
Treatments for Pulmonary Ricin Intoxication: Current Aspects and Future Prospects

 ,
,
Abstract
1. Introduction
2. Ricin-Induced Cytotoxicity and Pathophysiology of Pulmonary Ricinosis
2.1. Pathogenesis
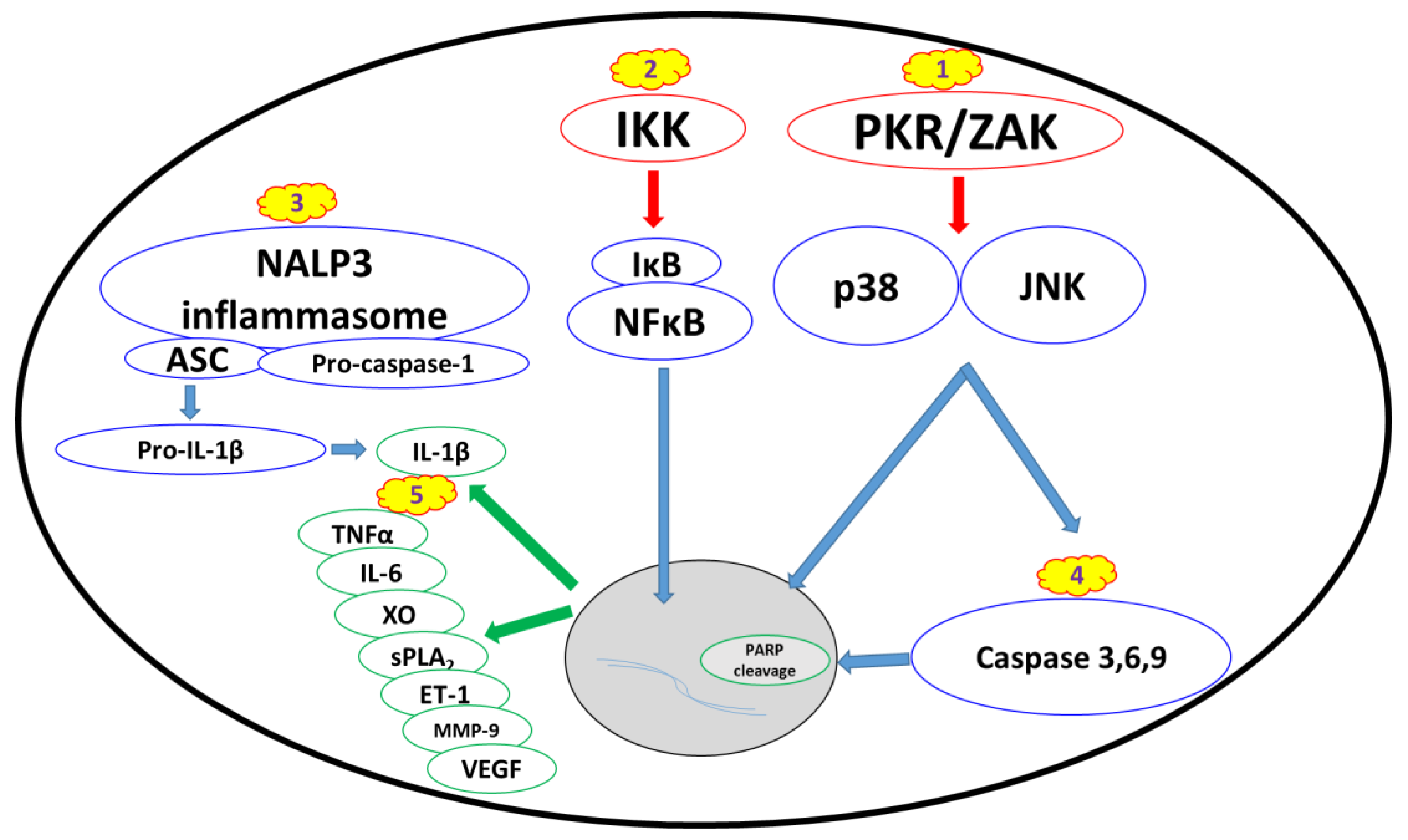
2.2. Biomarkers and Cellular Stress Pathways
2.2.1. Ribotoxic Stress Response
2.2.2. Nuclear Factor Kappa B Pathway
2.2.3. Proinflammatory Cytokines and Damage Mediators
2.2.4. Apoptosis and Changes in Cell Morphology
3. Countermeasures for Ricin Intoxication
3.1. Antitoxins
3.2. Disease-Modifying Countermeasures
3.2.1. Attenuation of Proinflammatory Cytokines and Damage Mediators
3.2.2. Ribotoxic Stress Response Inhibitors
3.2.3. NFкB Inhibitors
3.2.4. NALP3 Inflammasome Inhibitors
3.2.5. Compounds Counteracting Apoptosis and Cell Morphology Changes
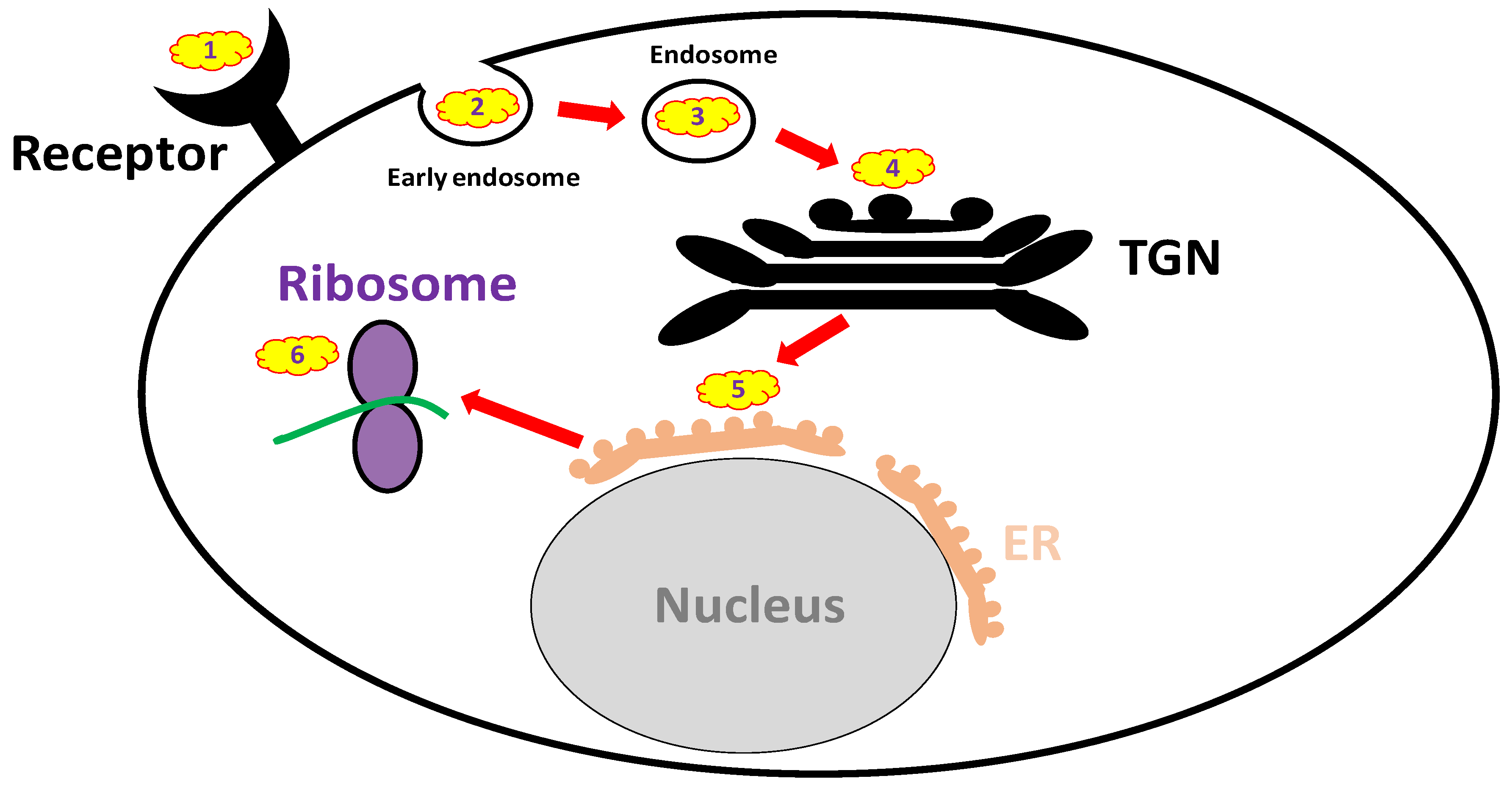
3.3. Anti-ricin Small Molecules
3.3.1. Receptor Mimicry (RTB Binders)
3.3.2. Endocytosis Blockers
3.3.3. Trafficking Blockers
3.3.4. Reductive Activation Inhibitors
3.3.5. Active Site and RTA Inhibitors
3.4. Multiple Pathway-Interfering Anti-Ricin Agents
3.5. Drug-Drug Combinational Treatment
3.6. Antitoxin-Drug Combinational Treatment
4. Summary and Future Prospects
Conflicts of Interest
References
- Olsnes, S.; Kozlov, J.V. Ricin. Toxicon 2001, 39, 1723–1728. [Google Scholar] [CrossRef]
- Endo, Y.; Mitsui, K.; Motizuki, M.; Tsurugi, K. The mechanism of action of ricin and related toxic lectins on eukaryotic ribosomes. The site and the characteristics of the modification in 28 s ribosomal rna caused by the toxins. J. Biol. Chem. 1987, 262, 5908–5912. [Google Scholar] [PubMed]
- Colombatti, M.; Johnson, V.G.; Skopicki, H.A.; Fendley, B.; Lewis, M.S.; Youle, R.J. Identification and characterization of a monoclonal antibody recognizing a galactose-binding domain of the toxin ricin. J. Immunol. 1987, 138, 3339–3344. [Google Scholar] [PubMed]
- Maddaloni, M.; Cooke, C.; Wilkinson, R.; Stout, A.V.; Eng, L.; Pincus, S.H. Immunological characteristics associated with the protective efficacy of antibodies to ricin. J. Immunol. 2004, 172, 6221–6228. [Google Scholar] [CrossRef] [PubMed]
- Noy-Porat, T.; Alcalay, R.; Epstein, E.; Sabo, T.; Kronman, C.; Mazor, O. Extended therapeutic window for post-exposure treatment of ricin intoxication conferred by the use of high-affinity antibodies. Toxicon 2017, 127, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Noy-Porat, T.; Rosenfeld, R.; Ariel, N.; Epstein, E.; Alcalay, R.; Zvi, A.; Kronman, C.; Ordentlich, A.; Mazor, O. Isolation of anti-ricin protective antibodies exhibiting high affinity from immunized non-human primates. Toxins 2016, 8, 64. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Noel, R.; Goudet, A.; Hinsinger, K.; Michau, A.; Pons, V.; Abdelkafi, H.; Secher, T.; Shima, A.; Shtanko, O.; et al. Inhibitors of retrograde trafficking active against ricin and shiga toxins also protect cells from several viruses, leishmania and chlamydiales. Chemico-Biol. Interact. 2017, 267, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Wahome, P.G.; Ahlawat, S.; Mantis, N.J. Identification of small molecules that suppress ricin-induced stress-activated signaling pathways. PLoS ONE 2012, 7, e49075. [Google Scholar] [CrossRef] [PubMed]
- Wahome, P.G.; Bai, Y.; Neal, L.M.; Robertus, J.D.; Mantis, N.J. Identification of small-molecule inhibitors of ricin and shiga toxin using a cell-based high-throughput screen. Toxicon 2010, 56, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Wahome, P.G.; Robertus, J.D.; Mantis, N.J. Small-molecule inhibitors of ricin and shiga toxins. Curr. Top. Microbiol. Immunol. 2012, 357, 179–207. [Google Scholar] [PubMed]
- Gal, Y.; Mazor, O.; Alcalay, R.; Seliger, N.; Aftalion, M.; Sapoznikov, A.; Falach, R.; Kronman, C.; Sabo, T. Antibody/doxycycline combined therapy for pulmonary ricinosis: Attenuation of inflammation improves survival of ricin-intoxicated mice. Toxicol. Rep. 2014, 1, 496–504. [Google Scholar] [CrossRef] [PubMed]
- Gal, Y.; Sapoznikov, A.; Falach, R.; Ehrlich, S.; Aftalion, M.; Sabo, T.; Kronman, C. Potent antiedematous and protective effects of ciprofloxacin in pulmonary ricinosis. Antimicrob. Agents Chemother. 2016, 60, 7153–7158. [Google Scholar] [PubMed]
- DaSilva, L.; Cote, D.; Roy, C.; Martinez, M.; Duniho, S.; Pitt, M.L.; Downey, T.; Dertzbaugh, M. Pulmonary gene expression profiling of inhaled ricin. Toxicon 2003, 41, 813–822. [Google Scholar] [CrossRef]
- Poli, M.A.; Rivera, V.R.; Pitt, M.L.; Vogel, P. Aerosolized specific antibody protects mice from lung injury associated with aerosolized ricin exposure. Toxicon 1996, 34, 1037–1044. [Google Scholar] [CrossRef]
- Roy, C.J.; Hale, M.; Hartings, J.M.; Pitt, L.; Duniho, S. Impact of inhalation exposure modality and particle size on the respiratory deposition of ricin in balb/c mice. Inhal. Toxicol. 2003, 15, 619–638. [Google Scholar] [CrossRef] [PubMed]
- Sapoznikov, A.; Falach, R.; Mazor, O.; Alcalay, R.; Gal, Y.; Seliger, N.; Sabo, T.; Kronman, C. Diverse profiles of ricin-cell interactions in the lung following intranasal exposure to ricin. Toxins 2015, 7, 4817–4831. [Google Scholar] [CrossRef] [PubMed]
- Wilhelmsen, C.L.; Pitt, M.L. Lesions of acute inhaled lethal ricin intoxication in rhesus monkeys. Vet. Pathol. 1996, 33, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Katalan, S.; Falach, R.; Rosner, A.; Goldvaser, M.; Brosh-Nissimov, T.; Dvir, A.; Mizrachi, A.; Goren, O.; Cohen, B.; Gal, Y.; et al. A novel swine model of ricin-induced acute respiratory distress syndrome. Dis. Model. Mech. 2017, 10, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Iordanov, M.S.; Pribnow, D.; Magun, J.L.; Dinh, T.H.; Pearson, J.A.; Chen, S.L.; Magun, B.E. Ribotoxic stress response: Activation of the stress-activated protein kinase jnk1 by inhibitors of the peptidyl transferase reaction and by sequence-specific rna damage to the alpha-sarcin/ricin loop in the 28s rrna. Mol. Cell. Biol. 1997, 17, 3373–3381. [Google Scholar] [CrossRef] [PubMed]
- Jetzt, A.E.; Cheng, J.S.; Tumer, N.E.; Cohick, W.S. Ricin a-chain requires c-jun n-terminal kinase to induce apoptosis in nontransformed epithelial cells. Int. J. Biochem. Cell Biol. 2009, 41, 2503–2510. [Google Scholar] [CrossRef] [PubMed]
- Lindauer, M.; Wong, J.; Magun, B. Ricin toxin activates the nalp3 inflammasome. Toxins 2010, 2, 1500–1514. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, S.; Tamura, T.; Oda, T. Cross-talk between the pathways leading to the induction of apoptosis and the secretion of tumor necrosis factor-alpha in ricin-treated raw 264.7 cells. J. Biochem. 2003, 134, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Jandhyala, D.M.; Ahluwalia, A.; Obrig, T.; Thorpe, C.M. Zak: A map3kinase that transduces shiga toxin- and ricin-induced proinflammatory cytokine expression. Cell. Microbiol. 2008, 10, 1468–1477. [Google Scholar] [CrossRef] [PubMed]
- Tamura, T.; Sadakata, N.; Oda, T.; Muramatsu, T. Role of zinc ions in ricin-induced apoptosis in u937 cells. Toxicol. Lett. 2002, 132, 141–151. [Google Scholar] [CrossRef]
- Jandhyala, D.M.; Wong, J.; Mantis, N.J.; Magun, B.E.; Leong, J.M.; Thorpe, C.M. A novel zak knockout mouse with a defective ribotoxic stress response. Toxins 2016, 8, 259. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.R.; He, K.; Landgraf, J.; Pan, X.; Pestka, J.J. Direct activation of ribosome-associated double-stranded rna-dependent protein kinase (pkr) by deoxynivalenol, anisomycin and ricin: A new model for ribotoxic stress response induction. Toxins 2014, 6, 3406–3425. [Google Scholar] [CrossRef] [PubMed]
- Chu, W.M.; Ostertag, D.; Li, Z.W.; Chang, L.; Chen, Y.; Hu, Y.; Williams, B.; Perrault, J.; Karin, M. Jnk2 and ikkbeta are required for activating the innate response to viral infection. Immunity 1999, 11, 721–731. [Google Scholar] [CrossRef]
