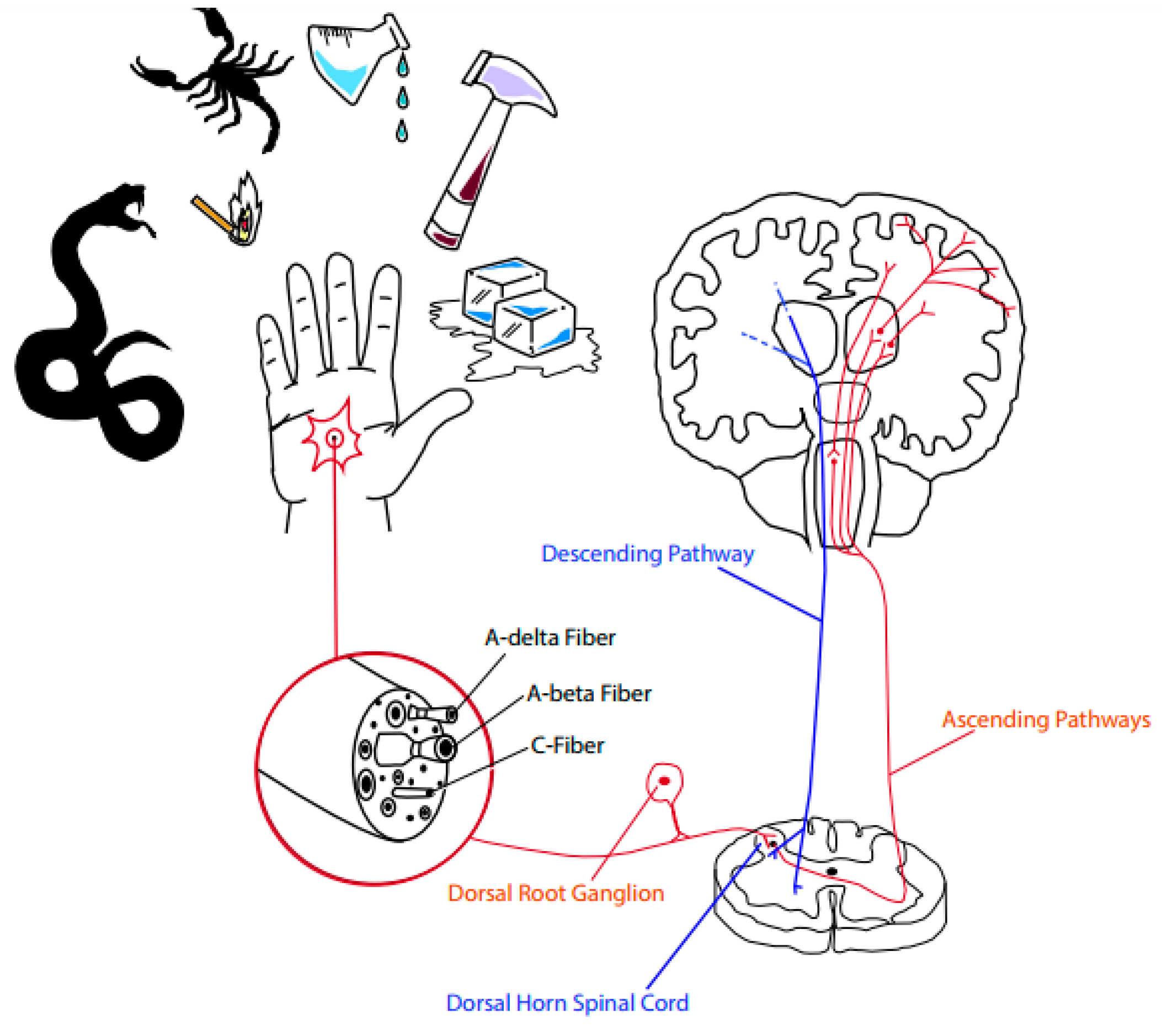
3. Pain and Analgesia: General Concepts
4. Crotoxin, a Heterodimeric Neurotoxin from Crotalus durissus terrificus Venom That Induces Analgesia
5. Antinociceptive Effect of Crotoxin
6. Antinociception Induced by Other Animal Venoms sPLA2s
6.1. Snake Venoms
6.2. Bee Venom
7. Animal Venom-Derived Inhibitors of Phospholipases A2 as Analgesics
8. Nociceptive Effects of Animal Venom sPLA2s
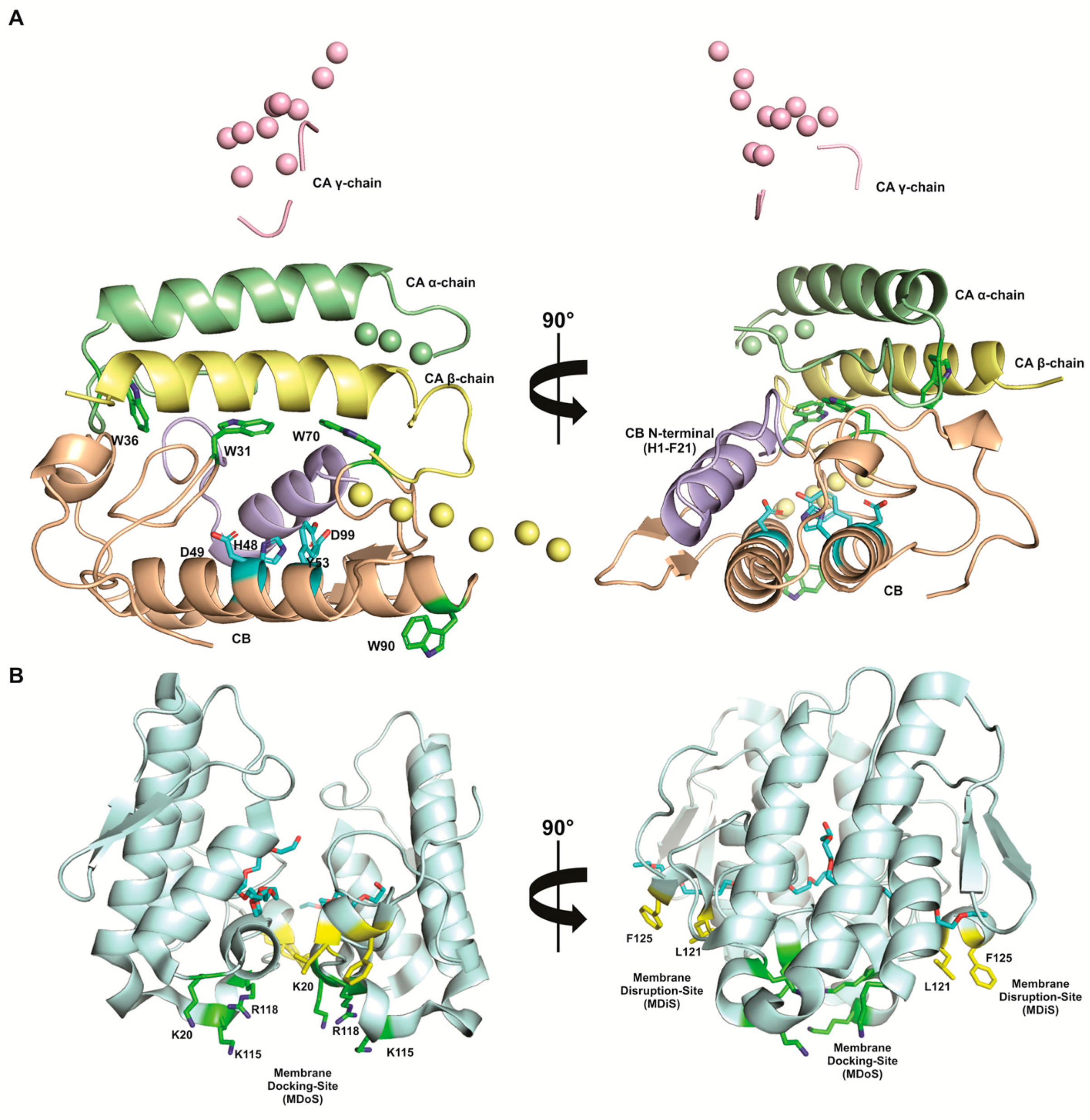
9. Lys49-PLA2s, a PLA2-Like Proteins Subgroup That Induces Hyperalgesia in a Catalytic Activity-Independent Way
- (i)
- Binding of a hydrophobic molecule in the hydrophobic channel recently opened. This event characterizes the transition between inactive and active states of the protein, being the active state the dimer with a hydrophobic molecule in the hydrophobic channel of each monomer. In the active state, MDoS and MDiS regions become exposed to the solvent and aligned in the same plane with a symmetric position for both monomers [33,152];
- (ii)
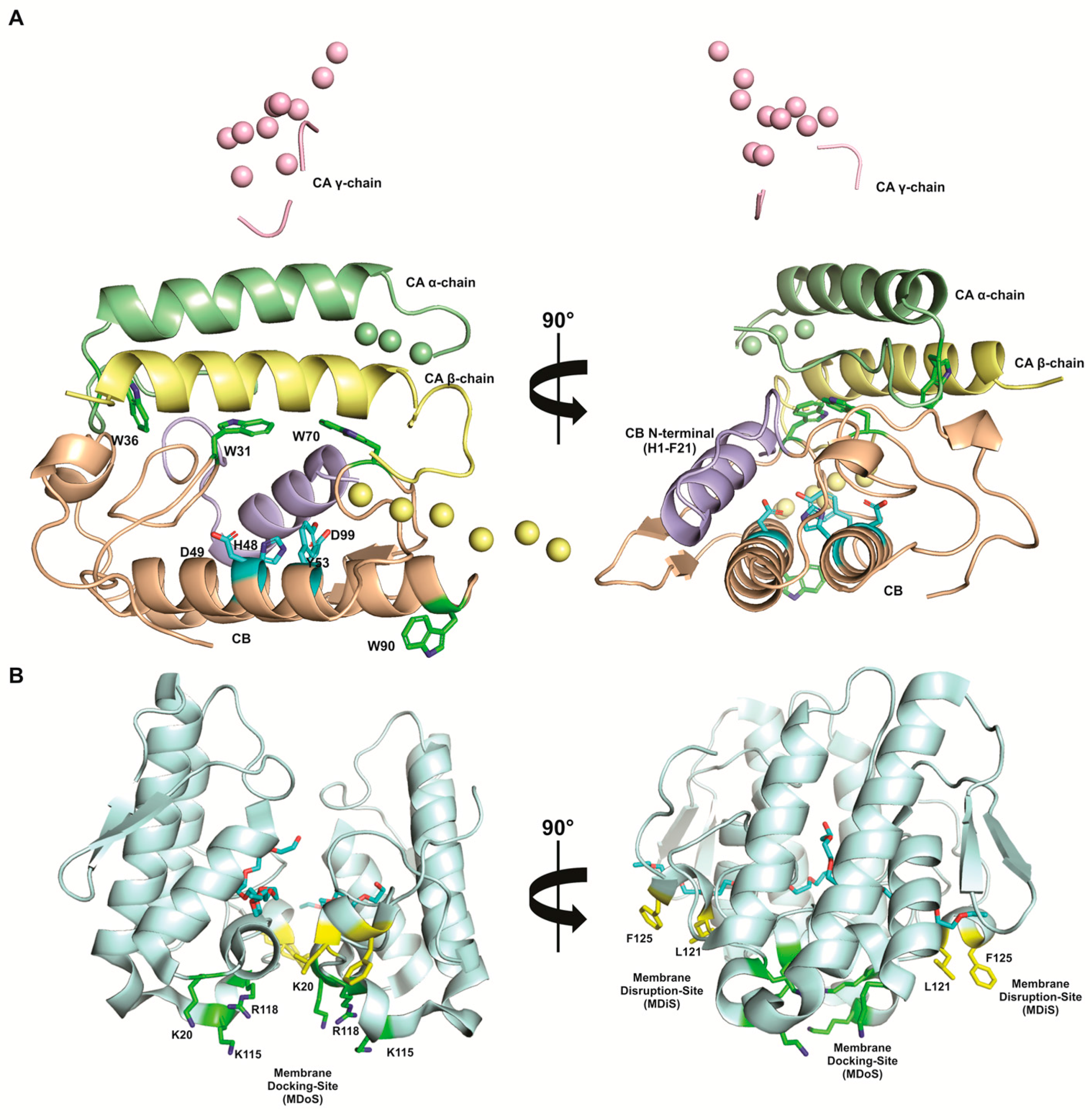
- Stabilization of the protein on the membrane by interaction of MDoS from both monomers and the phospholipid head groups on target cell membrane. MDoS is formed by K20, K115, and R118 cationic residues, but it can be aided by other positive and exposed residues, such as K80, K122, and K127 [33,149]. Indeed, several authors have shown that the cationic charge of these molecules is essential for their pharmacological properties, including hyperalgesia and inflammation [48]. Co-crystallization of inhibitors that bound to MDoS evidenced its involvement on PLA2s-like proteins activities [151,153];
- (iii)
- Membrane destabilization by the penetration of MDiS from both of the monomers into the target membrane. This insertion causes a disorganization of the lipid bilayer, causing an uncontrolled influx of ions (i.e., Ca2+ and Na+), and, consequently, cell death [33,149]. MDiS is formed by L121 and P125 residues, which are conserved in the majority of PLA2-like proteins [33,149]. Furthermore, L and P are residues with high hydrophobic indices and membrane permeability coefficient [154,155]. Co-crystallization of inhibitors that bound to MDiS evidenced its involvement on PLA2s-like proteins mechanism of action [153].
10. Concluding Remarks
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Harvey, A.L. Toxins and drug discovery. Toxicon 2014, 92, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Utkin, Y.N. Animal venom studies: Current benefits and future developments. World J. Biol. Chem. 2015, 6, 28. [Google Scholar] [CrossRef] [PubMed]
- Dennis, E.A.; Cao, J.; Hsu, Y.H.; Magrioti, V.; Kokotos, G. Phospholipase A2 enzymes: Physical structure, biological function, disease implication, chemical inhibition, and therapeutic intervention. Chem. Rev. 2011, 111, 6130–6185. [Google Scholar] [CrossRef] [PubMed]
- Calvete, J.J. Snake venomics: From the inventory of toxins to biology. Toxicon 2013, 75, 44–62. [Google Scholar] [CrossRef] [PubMed]
- Hirata, T.; Narumiya, S. Prostanoid receptors. Chem. Rev. 2011, 111, 6209–6230. [Google Scholar] [CrossRef] [PubMed]
- Moolenaar, W.H.; van Meeteren, L.A.; Giepmans, B.N.G. The ins and outs of lysophosphatidic acid signaling. Bioessays 2004, 26, 870–881. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.E.; Herr, D.R.; Chun, J. Lysophosphatidic acid (LPA) receptors: Signaling properties and disease relevance. Prostaglandins Other Lipid Mediat. 2010, 91, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Yost, C.C.; Weyrich, A.S.; Zimmerman, G.A. The platelet activating factor (PAF) signaling cascade in systemic inflammatory responses. Biochimie 2010, 92, 692–697. [Google Scholar] [CrossRef] [PubMed]
- Schaloske, R.H.; Dennis, E.A. The phospholipase A2 superfamily and its group numbering system. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2006, 1761, 1246–1259. [Google Scholar] [CrossRef] [PubMed]
- Su, M.J.; Chang, C.C. Presynaptic effects of snake venom toxins which have phospholipase A2 activity (β-bungarotoxin, taipoxin, crotoxin). Toxicon 1984, 22, 631–640. [Google Scholar] [CrossRef]
- Bon, C.; Changeux, J.; Jeng, T.; Fraenkel-Contat, H. Postsynaptic Effects of Crotoxin and of Its Isolated Subunits. Eur. J. Biochem. 1979, 99, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Lomonte, B.; Lundgren, J.; Johansson, B.; Bagge, U. The dynamics of local tissue damage induced by Bothrops asper snake venom and myotoxin II on the mouse cremaster muscle: An intravital and electron microscopic study. Toxicon 1994, 32, 41–55. [Google Scholar] [CrossRef]
- Barrington, P.L.; Soons, K.R.; Rosenberg, P. Cardiotoxicity of Naja nigricollis phospholipase A2 is not due to alterations in prostaglandin synthesis. Toxicon 1986, 24, 1107–1116. [Google Scholar] [CrossRef]
- Páramo, L.; Lomonte, B.; Pizarro-cerdá, J.; Bengoechea, J.A.; Gorvel, J.P.; Moreno, E. Bactericidal activity of Lys49 and Asp49 myotoxic phospholipases A2 from Bothrops asper snake venom: Synthetic Lys49 myotoxin II-(115-129)-peptide identifies its bactericidal region. Eur. J. Biochem. 1998, 253, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Jackson, S.P.; Mitchell, C.A.; Salem, H.H. Purification and characterisation of a snake venom phospholipase A2: A potent inhibitor of platelet aggregation. Thromb. Res. 1993, 70, 471–481. [Google Scholar] [CrossRef]
- Lloret, S.; Moreno, J.J. Oedema formation and degranulation of mast cells by phospholipase A2 purified from porcine pancreas and snake venoms. Toxicon 1993, 31, 949–956. [Google Scholar] [CrossRef]
- Condrea, E.; Yang, C.C.; Rosenberg, P. Lack of correlation between anticoagulant activity and phospholipid hydrolysis by snake venom phospholipases A2. Thromb. Haemost. 1981, 45, 82–85. [Google Scholar] [PubMed]
- Fletcher, J.E.; Rapuano, B.E.; Condrea, E.; Yang, C.C.; Ryan, M.; Rosenberg, P. Comparison of a relatively toxic phospholipase A2 from Naja nigricollis snake venom with that of a relatively non-toxic phospholipase A2 from Hemachatus haemachatus snake venom-II. Pharmacological properties in relationship to enzymatic activity. Biochem. Pharmacol. 1980, 29, 1565–1574. [Google Scholar] [CrossRef]
- Huang, H.C. Release of slow reacting substance from the guinea-pig lung by phospholipases A2 of Vipera russelli snake venom. Toxicon 1984, 22, 359–372. [Google Scholar] [CrossRef]
- Arni, R.K.; Ward, R.J. Phospholipase A2—A structural review. Toxicon 1996, 34, 827–841. [Google Scholar] [CrossRef]
- Six, D.A.; Dennis, E.A. The expanding superfamily of phospholipase A2 enzymes: Classification and characterization. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2000, 1488, 1–19. [Google Scholar] [CrossRef]
- Fernandes, C.A.H.; Marchi-Salvador, D.P.; Salvador, G.M.; Silva, M.C.O.; Costa, T.R.; Soares, A.M.; Fontes, M.R.M. Comparison between apo and complexed structures of Bothrops toxin-I reveals the role of Lys122 and Ca2+-binding loop region for the catalytically inactive Lys49-PL A2s. J. Struct. Biol. 2010, 171, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Corrêa, E.A.; Kayano, A.M.; Diniz-Sousa, R.; Setúbal, S.S.; Zanchi, F.B.; Zuliani, J.P.; Matos, N.B.; Almeida, J.R.; Resende, L.M.; Marangoni, S.; et al. Isolation, structural and functional characterization of a new Lys49 phospholipase A2 homologue from Bothrops neuwiedi urutu with bactericidal potential. Toxicon 2016, 115, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.L.; Otwinowski, Z.; Gelb, M.H.; Sigler, P.B. Crystal Structure of Bee-Venom Phospholipase A2 in a Complex with a Transition-State Analogue. Am. Assoc. Adv. Sci. 1990, 250, 1563–1566. [Google Scholar] [CrossRef]
- Scott, D.L.; White, S.P.; Otwinowski, Z.; Yuan, W.; Gelb, M.H.; Sigler, P.B. Interfacial catalysis: The mechanism of phospholipase A2. Science 1990, 250, 1541–1546. [Google Scholar] [CrossRef] [PubMed]
- Rogers, J.; Yu, B.Z.; Serves, S.V.; Tsivgoulis, G.M.; Sotiropoulos, D.N.; Ioannou, P.V.; Jain, M.K. Kinetic basis for the substrate specificity during hydrolysis of phospholipids by secreted phospholipase A2. Biochemistry 1996, 35, 9375–9384. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.Z.; Rogers, J.; Nicol, G.R.; Theopold, K.H.; Seshadri, K.; Vishweshwara, S.; Jain, M.K. Catalytic significance of the specificity of divalent cations as K(s)* and k(cat)* cofactors for secreted phospholipase A2. Biochemistry 1998, 37, 12576–12587. [Google Scholar] [CrossRef] [PubMed]
- Bahnson, B.J. Structure, function and interfacial allosterism in phospholipase A2: Insight from the anion-assisted dimer. Arch. Biochem. Biophys. 2005, 433, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.Z.; Jain, M.K.; Berg, O.G. The Divalent Cation Is Obligatory for the Binding of Ligands to the Catalytic Site of Secreted Phospholipase A2. Biochemistry 1993, 32, 6485–6492. [Google Scholar] [CrossRef] [PubMed]
- Berg, O.G.; Gelb, M.H.; Tsai, M.D.; Jain, M.K. Interfacial enzymology: The secreted phospholipase A2-paradigm. Chem. Rev. 2001, 101, 2613–2653. [Google Scholar] [CrossRef] [PubMed]
- Burke, J.E.; Karbarz, M.J.; Deems, R.A.; Li, S.; Woods, V.L.; Dennis, E.A. Interaction of group IA phospholipase A2 with metal ions and phospholipid vesicles probed with deuterium exchange mass spectrometry. Biochemistry 2008, 47, 6451–6459. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.T.; Gabdoulkhakov, A.; Genov, N.; Cintra, A.C.O.; Betzel, C.; Arni, R.K. Insights into metal ion binding in phospholipases A2: Ultra high-resolution crystal structures of an acidic phospholipase A2 in the Ca2+ free and bound states. Biochimie 2006, 88, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.A.H.; Borges, R.J.; Lomonte, B.; Fontes, M.R.M. A structure-based proposal for a comprehensive myotoxic mechanism of phospholipase A2-like proteins from viperid snake venoms. Biochim. Biophys. Acta Proteins Proteom. 2014, 1844, 2265–2276. [Google Scholar] [CrossRef] [PubMed]
- Bollinger, J.G.; Diraviyam, K.; Ghomashchi, F.; Murray, D.; Gelb, M.H. Interfacial binding of bee venom secreted phospholipase A2 to membranes occurs predominantly by a nonelectrostatic mechanism. Biochemistry 2004, 43, 13293–13304. [Google Scholar] [CrossRef] [PubMed]
- IASP. IASP Taxonomy. Available online: https://www.iasp-pain.org/Taxonomy (accessed on 26 October 2017).