- Koromilas, A.E.; Roy, S.; Barber, G.N.; Katze, M.G.; Sonenberg, N. Malignant transformation by a mutant of the ifn-inducible dsrna-dependent protein kinase. Science 1992, 257, 1685–1689. [Google Scholar] [CrossRef] [PubMed]
- Lengyel, P. Tumor-suppressor genes: News about the interferon connection. Proc. Natl. Acad. Sci. USA 1993, 90, 5893–5895. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Natoli, G.; Ghosh, G. Transcriptional regulation via the nf-kappab signaling module. Oncogene 2006, 25, 6706–6716. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. Nuclear factor-kappab in cancer development and progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.F.; Malik, A.B. Nf-kappa b activation as a pathological mechanism of septic shock and inflammation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L622–L645. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, T.S.; Holden, E.P.; Blackwell, T.R.; DeLarco, J.E.; Christman, J.W. Cytokine-induced neutrophil chemoattractant mediates neutrophilic alveolitis in rats: Association with nuclear factor kappa b activation. Am. J. Respir. Cell Mol. Biol. 1994, 11, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Haddad, E.B.; Salmon, M.; Koto, H.; Barnes, P.J.; Adcock, I.; Chung, K.F. Ozone induction of cytokine-induced neutrophil chemoattractant (cinc) and nuclear factor-kappa b in rat lung: Inhibition by corticosteroids. FEBS Lett. 1996, 379, 265–268. [Google Scholar] [CrossRef]
- Sacks, M.; Gordon, J.; Bylander, J.; Porter, D.; Shi, X.L.; Castranova, V.; Kaczmarczyk, W.; Van Dyke, K.; Reasor, M.J. Silica-induced pulmonary inflammation in rats: Activation of nf-kappa b and its suppression by dexamethasone. Biochem. Biophys. Res. Commun. 1998, 253, 181–184. [Google Scholar] [CrossRef] [PubMed]
- Sadikot, R.T.; Jansen, E.D.; Blackwell, T.R.; Zoia, O.; Yull, F.; Christman, J.W.; Blackwell, T.S. High-dose dexamethasone accentuates nuclear factor-kappa b activation in endotoxin-treated mice. Am. J. Respir. Crit. Care Med. 2001, 164, 873–878. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Korcheva, V.; Jacoby, D.B.; Magun, B. Intrapulmonary delivery of ricin at high dosage triggers a systemic inflammatory response and glomerular damage. Am. J. Pathol. 2007, 170, 1497–1510. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Korcheva, V.; Jacoby, D.B.; Magun, B.E. Proinflammatory responses of human airway cells to ricin involve stress-activated protein kinases and nf-kappab. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L1385–L1394. [Google Scholar] [CrossRef] [PubMed]
- Lindauer, M.L.; Wong, J.; Iwakura, Y.; Magun, B.E. Pulmonary inflammation triggered by ricin toxin requires macrophages and il-1 signaling. J. Immunol. 2009, 183, 1419–1426. [Google Scholar] [CrossRef] [PubMed]
- Finsterbusch, M.; Voisin, M.B.; Beyrau, M.; Williams, T.J.; Nourshargh, S. Neutrophils recruited by chemoattractants in vivo induce microvascular plasma protein leakage through secretion of tnf. J. Exp. Med. 2014, 211, 1307–1314. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Hoidal, J.R.; Mukherjee, T.K. Role of tnfalpha in pulmonary pathophysiology. Respir. Res. 2006, 7, 125. [Google Scholar] [CrossRef] [PubMed]
- Fattori, E.; Cappelletti, M.; Costa, P.; Sellitto, C.; Cantoni, L.; Carelli, M.; Faggioni, R.; Fantuzzi, G.; Ghezzi, P.; Poli, V. Defective inflammatory response in interleukin 6-deficient mice. J. Exp. Med. 1994, 180, 1243–1250. [Google Scholar] [CrossRef] [PubMed]
- Parsons, P.E.; Eisner, M.D.; Thompson, B.T.; Matthay, M.A.; Ancukiewicz, M.; Bernard, G.R.; Wheeler, A.P. Lower tidal volume ventilation and plasma cytokine markers of inflammation in patients with acute lung injury. Crit. Care Med. 2005, 33, 1–6, discussion 230–232. [Google Scholar] [CrossRef] [PubMed]
- Ranieri, V.M.; Suter, P.M.; Tortorella, C.; De Tullio, R.; Dayer, J.M.; Brienza, A.; Bruno, F.; Slutsky, A.S. Effect of mechanical ventilation on inflammatory mediators in patients with acute respiratory distress syndrome: A randomized controlled trial. JAMA 1999, 282, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Remick, D.G.; Bolgos, G.; Copeland, S.; Siddiqui, J. Role of interleukin-6 in mortality from and physiologic response to sepsis. Infect. Immun. 2005, 73, 2751–2757. [Google Scholar] [CrossRef] [PubMed]
- Arbibe, L.; Koumanov, K.; Vial, D.; Rougeot, C.; Faure, G.; Havet, N.; Longacre, S.; Vargaftig, B.B.; Bereziat, G.; Voelker, D.R.; et al. Generation of lyso-phospholipids from surfactant in acute lung injury is mediated by type-ii phospholipase a2 and inhibited by a direct surfactant protein a-phospholipase a2 protein interaction. J. Clin. Investig. 1998, 102, 1152–1160. [Google Scholar] [CrossRef] [PubMed]
- Holm, B.A.; Keicher, L.; Liu, M.Y.; Sokolowski, J.; Enhorning, G. Inhibition of pulmonary surfactant function by phospholipases. J. Appl. Physiol. 1991, 71, 317–321. [Google Scholar] [PubMed]
- Touqui, L.; Wu, Y.Z. Interaction of secreted phospholipase a2 and pulmonary surfactant and its pathophysiological relevance in acute respiratory distress syndrome. Acta Pharmacol. Sin. 2003, 24, 1292–1296. [Google Scholar] [PubMed]
- Lee, Y.M.; Hybertson, B.M.; Terada, L.S.; Repine, A.J.; Cho, H.G.; Repine, J.E. Mepacrine decreases lung leak in rats given interleukin-1 intratracheally. Am. J. Respir. Crit. Care Med. 1997, 155, 1624–1628. [Google Scholar] [CrossRef] [PubMed]
- Munoz, N.M.; Meliton, A.Y.; Meliton, L.N.; Dudek, S.M.; Leff, A.R. Secretory group v phospholipase a2 regulates acute lung injury and neutrophilic inflammation caused by lps in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, L879–L887. [Google Scholar] [CrossRef] [PubMed]
- Kaner, R.J.; Ladetto, J.V.; Singh, R.; Fukuda, N.; Matthay, M.A.; Crystal, R.G. Lung overexpression of the vascular endothelial growth factor gene induces pulmonary edema. Am. J. Respir. Cell Mol. Biol. 2000, 22, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Carney, D.E.; McCann, U.G.; Schiller, H.J.; Gatto, L.A.; Steinberg, J.; Picone, A.L.; Nieman, G.F. Metalloproteinase inhibition prevents acute respiratory distress syndrome. J. Surg. Res. 2001, 99, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Soccal, P.M.; Gasche, Y.; Pache, J.C.; Schneuwly, O.; Slosman, D.O.; Morel, D.R.; Spiliopoulos, A.; Suter, P.M.; Nicod, L.P. Matrix metalloproteinases correlate with alveolar-capillary permeability alteration in lung ischemia-reperfusion injury. Transplantation 2000, 70, 998–1005. [Google Scholar] [CrossRef] [PubMed]
- Grosso, M.A.; Brown, J.M.; Viders, D.E.; Mulvin, D.W.; Banerjee, A.; Velasco, S.E.; Repine, J.E.; Harken, A.H. Xanthine oxidase-derived oxygen radicals induce pulmonary edema via direct endothelial cell injury. J. Surg. Res. 1989, 46, 355–360. [Google Scholar] [CrossRef]
- Komaki, Y.; Sugiura, H.; Koarai, A.; Tomaki, M.; Ogawa, H.; Akita, T.; Hattori, T.; Ichinose, M. Cytokine-mediated xanthine oxidase upregulation in chronic obstructive pulmonary disease’s airways. Pulm. Pharmacol. Ther. 2005, 18, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Wright, R.M.; Ginger, L.A.; Kosila, N.; Elkins, N.D.; Essary, B.; McManaman, J.L.; Repine, J.E. Mononuclear phagocyte xanthine oxidoreductase contributes to cytokine-induced acute lung injury. Am. J. Respir. Cell Mol. Biol. 2004, 30, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Kumar, O.; Sugendran, K.; Vijayaraghavan, R. Oxidative stress associated hepatic and renal toxicity induced by ricin in mice. Toxicon 2003, 41, 333–338. [Google Scholar] [CrossRef]
- Muldoon, D.F.; Bagchi, D.; Hassoun, E.A.; Stohs, S.J. The modulating effects of tumor necrosis factor alpha antibody on ricin-induced oxidative stress in mice. J. Biochem. Toxicol. 1994, 9, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Muldoon, D.F.; Hassoun, E.A.; Stohs, S.J. Ricin-induced hepatic lipid peroxidation, glutathione depletion, and DNA single-strand breaks in mice. Toxicon 1992, 30, 977–984. [Google Scholar] [CrossRef]
- Muldoon, D.F.; Hassoun, E.A.; Stohs, S.J. Role of iron in ricin-induced lipid peroxidation and superoxide production. Res. Commun. Mol. Pathol. Pharmacol. 1996, 92, 107–118. [Google Scholar] [PubMed]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Oda, T.; Iwaoka, J.; Komatsu, N.; Muramatsu, T. Involvement of n-acetylcysteine-sensitive pathways in ricin-induced apoptotic cell death in u937 cells. Biosci. Biotechnol. Biochem. 1999, 63, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Blouquit, S.; Sari, A.; Lombet, A.; D’Herbomez, M.; Naline, E.; Matran, R.; Chinet, T. Effects of endothelin-1 on epithelial ion transport in human airways. Am. J. Respir. Cell Mol. Biol. 2003, 29, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Comellas, A.P.; Briva, A.; Dada, L.A.; Butti, M.L.; Trejo, H.E.; Yshii, C.; Azzam, Z.S.; Litvan, J.; Chen, J.; Lecuona, E.; et al. Endothelin-1 impairs alveolar epithelial function via endothelial etb receptor. Am. J. Respir. Crit. Care Med. 2009, 179, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Shaul, P.W.; Borok, Z.; Willis, B.C. Endothelin-1 induces alveolar epithelial-mesenchymal transition through endothelin type a receptor-mediated production of tgf-beta1. Am. J. Respir. Cell Mol. Biol. 2007, 37, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Frank, J.A.; Briot, R.; Lee, J.W.; Ishizaka, A.; Uchida, T.; Matthay, M.A. Physiological and biochemical markers of alveolar epithelial barrier dysfunction in perfused human lungs. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L52–L59. [Google Scholar] [CrossRef] [PubMed]
- Marasciulo, F.L.; Montagnani, M.; Potenza, M.A. Endothelin-1: The yin and yang on vascular function. Curr. Med. Chem. 2006, 13, 1655–1665. [Google Scholar] [CrossRef] [PubMed]
- Nakano, Y.; Tasaka, S.; Saito, F.; Yamada, W.; Shiraishi, Y.; Ogawa, Y.; Koh, H.; Hasegawa, N.; Fujishima, S.; Hashimoto, S.; et al. Endothelin-1 level in epithelial lining fluid of patients with acute respiratory distress syndrome. Respirology 2007, 12, 740–743. [Google Scholar] [CrossRef] [PubMed]
- Kuzkov, V.V.; Kirov, M.Y.; Sovershaev, M.A.; Kuklin, V.N.; Suborov, E.V.; Waerhaug, K.; Bjertnaes, L.J. Extravascular lung water determined with single transpulmonary thermodilution correlates with the severity of sepsis-induced acute lung injury. Crit. Care Med. 2006, 34, 1647–1653. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.M.; Rozendal, C.S.; Schieber, C.; Dehler, M.; Zugel, S.; Bardenheuer, H.J.; Bartsch, P.; Mairbaurl, H. The effect of endothelin-1 on alveolar fluid clearance and pulmonary edema formation in the rat. Anesthesia Analg. 2009, 108, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.F.; White, D.E. Ultrastructure of rat lung following inhalation of ricin aerosol. Int. J. Exp. Pathol. 1997, 78, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.N.; Lindsay, C.D.; Griffiths, G.D. Morphology of ricin and abrin exposed endothelial cells is consistent with apoptotic cell death. Hum. Exp. Toxicol. 1996, 15, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Lindstrom, A.L.; Erlandsen, S.L.; Kersey, J.H.; Pennell, C.A. An in vitro model for toxin-mediated vascular leak syndrome: Ricin toxin a chain increases the permeability of human endothelial cell monolayers. Blood 1997, 90, 2323–2334. [Google Scholar] [PubMed]
- Gonzalez, T.V.; Farrant, S.A.; Mantis, N.J. Ricin induces il-8 secretion from human monocyte/macrophages by activating the p38 map kinase pathway. Mol. Immunol. 2006, 43, 1920–1923. [Google Scholar] [CrossRef] [PubMed]
- Hassoun, E.; Wang, X. Ricin-induced toxicity in the macrophage j744a.1 cells: The role of tnf-alpha and the modulation effects of tnf-alpha polyclonal antibody. J. Biochem. Mol. Toxicol. 2000, 14, 95–101. [Google Scholar] [CrossRef]
- Hu, R.; Zhai, Q.; Liu, W.; Liu, X. An insight into the mechanism of cytotoxicity of ricin to hepatoma cell: Roles of bcl-2 family proteins, caspases, ca(2+)-dependent proteases and protein kinase c. J. Cell. Biochem. 2001, 81, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.H.; Shih, S.F.; Lin, J.Y. Ricin triggers apoptotic morphological changes through caspase-3 cleavage of bat3. J. Biol. Chem. 2004, 279, 19264–19275. [Google Scholar] [CrossRef] [PubMed]
- Soler-Rodriguez, A.M.; Ghetie, M.A.; Oppenheimer-Marks, N.; Uhr, J.W.; Vitetta, E.S. Ricin a-chain and ricin a-chain immunotoxins rapidly damage human endothelial cells: Implications for vascular leak syndrome. Exp. Cell Res. 1993, 206, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Yermakova, A.; Mantis, N.J. Protective immunity to ricin toxin conferred by antibodies against the toxin’s binding subunit (rtb). Vaccine 2011, 29, 7925–7935. [Google Scholar] [CrossRef] [PubMed]
- Neal, L.M.; O’Hara, J.; Brey, R.N., 3rd; Mantis, N.J. A monoclonal immunoglobulin g antibody directed against an immunodominant linear epitope on the ricin a chain confers systemic and mucosal immunity to ricin. Infect. Immun. 2010, 78, 552–561. [Google Scholar] [CrossRef] [PubMed]
- Li, X.P.; Chiou, J.C.; Remacha, M.; Ballesta, J.P.; Tumer, N.E. A two-step binding model proposed for the electrostatic interactions of ricin a chain with ribosomes. Biochemistry 2009, 48, 3853–3863. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, J.M.; Yermakova, A.; Mantis, N.J. Immunity to ricin: Fundamental insights into toxin-antibody interactions. In Ricin and Shiga Toxins; Springer: Berlin, Germany, 2011; Volume 357, pp. 209–241. [Google Scholar]
- Griffiths, G.D.; Phillips, G.J.; Holley, J. Inhalation toxicology of ricin preparations: Animal models, prophylactic and therapeutic approaches to protection. Inhal. Toxicol. 2007, 19, 873–887. [Google Scholar] [CrossRef] [PubMed]
- Pratt, T.S.; Pincus, S.H.; Hale, M.L.; Moreira, A.L.; Roy, C.J.; Tchou-Wong, K.M. Oropharyngeal aspiration of ricin as a lung challenge model for evaluation of the therapeutic index of antibodies against ricin a-chain for post-exposure treatment. Exp. Lung Res. 2007, 33, 459–481. [Google Scholar] [CrossRef] [PubMed]
- Israeli, O.; Falach, R.; Sapoznikov, A.; Gal, Y.; Shifman, O.; Ehrlich, S.; Aftalion, M.; Beth-Din, A.; Kronman, C.; Sabo, T. Determination of ricin intoxication in biological samples by monitoring depurinated 28s rrna in a unique reverse transcription-ligase-polymerase chain reaction assay. Forensic Toxicol. 2017, in press. [Google Scholar] [CrossRef]
- Pincus, S.H.; Das, A.; Song, K.; Maresh, G.A.; Corti, M.; Berry, J. Role of fc in antibody-mediated protection from ricin toxin. Toxins 2014, 6, 1512–1525. [Google Scholar] [CrossRef] [PubMed]
- Sully, E.K.; Whaley, K.J.; Bohorova, N.; Bohorov, O.; Goodman, C.; Kim, D.H.; Pauly, M.H.; Velasco, J.; Hiatt, E.; Morton, J.; et al. Chimeric plantibody passively protects mice against aerosolized ricin challenge. Clin. Vaccine Immunol. CVI 2014, 21, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Prigent, J.; Panigai, L.; Lamourette, P.; Sauvaire, D.; Devilliers, K.; Plaisance, M.; Volland, H.; Creminon, C.; Simon, S. Neutralising antibodies against ricin toxin. PLoS ONE 2011, 6, e20166. [Google Scholar] [CrossRef] [PubMed]
- Respaud, R.; Marchand, D.; Pelat, T.; Tchou-Wong, K.M.; Roy, C.J.; Parent, C.; Cabrera, M.; Guillemain, J.; Mac Loughlin, R.; Levacher, E.; et al. Development of a drug delivery system for efficient alveolar delivery of a neutralizing monoclonal antibody to treat pulmonary intoxication to ricin. J. Control. Release 2016, 234, 21–32. [Google Scholar] [CrossRef] [PubMed]