- Basbaum, A.I.; Bautista, D.M.; Scherrer, G.; Julius, D. Cellular and molecular mechanisms of pain. Cell 2009, 139, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Dubin, A.E.; Patapoutian, A. Nociceptors: The sensors of the pain pathway. J. Clin. Invest. 2010, 120, 3760–3772. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, C.; Cury, Y.; Moreira, V.; Picolo, G.; Chaves, F. Inflammation induced by Bothrops asper venom. Toxicon 2009, 54, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Ji, R.-R.; Xu, Z.-Z.; Gao, Y.-J. Emerging targets in neuroinflammation-driven chronic pain. Nat. Rev. Drug Discov. 2014, 13, 533–548. [Google Scholar] [CrossRef] [PubMed]
- Zambelli, V.O.; Gross, E.R.; Chen, C.-H.; Gutierrez, V.P.; Cury, Y.; Mochly-Rosen, D. Aldehyde dehydrogenase-2 regulates nociception in rodent models of acute inflammatory pain. Sci. Transl. Med. 2014, 6, 251ra118. [Google Scholar] [CrossRef] [PubMed]
- Julius, D.; Basbaum, A.I. Molecular mechanisms of nociception. Nature 2001, 413, 203–210. [Google Scholar] [CrossRef] [PubMed]
- De Angelis, F.; Tata, A.M. Analgesic Effects Mediated by Muscarinic Receptors: Mechanisms and Pharmacological Approaches. Cent. Nerv. Syst. Agents Med. Chem. 2016, 16, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Yaksh, T. Pharmacology of spinal adrenergic systems which modulate spinal nociceptive processing. Pharmacol. Biochem. Behav. 1985, 22, 845–858. [Google Scholar] [CrossRef]
- Cui, M.; Feng, Y.; McAdoo, D.J.; Willis, W.D. Periaqueductal gray stimulation-induced inhibition of nociceptive dorsal horn neurons in rats is associated with the release of norepinephrine, serotonin, and amino acids. J. Pharmacol. Exp. Ther. 1999, 289, 868–876. [Google Scholar] [PubMed]
- Bannister, K.; Lockwood, S.; Goncalves, L.; Patel, R.; Dickenson, A.H. An investigation into the inhibitory function of serotonin in diffuse noxious inhibitory controls in the neuropathic rat. Eur. J. Pain 2017, 21, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, C.F.P.; Landucci, E.C.T.; Antunes, E.; Chacur, M.; Cury, Y. Inflammatory effects of snake venom myotoxic phospholipases A2. Toxicon 2003, 42, 947–962. [Google Scholar] [CrossRef] [PubMed]
- Costa, S.K.P.; Camargo, E.A.; Antunes, E. Inflammatory Action of Secretory PLA2 from Snake Venoms. In Toxins and Drug Discovery; Springer: Dordrecht, The Netherlands, 2015; pp. 1–18. [Google Scholar]
- Chacur, M.; Longo, I.; Picolo, G.; Gutiérrez, J.M.; Lomonte, B.; Guerra, J.L.; Teixeira, C.F.P.; Cury, Y. Hyperalgesia induced by Asp49 and Lys49 phospholipases A2 from Bothrops asper snake venom: Pharmacological mediation and molecular determinants. Toxicon 2003, 41, 667–678. [Google Scholar] [CrossRef]
- Cintra-Francischinelli, M.; Caccin, P.; Chiavegato, A.; Pizzo, P.; Carmignoto, G.; Angulo, Y.; Lomonte, B.; Gutierrez, J.M.; Montecucco, C. Bothrops snake myotoxins induce a large efflux of ATP and potassium with spreading of cell damage and pain. Proc. Natl. Acad. Sci. USA 2010, 107, 14140–14145. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G. Purines and sensory nerves. Handb. Exp. Pharmacol. 2009, 194, 333–392. [Google Scholar] [CrossRef]
- Svensson, C.I.; Lucas, K.K.; Hua, X.-Y.; Powell, H.C.; Dennis, E.A.; Yaksh, T.L. Spinal phospholipase A2 in inflammatory hyperalgesia: Role of the small, secretory phospholipase A2. Neuroscience 2005, 133, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Lucas, K.K.; Svensson, C.I.; Hua, X.-Y.; Yaksh, T.L.; Dennis, E.A. Spinal phospholipase A2 in inflammatory hyperalgesia: Role of group IVA cPLA2. Br. J. Pharmacol. 2005, 144, 940–952. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Fitzsimmons, B.; Hefferan, M.P.; Svensson, C.I.; Wancewicz, E.; Monia, B.P.; Hung, G.; Butler, M.; Marsala, M.; Hua, X.-Y.; et al. Inhibition of spinal cytosolic phospholipase A(2) expression by an antisense oligonucleotide attenuates tissue injury-induced hyperalgesia. Neuroscience 2008, 154, 1077–1087. [Google Scholar] [CrossRef] [PubMed]
- Min, S.-H.; Soh, J.-S.; Park, J.-Y.; Choi, S.-U.; Lee, H.-W.; Lee, J.-J.; Kim, J.-H. Epidural dexamethasone decreased inflammatory hyperalgesia and spinal cPLA expression in a rat formalin test. Yonsei Med. J. 2014, 55, 1631–1639. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, S.; Lippross, S.; Neuhuber, W.L.; Zeilhofer, H.U. PGE(2) selectively blocks inhibitory glycinergic neurotransmission onto rat superficial dorsal horn neurons. Nat. Neurosci. 2002, 5, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Shunmugavel, A.; Dhammu, T.S.; Matsuda, F.; Singh, A.K.; Singh, I. Oral administration of cytosolic PLA2 inhibitor arachidonyl trifluoromethyl ketone ameliorates cauda equina compression injury in rats. J. Neuroinflamm. 2015, 12, 94. [Google Scholar] [CrossRef] [PubMed]
- Kokotou, M.G.; Galiatsatou, G.; Magrioti, V.; Koutoulogenis, G.; Barbayianni, E.; Limnios, D.; Mouchlis, V.D.; Satpathy, B.; Navratil, A.; Dennis, E.A.; et al. 2-Oxoesters: A Novel Class of Potent and Selective Inhibitors of Cytosolic Group IVA Phospholipase A2. Sci. Rep. 2017, 7, 7025. [Google Scholar] [CrossRef] [PubMed]
- Lewin, M.; Samuel, S.; Merkel, J.; Bickler, P. Varespladib (LY315920) Appears to Be a Potent, Broad-Spectrum, Inhibitor of Snake Venom Phospholipase A2 and a Possible Pre-Referral Treatment for Envenomation. Toxins 2016, 8, 248. [Google Scholar] [CrossRef] [PubMed]
- Brazil, O.V. Pharmacology of crystalline crotoxin. II. Neuromuscular blocking action. Mem. Inst. Butantan 1966, 33, 981–992. [Google Scholar] [PubMed]
- Chang, C.C.; Lee, J.D. Crotoxin, the neurotoxin of South American rattlesnake venom, is a presynaptic toxin acting like β-bungarotoxin. Naunyn. Schmiedeberg’s Arch. Pharmacol. 1977, 296, 159–168. [Google Scholar] [CrossRef]
- Santos, P.E.; Souza, S.D.; Freire-Maia, L.; Almeida, A.P. Effects of crotoxin on the isolated guinea pig heart. Toxicon 1990, 28, 215–224. [Google Scholar] [CrossRef]