- O’Hara, J.M.; Whaley, K.; Pauly, M.; Zeitlin, L.; Mantis, N.J. Plant-based expression of a partially humanized neutralizing monoclonal igg directed against an immunodominant epitope on the ricin toxin a subunit. Vaccine 2012, 30, 1239–1243. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.G.; Yin, J.; Chau, D.; Hu, C.C.; Lillico, D.; Yu, J.; Negrych, L.M.; Cherwonogrodzky, J.W. Conformation-dependent high-affinity potent ricin-neutralizing monoclonal antibodies. BioMed Res. Int. 2013, 2013, 471346. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.G.; Yin, J.; Chau, D.; Negrych, L.M.; Cherwonogrodzky, J.W. Humanization and characterization of an anti-ricin neutralization monoclonal antibody. PLoS ONE 2012, 7, e45595. [Google Scholar] [CrossRef] [PubMed]
- Rider, P.; Carmi, Y.; Cohen, I. Biologics for targeting inflammatory cytokines, clinical uses, and limitations. Int. J. Cell Biol. 2016, 2016, 9259646. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; Scheller, J.; Rose-John, S. Therapeutic strategies for the clinical blockade of il-6/gp130 signaling. J. Clin. Investig. 2011, 121, 3375–3383. [Google Scholar] [CrossRef] [PubMed]
- Ueda, O.; Tateishi, H.; Higuchi, Y.; Fujii, E.; Kato, A.; Kawase, Y.; Wada, N.A.; Tachibe, T.; Kakefuda, M.; Goto, C.; et al. Novel genetically-humanized mouse model established to evaluate efficacy of therapeutic agents to human interleukin-6 receptor. Sci. Rep. 2013, 3, 1196. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, R.; Janssen, W.; Altmann, C.; Andres-Hernando, A.; Okamura, K.; Vandivier, R.W.; Ahuja, N.; Faubel, S. Intratracheal il-6 protects against lung inflammation in direct, but not indirect, causes of acute lung injury in mice. PLoS ONE 2013, 8, e61405. [Google Scholar] [CrossRef] [PubMed]
- Wolters, P.J.; Wray, C.; Sutherland, R.E.; Kim, S.S.; Koff, J.; Mao, Y.; Frank, J.A. Neutrophil-derived il-6 limits alveolar barrier disruption in experimental ventilator-induced lung injury. J. Immunol. 2009, 182, 8056–8062. [Google Scholar] [CrossRef] [PubMed]
- Leite, L.M.; Carvalho, A.G.; Ferreira, P.L.; Pessoa, I.X.; Goncalves, D.O.; Lopes Ade, A.; Goes, J.G.; Alves, V.C.; Leal, L.K.; Brito, G.A.; et al. Anti-inflammatory properties of doxycycline and minocycline in experimental models: An in vivo and in vitro comparative study. Inflammopharmacology 2011, 19, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Zidovetzki, R.; Sherman, I.W.; Prudhomme, J.; Crawford, J. Inhibition of plasmodium falciparum lysophospholipase by anti-malarial drugs and sulphydryl reagents. Parasitology 1994, 108 Pt 3, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Ram, A.; Mabalirajan, U.; Singh, S.K.; Singh, V.P.; Ghosh, B. Mepacrine alleviates airway hyperresponsiveness and airway inflammation in a mouse model of asthma. Int. Immunopharmacol. 2008, 8, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Kuipers, M.T.; Aslami, H.; Vlaar, A.P.; Juffermans, N.P.; Tuip-de Boer, A.M.; Hegeman, M.A.; Jongsma, G.; Roelofs, J.J.; van der Poll, T.; Schultz, M.J.; et al. Pre-treatment with allopurinol or uricase attenuates barrier dysfunction but not inflammation during murine ventilator-induced lung injury. PLoS ONE 2012, 7, e50559. [Google Scholar] [CrossRef] [PubMed]
- Gasse, P.; Riteau, N.; Charron, S.; Girre, S.; Fick, L.; Petrilli, V.; Tschopp, J.; Lagente, V.; Quesniaux, V.F.; Ryffel, B.; et al. Uric acid is a danger signal activating nalp3 inflammasome in lung injury inflammation and fibrosis. Am. J. Respir. Crit. Care Med. 2009, 179, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Fahmi, A.N.; Shehatou, G.S.; Shebl, A.M.; Salem, H.A. Febuxostat protects rats against lipopolysaccharide-induced lung inflammation in a dose-dependent manner. Naunyn Schmiedebergs Arch. Pharmacol. 2016, 389, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, H.; Yang, K.; Rock, K.L. The xanthine oxidase inhibitor febuxostat reduces tissue uric acid content and inhibits injury-induced inflammation in the liver and lung. Eur. J. Pharmacol. 2015, 746, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Chow, C.W.; Herrera Abreu, M.T.; Suzuki, T.; Downey, G.P. Oxidative stress and acute lung injury. Am. J. Respir. Cell Mol. Biol. 2003, 29, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Cherubin, P.; Garcia, M.C.; Curtis, D.; Britt, C.B.; Craft, J.W., Jr.; Burress, H.; Berndt, C.; Reddy, S.; Guyette, J.; Zheng, T.; et al. Inhibition of cholera toxin and other ab toxins by polyphenolic compounds. PLoS ONE 2016, 11, e0166477. [Google Scholar] [CrossRef] [PubMed]
- Dyer, P.D.; Kotha, A.K.; Gollings, A.S.; Shorter, S.A.; Shepherd, T.R.; Pettit, M.W.; Alexander, B.D.; Getti, G.T.; El-Daher, S.; Baillie, L.; et al. An in vitro evaluation of epigallocatechin gallate (egcg) as a biocompatible inhibitor of ricin toxin. Biochim. Biophys. Acta 2016, 1860, 1541–1550. [Google Scholar] [CrossRef] [PubMed]
- Rocksen, D.; Ekstrand-Hammarstrom, B.; Johansson, L.; Bucht, A. Vitamin e reduces transendothelial migration of neutrophils and prevents lung injury in endotoxin-induced airway inflammation. Am. J. Respir. Cell Mol. Biol. 2003, 28, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Bae, H.B.; Li, M.; Kim, J.P.; Kim, S.J.; Jeong, C.W.; Lee, H.G.; Kim, W.M.; Kim, H.S.; Kwak, S.H. The effect of epigallocatechin gallate on lipopolysaccharide-induced acute lung injury in a murine model. Inflammation 2010, 33, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Dong, M.; Bo, L.; Li, C.; Liu, Q.; Li, Y.; Ma, L.; Xie, Y.; Fu, E.; Mu, D.; et al. Epigallocatechin-3-gallate ameliorates seawater aspiration-induced acute lung injury via regulating inflammatory cytokines and inhibiting jak/stat1 pathway in rats. Mediat. Inflamm. 2014, 2014, 612593. [Google Scholar] [CrossRef] [PubMed]
- Thickett, D.R.; Armstrong, L.; Christie, S.J.; Millar, A.B. Vascular endothelial growth factor may contribute to increased vascular permeability in acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 2001, 164, 1601–1605. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of vegf and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Holash, J.; Davis, S.; Papadopoulos, N.; Croll, S.D.; Ho, L.; Russell, M.; Boland, P.; Leidich, R.; Hylton, D.; Burova, E.; et al. Vegf-trap: A vegf blocker with potent antitumor effects. Proc. Natl. Acad. Sci. USA 2002, 99, 11393–11398. [Google Scholar] [CrossRef] [PubMed]
- Corne, J.; Chupp, G.; Lee, C.G.; Homer, R.J.; Zhu, Z.; Chen, Q.; Ma, B.; Du, Y.; Roux, F.; McArdle, J.; et al. Il-13 stimulates vascular endothelial cell growth factor and protects against hyperoxic acute lung injury. J. Clin. Investig. 2000, 106, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Channick, R.N.; Sitbon, O.; Barst, R.J.; Manes, A.; Rubin, L.J. Endothelin receptor antagonists in pulmonary arterial hypertension. J. Am. Coll. Cardiol. 2004, 43, 62S–67S. [Google Scholar] [CrossRef] [PubMed]
- Kuklin, V.N.; Kirov, M.Y.; Evgenov, O.V.; Sovershaev, M.A.; Sjoberg, J.; Kirova, S.S.; Bjertnaes, L.J. Novel endothelin receptor antagonist attenuates endotoxin-induced lung injury in sheep. Crit. Care Med. 2004, 32, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Birukova, A.A.; Wu, T.; Tian, Y.; Meliton, A.; Sarich, N.; Tian, X.; Leff, A.; Birukov, K.G. Iloprost improves endothelial barrier function in lipopolysaccharide-induced lung injury. Eur. Respir. J. 2013, 41, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, M.G.; Armstrong, S.M.; Wang, C.; Hwang, D.; Leong-Poi, H.; Advani, A.; Advani, S.; Zhang, H.; Szaszi, K.; Tabuchi, A.; et al. The tie2-agonist vasculotide rescues mice from influenza virus infection. Sci. Rep. 2015, 5, 11030. [Google Scholar] [CrossRef] [PubMed]
- Maris, N.A.; de Vos, A.F.; Dessing, M.C.; Spek, C.A.; Lutter, R.; Jansen, H.M.; van der Zee, J.S.; Bresser, P.; van der Poll, T. Antiinflammatory effects of salmeterol after inhalation of lipopolysaccharide by healthy volunteers. Am. J. Respir. Crit. Care Med. 2005, 172, 878–884. [Google Scholar] [CrossRef] [PubMed]
- Perkins, G.D.; McAuley, D.F.; Richter, A.; Thickett, D.R.; Gao, F. Bench-to-bedside review: Beta2-agonists and the acute respiratory distress syndrome. Crit. Care 2004, 8, 25–32. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Perkins, G.D.; McAuley, D.F.; Thickett, D.R.; Gao, F. The beta-agonist lung injury trial (balti): A randomized placebo-controlled clinical trial. Am. J. Respir. Crit. Care Med. 2006, 173, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Wigenstam, E.; Koch, B.; Bucht, A.; Jonasson, S. N-acetyl cysteine improves the effects of corticosteroids in a mouse model of chlorine-induced acute lung injury. Toxicology 2015, 328, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Ritter, C.; da Cunha, A.A.; Echer, I.C.; Andrades, M.; Reinke, A.; Lucchiari, N.; Rocha, J.; Streck, E.L.; Menna-Barreto, S.; Moreira, J.C.; et al. Effects of n-acetylcysteine plus deferoxamine in lipopolysaccharide-induced acute lung injury in the rat. Crit. Care Med. 2006, 34, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.G.; Tulapurkar, M.E.; Ramarathnam, A.; Brophy, A.; Martinez, R., 3rd; Hom, K.; Hodges, T.; Samadani, R.; Singh, I.S.; MacKerell, A.D., Jr.; et al. Novel noncatalytic substrate-selective p38alpha-specific mapk inhibitors with endothelial-stabilizing and anti-inflammatory activity. J. Immunol. 2017, 198, 3296–3306. [Google Scholar] [CrossRef] [PubMed]
- Cicenas, J. Jnk inhibitors: Is there a future? MAP Kinase 2015, 4, 31–37. [Google Scholar] [CrossRef]
- Ishii, M.; Suzuki, Y.; Takeshita, K.; Miyao, N.; Kudo, H.; Hiraoka, R.; Nishio, K.; Sato, N.; Naoki, K.; Aoki, T.; et al. Inhibition of c-jun nh2-terminal kinase activity improves ischemia/reperfusion injury in rat lungs. J. Immunol. 2004, 172, 2569–2577. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Kim, H.J.; Moon, C.S.; Chong, Y.H.; Kang, J.L. Inhibition of c-jun nh2-terminal kinase or extracellular signal-regulated kinase improves lung injury. Respir. Res. 2004, 5, 23. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Wu, N.; Wang, Y.; Han, X.; Zheng, Q.; Cai, X.; Zhang, H.; Zhao, M. Jnk inhibitor sp600125 attenuates paraquat-induced acute lung injury: An in vivo and in vitro study. Inflammation 2017, 40, 1319–1330. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Zhang, M.; Zhao, Y.; Chen, J.; Li, B.; Cai, W. Jnk inhibitor sp600125 protects against lipopolysaccharide-induced acute lung injury via upregulation of claudin-4. Exp. Ther. Med. 2014, 8, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Jammi, N.V.; Whitby, L.R.; Beal, P.A. Small molecule inhibitors of the rna-dependent protein kinase. Biochem. Biophys. Res. Commun. 2003, 308, 50–57. [Google Scholar] [CrossRef]
- Gray, J.S.; Bae, H.K.; Li, J.C.; Lau, A.S.; Pestka, J.J. Double-stranded rna-activated protein kinase mediates induction of interleukin-8 expression by deoxynivalenol, shiga toxin 1, and ricin in monocytes. Toxicol. Sci. 2008, 105, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.J.; Huang, W.; Kalikulov, D.; Yoo, J.W.; Placzek, A.N.; Stoica, L.; Zhou, H.; Bell, J.C.; Friedlander, M.J.; Krnjevic, K.; et al. Suppression of pkr promotes network excitability and enhanced cognition by interferon-gamma-mediated disinhibition. Cell 2011, 147, 1384–1396. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Arduini, A.; Baccaro, B.; Furuhashi, M.; Hotamisligil, G.S. Small-molecule inhibitors of pkr improve glucose homeostasis in obese diabetic mice. Diabetes 2014, 63, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Miller, S.C.; Huang, R.; Sakamuru, S.; Shukla, S.J.; Attene-Ramos, M.S.; Shinn, P.; Van Leer, D.; Leister, W.; Austin, C.P.; Xia, M. Identification of known drugs that act as inhibitors of nf-kappab signaling and their mechanism of action. Biochem. Pharmacol. 2010, 79, 1272–1280. [Google Scholar] [CrossRef] [PubMed]
- Burgos, R.A.; Hidalgo, M.A.; Figueroa, C.D.; Conejeros, I.; Hancke, J.L. New potential targets to modulate neutrophil function in inflammation. Mini Rev. Med. Chem. 2009, 9, 153–168. [Google Scholar] [CrossRef] [PubMed]
- Gamble, C.; McIntosh, K.; Scott, R.; Ho, K.H.; Plevin, R.; Paul, A. Inhibitory kappa b kinases as targets for pharmacological regulation. Br. J. Pharmacol. 2012, 165, 802–819. [Google Scholar] [CrossRef] [PubMed]
- Ziegelbauer, K.; Gantner, F.; Lukacs, N.W.; Berlin, A.; Fuchikami, K.; Niki, T.; Sakai, K.; Inbe, H.; Takeshita, K.; Ishimori, M.; et al. A selective novel low-molecular-weight inhibitor of ikappab kinase-beta (ikk-beta) prevents pulmonary inflammation and shows broad anti-inflammatory activity. Br. J. Pharmacol. 2005, 145, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Everhart, M.B.; Han, W.; Sherrill, T.P.; Arutiunov, M.; Polosukhin, V.V.; Burke, J.R.; Sadikot, R.T.; Christman, J.W.; Yull, F.E.; Blackwell, T.S. Duration and intensity of nf-kappab activity determine the severity of endotoxin-induced acute lung injury. J. Immunol. 2006, 176, 4995–5005. [Google Scholar] [CrossRef] [PubMed]
- Riteau, N.; Gasse, P.; Fauconnier, L.; Gombault, A.; Couegnat, M.; Fick, L.; Kanellopoulos, J.; Quesniaux, V.F.; Marchand-Adam, S.; Crestani, B.; et al. Extracellular atp is a danger signal activating p2x7 receptor in lung inflammation and fibrosis. Am. J. Respir. Crit. Care Med. 2010, 182, 774–783. [Google Scholar] [CrossRef] [PubMed]
- Grailer, J.J.; Canning, B.A.; Kalbitz, M.; Haggadone, M.D.; Dhond, R.M.; Andjelkovic, A.V.; Zetoune, F.S.; Ward, P.A. Critical role for the nlrp3 inflammasome during acute lung injury. J. Immunol. 2014, 192, 5974–5983. [Google Scholar] [CrossRef] [PubMed]
- Honda, H.; Nagai, Y.; Matsunaga, T.; Okamoto, N.; Watanabe, Y.; Tsuneyama, K.; Hayashi, H.; Fujii, I.; Ikutani, M.; Hirai, Y.; et al. Isoliquiritigenin is a potent inhibitor of nlrp3 inflammasome activation and diet-induced adipose tissue inflammation. J. Leukoc. Biol. 2014, 96, 1087–1100. [Google Scholar] [CrossRef] [PubMed]
- Juliana, C.; Fernandes-Alnemri, T.; Wu, J.; Datta, P.; Solorzano, L.; Yu, J.W.; Meng, R.; Quong, A.A.; Latz, E.; Scott, C.P.; et al. Anti-inflammatory compounds parthenolide and bay 11-7082 are direct inhibitors of the inflammasome. J. Biol. Chem. 2010, 285, 9792–9802. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Mueller, J.L.; Vitari, A.C.; Misaghi, S.; Fedorova, A.; Deshayes, K.; Lee, W.P.; Hoffman, H.M.; Dixit, V.M. Glyburide inhibits the cryopyrin/nalp3 inflammasome. J. Cell Biol. 2009, 187, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, C.; Chojnacki, J.; Toldo, S.; Mezzaroma, E.; Tranchida, N.; Rose, S.W.; Federici, M.; Van Tassell, B.W.; Zhang, S.; Abbate, A. A novel pharmacologic inhibitor of the nlrp3 inflammasome limits myocardial injury after ischemia-reperfusion in the mouse. J. Cardiovasc. Pharmacol. 2014, 63, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Coll, R.C.; Robertson, A.A.; Chae, J.J.; Higgins, S.C.; Munoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the nlrp3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Youm, Y.H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The ketone metabolite beta-hydroxybutyrate blocks nlrp3 inflammasome-mediated inflammatory disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [PubMed]