- Monteiro, H.S.A.; Da Silva, I.M.S.C.; Martins, A.M.C.; Fonteles, M.C. Actions of Crotalus durissus terrificus venom and crotoxin on the isolated rat kidney. Braz. J. Med. Biol. Res. 2001, 34, 1347–1352. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Wu, D.C.; Zhou, X.P.; Gong, S.; Cheng, B.C.; Qin, Z.H.; Reid, P.F.; Yin, Q.Z.; Jiang, X.H. Inhibitory effect of crotoxin on the pain-evoked discharge of neurons in thalamic parafascicular nucleus in rats. Toxicon 2008, 51, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.L.; Han, R.; Chen, Z.X.; Chen, B.W.; Gu, Z.L.; Reid, P.F.; Raymond, L.N.; Qin, Z.H. Opiate and acetylcholine-independent analgesic actions of crotoxin isolated from Crotalus durissus terrificus venom. Toxicon 2006, 48, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Nogueira-Neto, F.d.S.; Amorim, R.L.; Brigatte, P.; Picolo, G.; Ferreira, W.A.; Gutierrez, V.P.; Conceição, I.M.; Della-Casa, M.S.; Takahira, R.K.; Nicoletti, J.L.M.; et al. The analgesic effect of crotoxin on neuropathic pain is mediated by central muscarinic receptors and 5-lipoxygenase-derived mediators. Pharmacol. Biochem. Behav. 2008, 91, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Faure, G.; Xu, H.; Saul, F.A. Crystal structure of crotoxin reveals key residues involved in the stability and toxicity of this potent heterodimeric β-neurotoxin. J. Mol. Biol. 2011, 412, 176–191. [Google Scholar] [CrossRef] [PubMed]
- Hendon, R.A.; Fraenkel-Conrat, H. Biological Roles of the Two Components of Crotoxin. Proc. Natl. Acad. Sci. USA 1971, 68, 1560–1563. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.A.H.; Pazin, W.M.; Dreyer, T.R.; Bicev, R.N.; Cavalcante, W.L.G.; Fortes-Dias, C.L.; Ito, A.S.; Oliveira, C.L.P.; Fernandez, R.M.; Fontes, M.R.M. Biophysical studies suggest a new structural arrangement of crotoxin and provide insights into its toxic mechanism. Sci. Rep. 2017, 7, 43885. [Google Scholar] [CrossRef] [PubMed]
- Faure, G.; Saliou, B.; Bon, C.; Guillaume, J.L.; Camoin, L. Multiplicity of Acidic Subunit Isoforms of Crotoxin, the Phospholipase A2 Neurotoxin from Crotalus durissus terrificus Venom, Results from Posttranslational Modifications. Biochemistry 1991, 30, 8074–8083. [Google Scholar] [CrossRef] [PubMed]
- Kouyoumdjian, J.A.; Harris, J.B.; Johnson, M.A. Muscle necrosis caused by the sub-units of crotoxin. Toxicon 1986, 24, 575–583. [Google Scholar] [CrossRef]
- Faure, G.; Harvey, A.L.; Thomson, E.; Saliou, B.; Radvany, F.; Bon, C. Comparison of crotoxin isoforms reveals that stability of the complex plays a major role in its pharmacological action. Eur. J. Biochem. 1993, 214, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Cavalcante, W.L.G.; Noronha-Matos, J.B.; Timóteo, M.A.; Fontes, M.R.M.; Gallacci, M.; Correia-de-Sá, P. Neuromuscular paralysis by the basic phospholipase A2 subunit of crotoxin from Crotalus durissus terrificus snake venom needs its acid chaperone to concurrently inhibit acetylcholine release and produce muscle blockage. Toxicol. Appl. Pharmacol. 2017, 334, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Čurin-Šerbec, V.; Délot, E.; Faure, G.; Saliou, B.; Gubenšek, F.; Bon, C.; Choumet, V. Antipeptide antibodies directed to the C-terminal part of ammodytoxin A react with the PLA2 subunit of crotoxin and neutralize its pharmacological activity. Toxicon 1994, 32, 1337–1348. [Google Scholar] [CrossRef]
- Fortes-Dias, C.L.; dos Santos, R.M.M.; Magro, A.J.; Fontes, M.R.d.M.; Chávez-Olórtegui, C.; Granier, C. Identification of continuous interaction sites in PL A2-based protein complexes by peptide arrays. Biochimie 2009, 91, 1482–1492. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, M.C.; Yen, C.H.; Hseu, M.J.; Tseng, C.C.; Tsai, M.D.; Dupureur, C.M. Binding proteins on synaptic membranes for crotoxin and taipoxin, two phospholipases A2 with neurotoxicity. Toxicon 1995, 33, 451–457. [Google Scholar] [CrossRef]
- Snitko, Y.; Koduri, R.S.; Han, S.K.; Othman, R.; Baker, S.F.; Molini, B.J.; Wilton, D.C.; Gelb, M.H.; Cho, W. Mapping the interfacial binding surface of human secretory group IIa phospholipase A2. Biochemistry 1997, 36, 14325–14333. [Google Scholar] [CrossRef] [PubMed]
- Winget, J.M.; Pan, Y.H.; Bahnson, B.J. The interfacial binding surface of phospholipase A2s. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2006, 1761, 1260–1269. [Google Scholar] [CrossRef] [PubMed]
- Faure, G.; Čopič, A.; Le Porrier, S.; Gubenšek, F.; Bon, C.; Križaj, I. Crotoxin acceptor protein isolated from Torpedo electric organ: Binding properties to crotoxin by surface plasmon resonance. Toxicon 2003, 41, 509–517. [Google Scholar] [CrossRef]
- Montecucco, C.; Rossetto, O. How do presynaptic PLA2 neurotoxins block nerve terminals? Trends Biochem. Sci. 2000, 25, 266–270. [Google Scholar] [CrossRef]
- Lomeo, R.D.S.; Gonçalves, A.P.D.F.; Da Silva, C.N.; De Paula, A.T.; Costa Santos, D.O.; Fortes-Dias, C.L.; Gomes, D.A.; De Lima, M.E. Crotoxin from Crotalus durissus terrificus snake venom induces the release of glutamate from cerebrocortical synaptosomes via N and P/Q calcium channels. Toxicon 2014, 85, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Vulfius, C.A.; Kasheverov, I.E.; Kryukova, E.V.; Spirova, E.N.; Shelukhina, I.V.; Starkov, V.G.; Andreeva, T.V.; Faure, G.; Zouridakis, M.; Tsetlin, V.I.; et al. Pancreatic and snake venom presynaptically active phospholipases A2 inhibit nicotinic acetylcholine receptors. PLoS ONE 2017, 12, e0186206. [Google Scholar] [CrossRef] [PubMed]
- Ollero, M.; Bakouh, N.; Lourdel, S.; Odolczyk, N.; Premchandar, A.; Servel, N.; Hatton, A.; Ostrowski, M.K.; Xu, H.; Saul, F.A.; et al. Rattlesnake Phospholipase A2 Increases CFTR-Chloride Channel Current and Corrects ∆ F508CFTR Dysfunction: Impact in Cystic Fibrosis. J. Mol. Biol. 2016, 428, 2898–2915. [Google Scholar] [CrossRef]
- Ostrowski, M.; Porowinska, D.; Prochnicki, T.; Prevost, M.; Raynal, B.; Baron, B.; Sauguet, L.; Corringer, P.J.; Faure, G. Neurotoxic phospholipase A2 from rattlesnake as a new ligand and new regulator of prokaryotic receptor GLIC (proton-gated ion channel from G. violaceus). Toxicon 2016, 116, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Faure, G.; Bon, C. Several isoforms of crotoxin are present in individual venoms from the South American rattlesnake Crotalus durissus terrificus. Toxicon 1987, 25, 229–234. [Google Scholar] [CrossRef]
- Slotta, K.H.; Fraenkel-Conrat, H. Schlangengifte III: Mitteilung: Reinigung und Krystallisation des Klapperschlangen-Giftes. Ber. Dtsch. Chem. Ges. 1938, 71, 1076–1081. [Google Scholar] [CrossRef]
- Marchi-Salvador, D.P.; Corrêa, L.C.; Magro, A.J.; Oliveira, C.Z.; Soares, A.M.; Fontes, M.R.M. Insights into the role of oligomeric state on the biological activities of crotoxin: Crystal structure of a tetrameric phospholipase A2 formed by two isoforms of crotoxin B from Crotalus durissus terrificus venom. Proteins Struct. Funct. Genet. 2008, 72, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Magro, A.J.; Fernandes, C.A.H.; dos Santos, J.I.; Fontes, M.R.M. Influence of quaternary conformation on the biological activities of the Asp49-phospholipases A2s from snake venoms. Protein Pept. Lett. 2009, 16, 852–859. [Google Scholar] [CrossRef] [PubMed]