- Shao, B.Z.; Xu, Z.Q.; Han, B.Z.; Su, D.F.; Liu, C. Nlrp3 inflammasome and its inhibitors: A review. Front. Pharmacol. 2015, 6, 262. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Bian, J.; Ge, Y. Zinc-deficient diet aggravates ventilation-induced lung injury in rats. J. Biomed. Res. 2012, 26, 59–65. [Google Scholar] [CrossRef]
- Gomez, N.N.; Davicino, R.C.; Biaggio, V.S.; Bianco, G.A.; Alvarez, S.M.; Fischer, P.; Masnatta, L.; Rabinovich, G.A.; Gimenez, M.S. Overexpression of inducible nitric oxide synthase and cyclooxygenase-2 in rat zinc-deficient lung: Involvement of a nf-kappab dependent pathway. Nitric Oxide 2006, 14, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Joshi, P.C.; Mehta, A.; Jabber, W.S.; Fan, X.; Guidot, D.M. Zinc deficiency mediates alcohol-induced alveolar epithelial and macrophage dysfunction in rats. Am. J. Respir. Cell Mol. Biol. 2009, 41, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Liu, M.J.; Lee, B.; Besecker, B.; Lai, J.P.; Guttridge, D.C.; Knoell, D.L. Zinc modulates the innate immune response in vivo to polymicrobial sepsis through regulation of nf-kappab. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 298, L744–L754. [Google Scholar] [CrossRef] [PubMed]
- Turut, H.; Kurutas, E.B.; Bulbuloglu, E.; Yasim, A.; Ozkaya, M.; Onder, A.; Imrek, S.S. Zinc aspartate alleviates lung injury induced by intestinal ischemia-reperfusion in rats. J. Surg. Res. 2009, 151, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Knoell, D.L. Zinc modulates cytokine-induced lung epithelial cell barrier permeability. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L1132–L1141. [Google Scholar] [CrossRef] [PubMed]
- Bouchier-Hayes, L.; Lartigue, L.; Newmeyer, D.D. Mitochondria: Pharmacological manipulation of cell death. J. Clin. Investig. 2005, 115, 2640–2647. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Kroemer, G. Pharmacological manipulation of cell death: Clinical applications in sight? J. Clin. Investig. 2005, 115, 2610–2617. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, W.; Zilbermintz, L.; Cheng, L.W.; Zozaya, J.; Tran, S.H.; Elliott, J.H.; Polukhina, K.; Manasherob, R.; Li, A.; Chi, X.; et al. Bithionol blocks pathogenicity of bacterial toxins, ricin, and zika virus. Sci. Rep. 2016, 6, 34475. [Google Scholar] [CrossRef] [PubMed]
- Dawson, R.M.; Paddle, B.M.; Alderton, M.R. Characterization of the asialofetuin microtitre plate-binding assay for evaluating inhibitors of ricin lectin activity. J. Appl. Toxicol. JAT 1999, 19, 307–312. [Google Scholar] [CrossRef]
- Spiro, R.G.; Bhoyroo, V.D. Structure of the o-glycosidically linked carbohydrate units of fetuin. J. Biol. Chem. 1974, 249, 5704–5717. [Google Scholar] [PubMed]
- Baenziger, J.U.; Fiete, D. Structural determinants of ricinus communis agglutinin and toxin specificity for oligosaccharides. J. Biol. Chem. 1979, 254, 9795–9799. [Google Scholar] [PubMed]
- Tonevitsky, A.G.; Zhukova, O.S.; Mirimanova, N.V.; Omelyanenko, V.G.; Timofeeva, N.V.; Bergelson, L.D. Effect of gangliosides on binding, internalization and cytotoxic activity of ricin. FEBS Lett. 1990, 264, 249–252. [Google Scholar] [CrossRef]
- Blome, M.C.; Schengrund, C.L. Multivalent binding of ricin to bovine serum albumin-based neoglycoconjugates. Toxicon 2008, 51, 1214–1224. [Google Scholar] [CrossRef] [PubMed]
- Uzawa, H.; Ohga, K.; Shinozaki, Y.; Ohsawa, I.; Nagatsuka, T.; Seto, Y.; Nishida, Y. A novel sugar-probe biosensor for the deadly plant proteinous toxin, ricin. Biosens. Bioelectron. 2008, 24, 929–933. [Google Scholar] [CrossRef] [PubMed]
- Nagatsuka, T.; Uzawa, H.; Ohsawa, I.; Seto, Y.; Nishida, Y. Use of lactose against the deadly biological toxin ricin. ACS Appl. Mater. Interfaces 2010, 2, 1081–1085. [Google Scholar] [CrossRef] [PubMed]
- Nagatsuka, T.; Uzawa, H.; Sato, K.; Ohsawa, I.; Seto, Y.; Nishida, Y. Glycotechnology for decontamination of biological agents: A model study using ricin and biotin-tagged synthetic glycopolymers. ACS Appl. Mater. Interfaces 2012, 4, 832–837. [Google Scholar] [CrossRef] [PubMed]
- Dawson, R.M.; Alderton, M.R.; Wells, D.; Hartley, P.G. Monovalent and polyvalent carbohydrate inhibitors of ricin binding to a model of the cell-surface receptor. J. Appl. Toxicol. JAT 2006, 26, 247–252. [Google Scholar] [CrossRef] [PubMed]
- Ganguly, D.; Mukhopadhyay, C. Extended binding site of ricin b lectin for oligosaccharide recognition. Biopolymers 2007, 86, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.M.; McIntyre, G.; Gauthier, M.N.; Zullo, D.; Rao, V.; Steeves, R.M.; Goldmacher, V.S.; Blattler, W.A. The galactose-binding sites of the cytotoxic lectin ricin can be chemically blocked in high yield with reactive ligands prepared by chemical modification of glycopeptides containing triantennary n-linked oligosaccharides. Biochemistry 1991, 30, 3234–3247. [Google Scholar] [CrossRef] [PubMed]
- Itakura, Y.; Nakamura-Tsuruta, S.; Kominami, J.; Sharon, N.; Kasai, K.; Hirabayashi, J. Systematic comparison of oligosaccharide specificity of ricinus communis agglutinin i and erythrina lectins: A search by frontal affinity chromatography. J. Biochem. 2007, 142, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K.; Olsnes, S. Entry of the toxic proteins abrin, modeccin, ricin, and diphtheria toxin into cells. Ii. Effect of ph, metabolic inhibitors, and ionophores and evidence for toxin penetration from endocytotic vesicles. J. Biol. Chem. 1982, 257, 7504–7513. [Google Scholar] [PubMed]
- Sandvig, K.; van Deurs, B. Selective modulation of the endocytic uptake of ricin and fluid phase markers without alteration in transferrin endocytosis. J. Biol. Chem. 1990, 265, 6382–6388. [Google Scholar] [PubMed]
- Yoshida, T.; Chen, C.C.; Zhang, M.S.; Wu, H.C. Disruption of the golgi apparatus by brefeldin a inhibits the cytotoxicity of ricin, modeccin, and pseudomonas toxin. Exp. Cell Res. 1991, 192, 389–395. [Google Scholar] [CrossRef]
- Wellner, R.B.; Pless, D.D.; Thompson, W.L. Characterization of 3′-azido-3′-deoxythymidine inhibition of ricin and pseudomonas exotoxin a toxicity in cho and vero cells. J. Cell. Physiol. 1994, 159, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Nambiar, M.P.; Murugesan, R.; Wu, H.C. Inhibition of the cytotoxicity of protein toxins by a novel plant metabolite, mansonone-d. J. Cell. Physiol. 1998, 176, 40–49. [Google Scholar] [CrossRef]
- Okimoto, T.; Seguchi, T.; Ono, M.; Nakayama, Y.; Funatsu, G.; Fujiwara, T.; Ikehara, Y.; Kuwano, M. Brefeldin a protects ricin-induced cytotoxicity in human cancer kb cell line, but not in its resistant counterpart with altered golgi structures. Cell Struct. Funct. 1993, 18, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Sandvig, K.; Prydz, K.; Hansen, S.H.; van Deurs, B. Ricin transport in brefeldin a-treated cells: Correlation between golgi structure and toxic effect. J. Cell Biol. 1991, 115, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Simm, R.; Kvalvaag, A.S.; van Deurs, B.; Lindback, T.; Sandvig, K. Benzyl alcohol induces a reversible fragmentation of the golgi apparatus and inhibits membrane trafficking between endosomes and the trans-golgi network. Exp. Cell Res. 2017, 357, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Redmann, V.; Gardner, T.; Lau, Z.; Morohashi, K.; Felsenfeld, D.; Tortorella, D. Novel class of potential therapeutics that target ricin retrograde translocation. Toxins 2013, 6, 33–53. [Google Scholar] [CrossRef] [PubMed]
- Stechmann, B.; Bai, S.K.; Gobbo, E.; Lopez, R.; Merer, G.; Pinchard, S.; Panigai, L.; Tenza, D.; Raposo, G.; Beaumelle, B.; et al. Inhibition of retrograde transport protects mice from lethal ricin challenge. Cell 2010, 141, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Park, J.G.; Kahn, J.N.; Tumer, N.E.; Pang, Y.P. Common pharmacophore of structurally distinct small-molecule inhibitors of intracellular retrograde trafficking of ribosome inactivating proteins. Sci. Rep. 2013, 3, 3397. [Google Scholar] [CrossRef] [PubMed]