- Wolz-Richter, S.; Esser, K.-H.; Hess, A. Antinociceptive activity of crotoxin in the central nervous system: A functional Magnetic Resonance Imaging study. Toxicon 2013, 74, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, S.C.; Hyslop, S.; Fontes, M.R.M.; Prado-Franceschi, J.; Zambelli, V.O.; Magro, A.J.; Brigatte, P.; Gutierrez, V.P.; Cury, Y. Crotoxin: Novel activities for a classic β-neurotoxin. Toxicon 2010, 55, 1045–1060. [Google Scholar] [CrossRef] [PubMed]
- Brigatte, P.; Faiad, O.J.; Ferreira Nocelli, R.C.; Landgraf, R.G.; Palma, M.S.; Cury, Y.; Curi, R.; Sampaio, S.C. Walker 256 Tumor Growth Suppression by Crotoxin Involves Formyl Peptide Receptors and Lipoxin A4. Mediat. Inflamm. 2016, 2016, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Cura, J.E.; Blanzaco, D.P.; Brisson, C.; Cura, M.A.; Cabrol, R.; Larrateguy, L.; Mendez, C.; Sechi, J.C.; Silveira, J.S.; Theiller, E.; et al. Phase I and pharmacokinetics study of crotoxin (cytotoxic PLA(2), NSC-624244) in patients with advanced cancer. Clin. Cancer Res. 2002, 8, 1033–1041. [Google Scholar] [PubMed]
- Costa, L.A.; Fornari, M.C.; Berardi, V.E.; Miles, H.A.; Diez, R.A. In vivo effect of snake phospholipase A2 (crotoxin + cardiotoxin) on serum IL-1α, TNF-α and IL-1ra level in humans. Immunol. Lett. 2001, 75, 137–141. [Google Scholar] [CrossRef]
- Dyachenko, I.A.; Murashev, A.N.; Andreeva, T.V.; Tsetlin, V.I.; Utkin, Y.N. Analysis of nociceptive effects of neurotoxic phospholipase A2 from Vipera nikolskii venom in mice. J. Venom Res. 2013, 4, 1–4. [Google Scholar] [PubMed]
- Kleronomos, C.A. Bee venom therapy: History, mechanisms and clinical considerations. Pain Pract. 2010, 20, 74–79. [Google Scholar]
- Roh, D.-H.; Kwon, Y.-B.; Kim, H.-W.; Ham, T.-W.; Yoon, S.-Y.; Kang, S.-Y.; Han, H.-J.; Lee, H.-J.; Beitz, A.J.; Lee, J.-H. Acupoint stimulation with diluted bee venom (apipuncture) alleviates thermal hyperalgesia in a rodent neuropathic pain model: Involvement of spinal alpha2-adrenoceptors. J. Pain 2004, 5, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Baek, Y.H.; Huh, J.E.; Lee, J.D.; Choi, D.Y.; Park, D.S. Antinociceptive effect and the mechanism of bee venom acupuncture (Apipuncture) on inflammatory pain in the rat model of collagen-induced arthritis: Mediation by α2-Adrenoceptors. Brain Res. 2006, 1073–1074, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.W.; Kwon, Y.B.; Han, H.J.; Yang, I.S.; Beitz, A.J.; Lee, J.H. Antinociceptive mechanisms associated with diluted bee venom acupuncture (apipuncture) in the rat formalin test: Involvement of descending adrenergic and serotonergic pathways. Pharmacol. Res. 2005, 51, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Li, D.X.; Yoon, H.; Go, D.; Quan, F.S.; Min, B.-I.; Kim, S.K. Serotonergic mechanism of the relieving effect of bee venom acupuncture on oxaliplatin-induced neuropathic cold allodynia in rats. BMC Complement. Altern. Med. 2014, 14, 471. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Kim, W.; Shin, D.; Jung, Y.; Bae, H.; Kim, S.K. Preventive Effects of Bee Venom Derived Phospholipase A2 on Oxaliplatin-Induced Neuropathic Pain in Mice. Toxins 2016, 8, 27. [Google Scholar] [CrossRef] [PubMed]
- Mann, J. Sponges to wipe away pain. Nature 1992, 358, 540. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Lee, Y.; Kim, W.; Lee, K.; Bae, H.; Kim, S.K. Analgesic effects of bee venom derived phospholipase A2 in a mouse model of oxaliplatin-induced neuropathic pain. Toxins 2015, 7, 2422–2434. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Wilce, J. Venom as a source of useful biologically active molecules. Emerg. Med. 2001, 13, 28–36. [Google Scholar] [CrossRef]
- Hakim, M.; Yang, S.; Lai, R. Centipede Venoms and Their Components: Resources for Potential Therapeutic Applications. Toxins 2015, 7, 4832–4851. [Google Scholar] [CrossRef] [PubMed]
- Utkin, Y.N. Modern trends in animal venom research—Omics and nanomaterials. World J. Biol. Chem. 2017, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Nevalainen, T.J.; Haapamäki, M.M.; Grönroos, J.M. Roles of secretory phospholipases A(2) in inflammatory diseases and trauma. Biochim. Biophys. Acta 2000, 1488, 83–90. [Google Scholar] [CrossRef]
- Rosenfeld, G. Symptomatology, pathology and treatment of snake bites in South America. In Venomous Animals and Their Venoms; Bücherl, W., Buckley, E.E., Eds.; Academic Press: New York, NY, USA, 1971; pp. 345–384. [Google Scholar]
- Ohsaka, A. Hemorrhagic, necrotizing and edema-forming effects of snake venoms. In Handbook of Experimental Pharmacology, Snake Venoms; Lee, C.Y., Ed.; Springer: Berlin, Germany, 1979; pp. 480–546. [Google Scholar]
- Gutiérrez, J.M.; Arroyo, O.; Bolaños, R. Myonecrosis, hemorrhage and edema induced by Bothrops asper venom in white mice (author’s transl). Toxicon 1980, 18, 603–610. [Google Scholar] [CrossRef]
- Gutiérrez, J.M.; Lomonte, B. Phospholipase A2 myotoxins from Bothrops snake venoms. Toxicon 1995, 33, 1405–1424. [Google Scholar] [CrossRef]
- Gutiérrez, J.M.; Chaves, F.; Cerdas, L. Inflammatory infiltrate in skeletal muscle injected with Bothrops asper venom. Rev. Biol. Trop. 1986, 34, 209–214. [Google Scholar] [PubMed]
- Arroyo, O.; Rojas, G.; Gutierrez, J. El Envenenamiento por Mordedura de Serpiente en Centroamérica; Acta Medica Costarricense: San José, Costa Rica, 1996. [Google Scholar]
- Otero, R.; Tobon, G.S.; Gomes, L.F.; Osorio, R.; Valderrama, R.; Hoyos, D.; Urreta, J.E.; Molina, S.; Arboleda, J.J. Acidente ofıdico in Antioquia y Choco. Aspectos clınicos y epidemiologicos. Acta Med. Colomb. 1992, 17, 229–249. [Google Scholar]
- White, J.; Meier, J. (Eds.) Clinical toxicology of snakebite in South America. In Handbook of Clinical Toxicology of Animal, Venoms and Poisons; CRC Press: Boca Raton, FL, USA, 1995. [Google Scholar]
- Trebien, H.A.; Calixto, J.B. Pharmacological evaluation of rat paw oedema induced by Bothrops jararaca venom. Agents Actions 1989, 26, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Cury, Y.; Teixeira, C.F.; Sudo, L.S. Edematogenic responses induced by Bothrops jararaca venom in rats: Role of lymphocytes. Toxicon 1994, 32, 1425–1431. [Google Scholar] [CrossRef]
- Teixeira, C.F.; Cury, Y.; Oga, S.; Jancar, S. Hyperalgesia induced by Bothrops jararaca venom in rats: Role of eicosanoids and platelet activating factor (PAF). Toxicon 1994, 32, 419–426. [Google Scholar] [CrossRef]
- Chaves, F.; Barboza, M.; Gutiérrez, J.M. Pharmacological study of edema induced by venom of the snake Bothrops asper (terciopelo) in mice. Toxicon 1995, 33, 31–39. [Google Scholar] [CrossRef]