- Barbier, J.; Bouclier, C.; Johannes, L.; Gillet, D. Inhibitors of the cellular trafficking of ricin. Toxins 2012, 4, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Bassik, M.C.; Kampmann, M.; Lebbink, R.J.; Wang, S.; Hein, M.Y.; Poser, I.; Weibezahn, J.; Horlbeck, M.A.; Chen, S.; Mann, M.; et al. A systematic mammalian genetic interaction map reveals pathways underlying ricin susceptibility. Cell 2013, 152, 909–922. [Google Scholar] [CrossRef] [PubMed]
- Bellisola, G.; Fracasso, G.; Ippoliti, R.; Menestrina, G.; Rosen, A.; Solda, S.; Udali, S.; Tomazzolli, R.; Tridente, G.; Colombatti, M. Reductive activation of ricin and ricin a-chain immunotoxins by protein disulfide isomerase and thioredoxin reductase. Biochem. Pharmacol. 2004, 67, 1721–1731. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, L.; Battelli, M.G.; Stirpe, F. Reduction of ricin and other plant toxins by thiol:Protein disulfide oxidoreductases. Arch. Biochem. Biophys. 1982, 216, 380–383. [Google Scholar] [CrossRef]
- Pasetto, M.; Barison, E.; Castagna, M.; Della Cristina, P.; Anselmi, C.; Colombatti, M. Reductive activation of type 2 ribosome-inactivating proteins is promoted by transmembrane thioredoxin-related protein. J. Biol. Chem. 2012, 287, 7367–7373. [Google Scholar] [CrossRef] [PubMed]
- Kean, W.F.; Hart, L.; Buchanan, W.W. Auranofin. Br. J. Rheumatol. 1997, 36, 560–572. [Google Scholar] [CrossRef] [PubMed]
- Dickerhof, N.; Kleffmann, T.; Jack, R.; McCormick, S. Bacitracin inhibits the reductive activity of protein disulfide isomerase by disulfide bond formation with free cysteines in the substrate-binding domain. FEBS J. 2011, 278, 2034–2043. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.P.; Park, J.G.; Wang, S.; Vummenthala, A.; Mishra, R.K.; McLaughlin, J.E.; Di, R.; Kahn, J.N.; Tumer, N.E.; Janosi, L.; et al. Small-molecule inhibitor leads of ribosome-inactivating proteins developed using the doorstop approach. PLoS ONE 2011, 6, e17883. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Wu, F.; Martiniuk, F.; Hale, M.L.; Ellington, A.D.; Tchou-Wong, K.M. Protective effects of anti-ricin a-chain rna aptamer against ricin toxicity. World J. Gastroenterol. 2008, 14, 6360–6365. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Watt, B.; Wahome, P.G.; Mantis, N.J.; Robertus, J.D. Identification of new classes of ricin toxin inhibitors by virtual screening. Toxicon 2010, 56, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Zhang, Y.; Chen, Y.; Niu, X.; Zhang, Y.; Li, R.; Yang, C.; Wang, Q.; Li, X.; Deng, X. Baicalin inhibits the lethality of ricin in mice by inducing protein oligomerization. J. Biol. Chem. 2015, 290, 12899–12907. [Google Scholar] [CrossRef] [PubMed]
- Jeon, K.I.; Jeong, J.Y.; Jue, D.M. Thiol-reactive metal compounds inhibit nf-kappa b activation by blocking i kappa b kinase. J. Immunol. 2000, 164, 5981–5989. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Gonzalez, J.J.; Guevara-Flores, A.; Rendon, J.L.; del Arenal, I.P. Auranofin-induced oxidative stress causes redistribution of the glutathione pool in taenia crassiceps cysticerci. Mol. Biochem. Parasitol. 2015, 201, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.A.; Zhang, R.; Piao, M.J.; Chae, S.; Kim, H.S.; Park, J.H.; Jung, K.S.; Hyun, J.W. Baicalein inhibits oxidative stress-induced cellular damage via antioxidant effects. Toxicol. Ind. Health 2012, 28, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Shieh, D.E.; Liu, L.T.; Lin, C.C. Antioxidant and free radical scavenging effects of baicalein, baicalin and wogonin. Anticancer Res. 2000, 20, 2861–2865. [Google Scholar] [PubMed]
- Deng, J.; Wang, D.X.; Liang, A.L.; Tang, J.; Xiang, D.K. Effects of baicalin on alveolar fluid clearance and alpha-enac expression in rats with lps-induced acute lung injury. Can. J. Physiol. Pharmacol. 2017, 95, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.M.; Pan, L.; Wang, Y.; Xu, Q.Z. Baicalin exerts protective effects against lipopolysaccharide-induced acute lung injury by regulating the crosstalk between the cx3cl1-cx3cr1 axis and nf-kappab pathway in cx3cl1-knockout mice. Int. J. Mol. Med. 2016, 37, 703–715. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.L.; Chen, C.S.; Hsu, C.W.; Li, M.H.; Chang, H.; Tsai, S.H.; Chu, S.J. Therapeutic effects of baicalin on lipopolysaccharide-induced acute lung injury in rats. Am. J. Chin. Med. 2008, 36, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Das, D.K.; Sato, M.; Ray, P.S.; Maulik, G.; Engelman, R.M.; Bertelli, A.A.; Bertelli, A. Cardioprotection of red wine: Role of polyphenolic antioxidants. Drugs Exp. Clin. Res. 1999, 25, 115–120. [Google Scholar] [PubMed]
- Cassel, S.L.; Eisenbarth, S.C.; Iyer, S.S.; Sadler, J.J.; Colegio, O.R.; Tephly, L.A.; Carter, A.B.; Rothman, P.B.; Flavell, R.A.; Sutterwala, F.S. The nalp3 inflammasome is essential for the development of silicosis. Proc. Natl. Acad. Sci. USA 2008, 105, 9035–9040. [Google Scholar] [CrossRef] [PubMed]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nunez, G.; Schnurr, M.; et al. Nlrp3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [PubMed]
- Dorr, R.T. Radioprotectants: Pharmacology and clinical applications of amifostine. Semin. Radiat. Oncol. 1998, 8, 10–13. [Google Scholar] [PubMed]
- Acosta, J.C.; Richard, C.; Delgado, M.D.; Horita, M.; Rizzo, M.G.; Fernandez-Luna, J.L.; Leon, J. Amifostine impairs p53-mediated apoptosis of human myeloid leukemia cells. Mol. Cancer Ther. 2003, 2, 893–900. [Google Scholar] [PubMed]
- Lee, E.J.; Gerhold, M.; Palmer, M.W.; Christen, R.D. P53 protein regulates the effects of amifostine on apoptosis, cell cycle progression, and cytoprotection. Br. J. Cancer 2003, 88, 754–759. [Google Scholar] [CrossRef] [PubMed]
- Bergen, L.G.; Borisy, G.G. Tubulin-colchicine complex (tc) inhibits microtubule depolymerization by a capping reaction exerted preferentially at the minus end. J. Cell. Biochem. 1986, 30, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Ghio, A.J.; Kennedy, T.P.; Hatch, G.E.; Tepper, J.S. Reduction of neutrophil influx diminishes lung injury and mortality following phosgene inhalation. J. Appl. Physiol. 1991, 71, 657–665. [Google Scholar] [PubMed]
- Choudhury, S.; Kandasamy, K.; Maruti, B.S.; Addison, M.P.; Kasa, J.K.; Darzi, S.A.; Singh, T.U.; Parida, S.; Dash, J.R.; Singh, V.; et al. Atorvastatin along with imipenem attenuates acute lung injury in sepsis through decrease in inflammatory mediators and bacterial load. Eur. J. Pharmacol. 2015, 765, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Siempos, I.I.; Maniatis, N.A.; Kopterides, P.; Magkou, C.; Glynos, C.; Roussos, C.; Armaganidis, A. Pretreatment with atorvastatin attenuates lung injury caused by high-stretch mechanical ventilation in an isolated rabbit lung model. Crit. Care Med. 2010, 38, 1321–1328. [Google Scholar]
- Singla, S.; Zhou, T.; Javaid, K.; Abbasi, T.; Casanova, N.; Zhang, W.; Ma, S.F.; Wade, M.S.; Noth, I.; Sweiss, N.J.; et al. Expression profiling elucidates a molecular gene signature for pulmonary hypertension in sarcoidosis. Pulm. Circ. 2016, 6, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Akamatsu, H.; Asada, M.; Komura, J.; Asada, Y.; Niwa, Y. Effect of doxycycline on the generation of reactive oxygen species: A possible mechanism of action of acne therapy with doxycycline. Acta Dermato-Venereol. 1992, 72, 178–179. [Google Scholar]