- Búrigo, A.C.; Calixto, J.B.; Medeiros, Y.S. Pharmacological profile of rat pleurisy induced by Bothrops jararaca venom. J. Pharm. Pharmacol. 1996, 48, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Farsky, S.H.; Walber, J.; Costa-Cruz, M.; Cury, Y.; Teixeira, C.F.; Curry, Y. Leukocyte response induced by Bothrops jararaca crude venom: In vivo and in vitro studies. Toxicon 1997, 35, 185–193. [Google Scholar] [CrossRef]
- Chacur, M.; Picolo, G.; Gutiérrez, J.M.; Teixeira, C.F.; Cury, Y. Pharmacological modulation of hyperalgesia induced by Bothrops asper (terciopelo) snake venom. Toxicon 2001, 39, 1173–1181. [Google Scholar] [CrossRef]
- Chacur, M.; Picolo, G.; Teixeira, C.F.P.; Cury, Y. Bradykinin is involved in hyperalgesia induced by Bothrops jararaca venom. Toxicon 2002, 40, 1047–1051. [Google Scholar] [CrossRef]
- Gammon, C.M.; Allen, A.C.; Morell, P. Bradykinin stimulates phosphoinositide hydrolysis and mobilization of arachidonic acid in dorsal root ganglion neurons. J. Neurochem. 1989, 53, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Allen, A.C.; Gammon, C.M.; Ousley, A.H.; McCarthy, K.D.; Morell, P. Bradykinin stimulates arachidonic acid release through the sequential actions of an sn-1 diacylglycerol lipase and a monoacylglycerol lipase. J. Neurochem. 1992, 58, 1130–1139. [Google Scholar] [CrossRef] [PubMed]
- Farmer, S.G.; Burch, R.M. Biochemical and Molecular Pharmacology of Kinin Receptors. Annu. Rev. Pharmacol. Toxicol. 1992, 32, 511–536. [Google Scholar] [CrossRef] [PubMed]
- Rueff, A.; Dray, A. Sensitization of peripheral afferent fibres in the in vitro neonatal rat spinal cord-tail by bradykinin and prostaglandins. Neuroscience 1993, 54, 527–535. [Google Scholar] [CrossRef]
- Messlinger, K.; Pawlak, M.; Schepelmann, K.; Schmidt, R.F. Responsiveness of slowly conducting articular afferents to bradykinin: Effects of an experimental arthritis. Pain 1994, 59, 335–343. [Google Scholar] [CrossRef]
- Chacur, M.; Milligan, E.D.; Sloan, E.M.; Wieseler-Frank, J.; Barrientos, R.M.; Martin, D.; Poole, S.; Lomonte, B.; Gutiérrez, J.M.; Maier, S.F.; et al. Snake venom phospholipase A2s (Asp49 and Lys49) induce mechanical allodynia upon peri-sciatic administration: Involvement of spinal cord glia, proinflammatory cytokines and nitric oxide. Pain 2004, 108, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Milligan, E.D.; Watkins, L.R. Pathological and protective roles of glia in chronic pain. Nat. Rev. Neurosci. 2009, 10, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Batti, L.; Sundukova, M.; Murana, E.; Pimpinella, S.; De Castro Reis, F.; Pagani, F.; Wang, H.; Pellegrino, E.; Perlas, E.; Di Angelantonio, S.; et al. TMEM16F Regulates Spinal Microglial Function in Neuropathic Pain States. Cell Rep. 2016, 15, 2608–2615. [Google Scholar] [CrossRef] [PubMed]
- Watkins, L.R.; Wieseler-Frank, J.; Milligan, E.D.; Johnston, I.; Maier, S.F. Chapter 22 Contribution of glia to pain processing in health and disease. Handb. Clin. Neurol. 2006, 81, 309–323. [Google Scholar] [PubMed]
- Chacur, M.; Gutiérrez, J.M.; Milligan, E.D.; Wieseler-Frank, J.; Britto, L.R.G.; Maier, S.F.; Watkins, L.R.; Cury, Y. Snake venom components enhance pain upon subcutaneous injection: An initial examination of spinal cord mediators. Pain 2004, 111, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Dutra, N.C.L.; Telles, M.P.C.; Dutra, D.L.; Silva Júnior, N.J. Genetic diversity in populations of the viper Bothrops moojeni Hoge, 1966 in Central Brazil using RAPD markers. Genet. Mol. Res. 2008, 7, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Mamede, C.C.N.; de Sousa, B.B.; Pereira, D.F.d.C.; Matias, M.S.; de Queiroz, M.R.; de Morais, N.C.G.; Vieira, S.A.P.B.; Stanziola, L.; de Oliveira, F. Comparative analysis of local effects caused by Bothrops alternatus and Bothrops moojeni snake venoms: Enzymatic contributions and inflammatory modulations. Toxicon 2016, 117, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Medzihradszky, K.F.; Sánchez, E.E.; Basbaum, A.I.; Julius, D. Lys49 myotoxin from the Brazilian lancehead pit viper elicits pain through regulated ATP release. Proc. Natl. Acad. Sci. USA 2017, 114, E2524–E2532. [Google Scholar] [CrossRef] [PubMed]
- Rong, L.; Zhang, C.; Lei, Q.; Hu, M.-M.; Feng, J.; Shu, H.-B.; Liu, Y.; Zhang, X.-Z. Hydrogen peroxide detection with high specificity in living cells and inflamed tissues. Regen. Biomater. 2016, 3, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Lomonte, B.; Moreno, E.; Tarkowski, A.; Hanson, L.Å.; Maccarana, M. Neutralizing interaction between heparins and myotoxin II, a lysine 49 phospholipase A2 from Bothrops asper snake venom: Identification of a heparin-binding and cytolytic toxin region by the use of synthetic peptides and molecular modeling. J. Biol. Chem. 1994, 269, 29867–29873. [Google Scholar] [PubMed]
- Núñez, C.E.; Angulo, Y.; Lomonte, B. Identification of the myotoxic site of the Lys49 phospholipase A(2) from Agkistrodon piscivorus piscivorus snake venom: Synthetic C-terminal peptides from Lys49, but not from Asp49 myotoxins, exert membrane-damaging activities. Toxicon 2001, 39, 1587–1594. [Google Scholar] [CrossRef]
- Zambelli, V.O.; Chioato, L.; Gutierrez, V.P.; Ward, R.J.; Cury, Y. Structural determinants of the hyperalgesic activity of myotoxic Lys49-phospholipase A2. J. Venom. Anim. Toxins Incl. Trop. Dis. 2017, 23, 7. [Google Scholar] [CrossRef] [PubMed]
- Chioato, L.; De Oliveira, A.H.C.; Ruller, R.; Sá, J.M.; Ward, R.J. Distinct sites for myotoxic and membrane-damaging activities in the C-terminal region of a Lys49-phospholipase A2. Biochem. J. 2002, 366, 971–976. [Google Scholar] [CrossRef] [PubMed]
- Cruciani, R.A.; Jain, S. Pancreatic Pain: A Mini Review. Pancreatology 2008, 8, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Camargo, E.A.; Santana, D.G.; Silva, C.I.; Teixeira, S.A.; Toyama, M.H.; Cotrim, C.; Landucci, E.C.T.; Antunes, E.; Muscara, M.N.; Costa, S.K.P. Inhibition of inducible nitric oxide synthase-derived nitric oxide as a therapeutical target for acute pancreatitis induced by secretory phospholipase A2. Eur. J. Pain 2014, 18, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Camargol, E.A.; Zanonil, C.I.; Toyamal, M.H.; Muscaral, M.N.; Dochertyl, R.J.; Costal, S.K.P. Abdominal hyperalgesia in secretory phospholipase A2—Induced rat pancreatitis: Distinct roles of NK1 receptors. Eur. J. Pain 2011, 15, 900–906. [Google Scholar] [CrossRef] [PubMed]
- Camargo, E.A.; Ferreira, T.; Ribela, M.T.C.P.; de Nucci, G.; Landucci, E.C.T.; Antunes, E. Role of Substance P and Bradykinin in Acute Pancreatitis Induced by Secretory Phospholipase A2. Pancreas 2008, 37, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Dias, R.G.; Sampaio, S.C.; Sant’Anna, M.B.; Cunha, F.Q.; Gutiérrez, J.M.; Lomonte, B.; Cury, Y.; Picolo, G. Articular inflammation induced by an enzymatically-inactive Lys49 phospholipase A2: Activation of endogenous phospholipases contributes to the pronociceptive effect. J. Venom. Anim. Toxins Incl. Trop. Dis. 2017, 23, 18. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.-I.; Brahn, E. Rheumatoid arthritis therapy: Advances from bench to bedside. Autoimmunity 2010, 43, 478–492. [Google Scholar] [CrossRef] [PubMed]
- Niedermeier, M.; Pap, T.; Korb, A. Therapeutic opportunities in fibroblasts in inflammatory arthritis. Best Pract. Res. Clin. Rheumatol. 2010, 24, 527–540. [Google Scholar] [CrossRef] [PubMed]
- Correa, D.; Lietman, S.A. Articular cartilage repair: Current needs, methods and research directions. Semin. Cell Dev. Biol. 2017, 62, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, J.S.; Schoones, J.W.; Huizinga, T.W.; Toes, R.E.; van der Helm-van Mil, A.H. Possibilities for preventive treatment in rheumatoid arthritis? Lessons from experimental animal models of arthritis: A systematic literature review and meta-analysis. Ann. Rheum. Dis. 2017, 76, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.A.H.; Comparetti, E.J.; Borges, R.J.; Huancahuire-Vega, S.; Ponce-Soto, L.A.; Marangoni, S.; Soares, A.M.; Fontes, M.R.M. Structural bases for a complete myotoxic mechanism: Crystal structures of two non-catalytic phospholipases A2-like from Bothrops brazili venom. Biochim. Biophys. Acta Proteins Proteom. 2013, 1834, 2772–2781. [Google Scholar] [CrossRef] [PubMed]
- Chioato, L.; Aragão, E.A.; Lopes Ferreira, T.; Ivo de Medeiros, A.; Faccioli, L.H.; Ward, R.J. Mapping of the structural determinants of artificial and biological membrane damaging activities of a Lys49 phospholipase A2 by scanning alanine mutagenesis. Biochim. Biophys. Acta Biomembr. 2007, 1768, 1247–1257. [Google Scholar] [CrossRef] [PubMed]
- De Lima, L.F.G.; Borges, R.J.; Viviescas, M.A.; Fernandes, C.A.H.; Fontes, M.R.M. Structural studies with BnSP-7 reveal an atypical oligomeric conformation compared to phospholipases A2-like toxins. Biochimie 2017, 142, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Borges, R.J.; Cardoso, F.F.; Fernandes, C.A.H.; Dreyer, T.R.; de Moraes, D.S.; Floriano, R.S.; Rodrigues-Simioni, L.; Fontes, M.R.M. Functional and structural studies of a Phospholipase A2-like protein complexed to zinc ions: Insights on its myotoxicity and inhibition mechanism. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 3199–3209. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.A.H.; Cardoso, F.F.; Cavalcante, W.G.L.; Soares, A.M.; Dal-Pai, M.; Gallacci, M.; Fontes, M.R.M. Structural basis for the inhibition of a phospholipase A2 like toxin by caffeic and aristolochic acids. PLoS ONE 2015, 10, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Wimley, W.C.; White, S.H. Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat. Struct. Mol. Biol. 1996, 3, 842–848. [Google Scholar] [CrossRef]
- Naoi, M.; Naoi, M.; Shimizu, T.; Malviya, A.N.; Yagi, K. Permeability of amino acids into liposomes. BBA Biomembr. 1977, 471, 305–310. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Animal | Species | Compound | Structure | Mechanism of Analgesia | Reference |
|---|---|---|---|---|---|
| Snake | Crotalus durissus terrificus | Crotoxin | Phospholipase A2 | Central muscarinic receptors, α-adrenoceptors, 5-HT receptors and lypoxin A4 release | [63,64,65,88,90,91] |
| Snake | Crotalus durissus terrificus and Naja naja atra | VRCTC-310-Onco composed of crotoxin from C. d. terrificus and cardiotoxin from N. n. atra, at equimolar ratio | Phospholipase A2 and a sixteen amino-acidpolypeptide | Increase on the plasma level of IL-1 receptor antagonist (IL-1ra) | [92] |
| Snake | Vipera nikolskii | HDP-2 | Phospholipase A2 | Not confirmed | [93] |
| Bee | Apis mellifera | bvPLA2 | Phospholipase A2 | α2-adrenegic receptors | [99,101] |
| Marine Sponge | Luffariella family and Cacospongia mollior | Manoalide, luffariellolide and scalaradial | Structure containing aldehyde groups | Phospholipase A2-inhibitor | [100] |
| sPLA2 | sPLA2 Subtype | Venom Source | Pain-Enhancing Effects | sPLA2’ Structural Determinants | Mechanisms | References |
|---|---|---|---|---|---|---|
| Myotoxin II | Lys-49 PLA2 | B. asper | Mechanical hyperalgesia (injected s.c., intra-articularly or around nerve) | C-terminal cationic/hydrophobic sequence 115–129 | Periphery: histamine, serotonin, sympathomimetic amines, endothelin, bradykinin, cytokines, prostaglandins; cellular influx (intraarticular injection) | [48,143] |
| Spinal cord: nitric oxide, prostanoids, IL-1 and IL-6 *, astrocytes and microglia | [48,127,131] | |||||
| Mechanical allodynia (injected around nerve) | ND & | Spinal cord: nitric oxide, prostanoid,; IL-1 and IL-6 *, astrocytes and microglia | [48,127] | |||
| Myotoxin III | Asp-49 PLA2 | B. asper | Mechanical hyperalgesia and allodynia (injected s.c. or around nerve) | Enzymatic activity | Periphery: Bradykinin | [48] |
| Spinal cord: nitric oxide, prostanoids; IL-1 and IL-6 *, astrocytes and microglia | [48,127,131] | |||||
| BomoTx | Asp-49 PLA2 | B. moojeni | nonneurogenic inflammatory pain, thermal hyperalgesia, mechanical allodynia (injected s.c.) | ND & | ATP release; P2X2 and P2X3 purinergic receptors activation (mechanical sensitization), involvement of TRPV1-fibers (thermal hypernociception) | [134] |
| BthTx-I | Lys-49 PLA2 | B. jararacussu | Mechanical hyperalgesia (injected s.c.) | K115; K116; R118; K122 (in the C-terminal) | ND & | [137] |
| CB # | Asp-49 PLA2 | C. d. terrificus | Abdominal hyperalgesia (injected into the common bile duct) | ND & | NK1 receptors | [141] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zambelli, V.O.; Picolo, G.; Fernandes, C.A.H.; Fontes, M.R.M.; Cury, Y. Secreted Phospholipases A2 from Animal Venoms in Pain and Analgesia. Toxins 2017, 9, 406. https://doi.org/10.3390/toxins9120406
Zambelli VO, Picolo G, Fernandes CAH, Fontes MRM, Cury Y. Secreted Phospholipases A2 from Animal Venoms in Pain and Analgesia. Toxins. 2017; 9(12):406. https://doi.org/10.3390/toxins9120406
Chicago/Turabian StyleZambelli, Vanessa O., Gisele Picolo, Carlos A. H. Fernandes, Marcos R. M. Fontes, and Yara Cury. 2017. "Secreted Phospholipases A2 from Animal Venoms in Pain and Analgesia" Toxins 9, no. 12: 406. https://doi.org/10.3390/toxins9120406
APA StyleZambelli, V. O., Picolo, G., Fernandes, C. A. H., Fontes, M. R. M., & Cury, Y. (2017). Secreted Phospholipases A2 from Animal Venoms in Pain and Analgesia. Toxins, 9(12), 406. https://doi.org/10.3390/toxins9120406