- Dalm, D.; Palm, G.J.; Aleksandrov, A.; Simonson, T.; Hinrichs, W. Nonantibiotic properties of tetracyclines: Structural basis for inhibition of secretory phospholipase a2. J. Mol. Biol. 2010, 398, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Fainaru, O.; Adini, I.; Benny, O.; Bazinet, L.; Pravda, E.; D’Amato, R.; Folkman, J. Doxycycline induces membrane expression of ve-cadherin on endothelial cells and prevents vascular hyperpermeability. FASEB J. 2008, 22, 3728–3735. [Google Scholar] [CrossRef] [PubMed]
- Golub, L.M.; Lee, H.M.; Ryan, M.E.; Giannobile, W.V.; Payne, J.; Sorsa, T. Tetracyclines inhibit connective tissue breakdown by multiple non-antimicrobial mechanisms. Adv. Dent. Res. 1998, 12, 12–26. [Google Scholar] [CrossRef] [PubMed]
- Krakauer, T.; Buckley, M. Doxycycline is anti-inflammatory and inhibits staphylococcal exotoxin-induced cytokines and chemokines. Antimicrob. Agents Chemother. 2003, 47, 3630–3633. [Google Scholar] [CrossRef] [PubMed]
- Dalhoff, A. Immunomodulatory activities of fluoroquinolones. Infection 2005, 33 (Suppl. 2), 55–70. [Google Scholar] [CrossRef] [PubMed]
- Bosma, K.J.; Lewis, J.F. Emerging therapies for treatment of acute lung injury and acute respiratory distress syndrome. Expert Opin. Emerg. Drugs 2007, 12, 461–477. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; DalNogare, A. Pharmacological therapy for acute respiratory distress syndrome. Mayo Clin. Proc. 2006, 81, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.D.; Matthay, M.A. Advances in critical care for the nephrologist: Acute lung injury/ards. Clin. J. Am. Soc. Nephrol. 2008, 3, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Raghavendran, K.; Pryhuber, G.S.; Chess, P.R.; Davidson, B.A.; Knight, P.R.; Notter, R.H. Pharmacotherapy of acute lung injury and acute respiratory distress syndrome. Curr. Med. Chem. 2008, 15, 1911–1924. [Google Scholar] [CrossRef] [PubMed]
- D’Elia, R.V.; Harrison, K.; Oyston, P.C.; Lukaszewski, R.A.; Clark, G.C. Targeting the “cytokine storm” for therapeutic benefit. Clin. Vaccine Immunol. CVI 2013, 20, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Mabley, J.G.; Pacher, P.; Szabo, C. Activation of the cholinergic antiinflammatory pathway reduces ricin-induced mortality and organ failure in mice. Mol. Med. 2009, 15, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Cara, D.C.; Kaur, J.; Forster, M.; McCafferty, D.M.; Kubes, P. Role of p38 mitogen-activated protein kinase in chemokine-induced emigration and chemotaxis in vivo. J. Immunol. 2001, 167, 6552–6558. [Google Scholar] [CrossRef] [PubMed]
- McDonald, B.; Pittman, K.; Menezes, G.B.; Hirota, S.A.; Slaba, I.; Waterhouse, C.C.; Beck, P.L.; Muruve, D.A.; Kubes, P. Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science 2010, 330, 362–366. [Google Scholar] [CrossRef] [PubMed]
- Druey, K.M.; Greipp, P.R. Narrative review: The systemic capillary leak syndrome. Ann. Intern. Med. 2010, 153, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Mathison, R.D.; Christie, E.; Davison, J.S. The tripeptide feg inhibits leukocyte adhesion. J. Inflamm. (Lond.) 2008, 5, 6. [Google Scholar] [CrossRef] [PubMed]
- Dery, R.E.; Ulanova, M.; Puttagunta, L.; Stenton, G.R.; James, D.; Merani, S.; Mathison, R.; Davison, J.; Befus, A.D. Frontline: Inhibition of allergen-induced pulmonary inflammation by the tripeptide feg: A mimetic of a neuro-endocrine pathway. Eur. J. Immunol. 2004, 34, 3315–3325. [Google Scholar] [CrossRef] [PubMed]
- Mathison, R.D.; Davison, J.S.; Befus, A.D.; Gingerich, D.A. Salivary gland derived peptides as a new class of anti-inflammatory agents: Review of preclinical pharmacology of c-terminal peptides of smr1 protein. J. Inflamm. (Lond.) 2010, 7, 49. [Google Scholar] [CrossRef] [PubMed]
- Elder, A.S.; Bersten, A.D.; Saccone, G.T.; Dixon, D.L. Prevention and amelioration of rodent endotoxin-induced lung injury with administration of a novel therapeutic tripeptide feg. Pulm. Pharmacol. Ther. 2013, 26, 167–171. [Google Scholar] [CrossRef] [PubMed]
- Panos, R.J.; Bak, P.M.; Simonet, W.S.; Rubin, J.S.; Smith, L.J. Intratracheal instillation of keratinocyte growth factor decreases hyperoxia-induced mortality in rats. J. Clin. Investig. 1995, 96, 2026–2033. [Google Scholar] [CrossRef] [PubMed]
- Yano, T.; Deterding, R.R.; Simonet, W.S.; Shannon, J.M.; Mason, R.J. Keratinocyte growth factor reduces lung damage due to acid instillation in rats. Am. J. Respir. Cell Mol. Biol. 1996, 15, 433–442. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.L.; Soeder, Y.; Dahlke, M.H. Concise review: Mesenchymal stromal cell-based approaches for the treatment of acute respiratory distress and sepsis syndromes. Stem Cells Transl. Med. 2017, 6, 1141–1151. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Antibody Name | Antibody Type | Target | Reference |
|---|---|---|---|
| RAC18 | murine; chimeric | RTA | [86] |
| PB10 | chimeric | [87] | |
| RA36 | murine | [88] | |
| 43RCA-G1 | humanized | [89] | |
| GD12 | murine; chimeric | [90] | |
| MH1 | chimeric | [6] | |
| MH36 | chimeric | [6] | |
| JB4 | chimeric | RTB | [87] |
| RB34 | murine | [88] | |
| RB37 | murine | [88] | |
| D9 | murine; humanized | [91,92] | |
| MH2 | chimeric | [6] | |
| MH73 | chimeric | [6] | |
| MH75 | chimeric | [6] | |
| MH77 | chimeric | [6] |
| Pathway | Target | Inhibitors |
|---|---|---|
| Proinflammatory cytokines | IL-1β | anakinra, immunomodulators |
| TNFα | anti-TNFα agents, immunomodulators | |
| IL-6 | tocilizumab, immunomodulators | |
| Damage mediators | XO | allopurinol, febuxostat, antioxidants |
| sPLA2 | Mepacrine | |
| ET-1 | bosentan, tezosentan | |
| MMP-9 | Doxycycline | |
| VEGF | bevacizumab, aflibercept | |
| NFкB pathway | NFкB | NFкB inhibitors, ‘Compound A’ |
| IKK | IкK inhibitors, auranofin, BMS-345541 | |
| MAP3K | PKR | 2-AP, C16, imoxine, PKRi |
| ZAK | sorafenib, nilotinib, DHP-2 | |
| MAPK | p38 | PW66, UM101, p38 inhibitors |
| JNK | PW66, SP600125, JNK inhibitors | |
| NALP3 inflammasome | NALP3 inflammasome | MCC950, parthenolide, glyburide, BHB, isoliquiritigenen |
| IL-1β | Anakinra | |
| Apoptosis | Apoptosis | antioxidants, zinc, apoptosis inhibitors |
| caspases 3, 6, 7, 9 | PW69, bithionol |
| Mechanism | Cell Target | Inhibitors |
|---|---|---|
| Receptor mimicry | RTB | Derivatives of glycosphingolipids, lactose and galactose |
| Endocytosis blockers | Early endosome | NaN3, cytochalasin D, colchicine |
| Endosome | ||
| Trafficking blockers | TGN | Retro-2, DA2MT, atorvastatin, brefeldin A, mansonone D |
| ER | benzyl alcohol, 3′-Azido-3′-Deoxythimidine | |
| Reductive activation inhibitors | PDI, TrxR, TMX, | auranofin, bacitracin |
| glutathione disulfide oxidoreductase | ||
| Active site and RTA inhibitors | Ribosomes | purine- pterin- and pyrimidine-based inhibitors, 4-fluorophenyl methyl 2-(furan-2-yl)quinolone-4-carboxylate, difluoromethylornithine, aptamers, RIP-α-sarcin/ricin loop interface blockers, baicalin |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gal, Y.; Mazor, O.; Falach, R.; Sapoznikov, A.; Kronman, C.; Sabo, T. Treatments for Pulmonary Ricin Intoxication: Current Aspects and Future Prospects. Toxins 2017, 9, 311. https://doi.org/10.3390/toxins9100311
Gal Y, Mazor O, Falach R, Sapoznikov A, Kronman C, Sabo T. Treatments for Pulmonary Ricin Intoxication: Current Aspects and Future Prospects. Toxins. 2017; 9(10):311. https://doi.org/10.3390/toxins9100311
Chicago/Turabian StyleGal, Yoav, Ohad Mazor, Reut Falach, Anita Sapoznikov, Chanoch Kronman, and Tamar Sabo. 2017. "Treatments for Pulmonary Ricin Intoxication: Current Aspects and Future Prospects" Toxins 9, no. 10: 311. https://doi.org/10.3390/toxins9100311
APA StyleGal, Y., Mazor, O., Falach, R., Sapoznikov, A., Kronman, C., & Sabo, T. (2017). Treatments for Pulmonary Ricin Intoxication: Current Aspects and Future Prospects. Toxins, 9(10), 311. https://doi.org/10.3390/toxins9100311