
Identification of an Essential Region for Translocation of Clostridium difficile Toxin B


Abstract
:
1. Introduction
2. Results
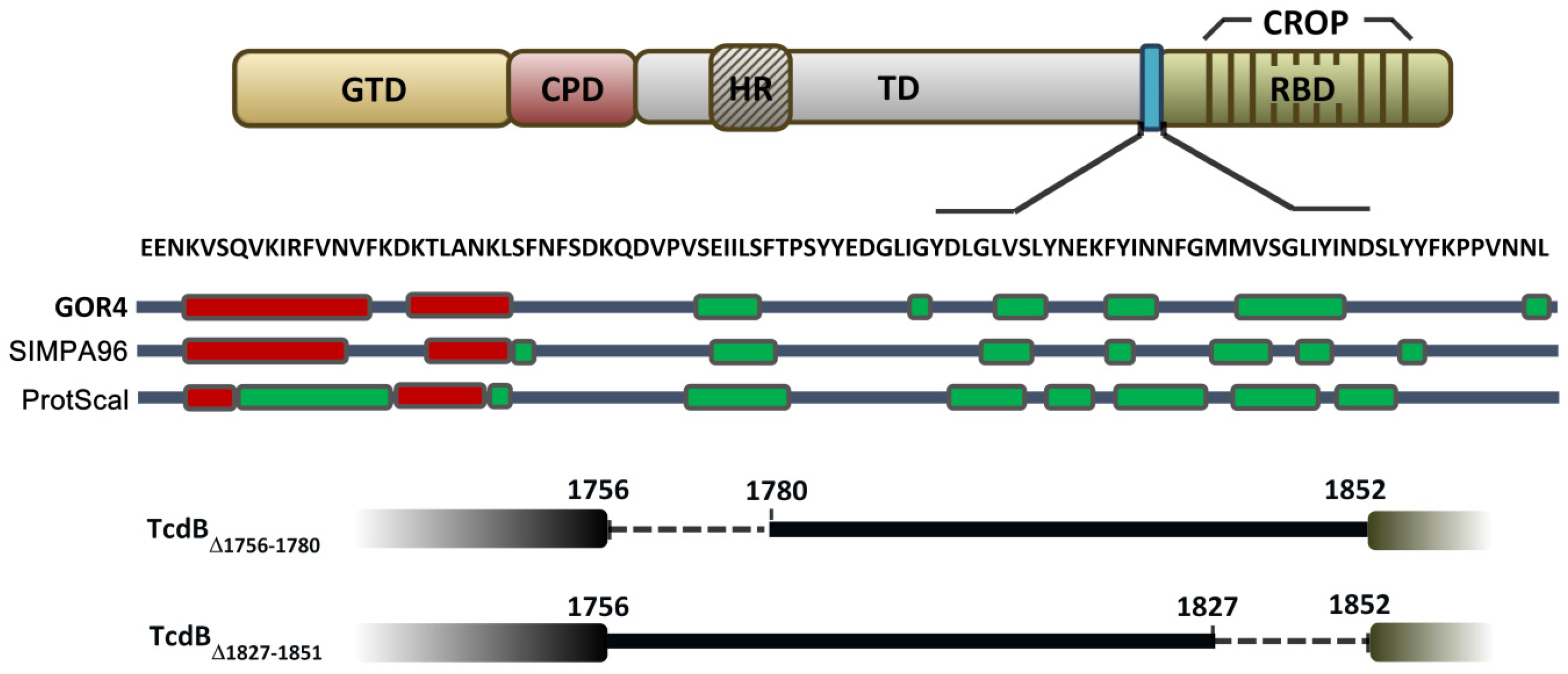
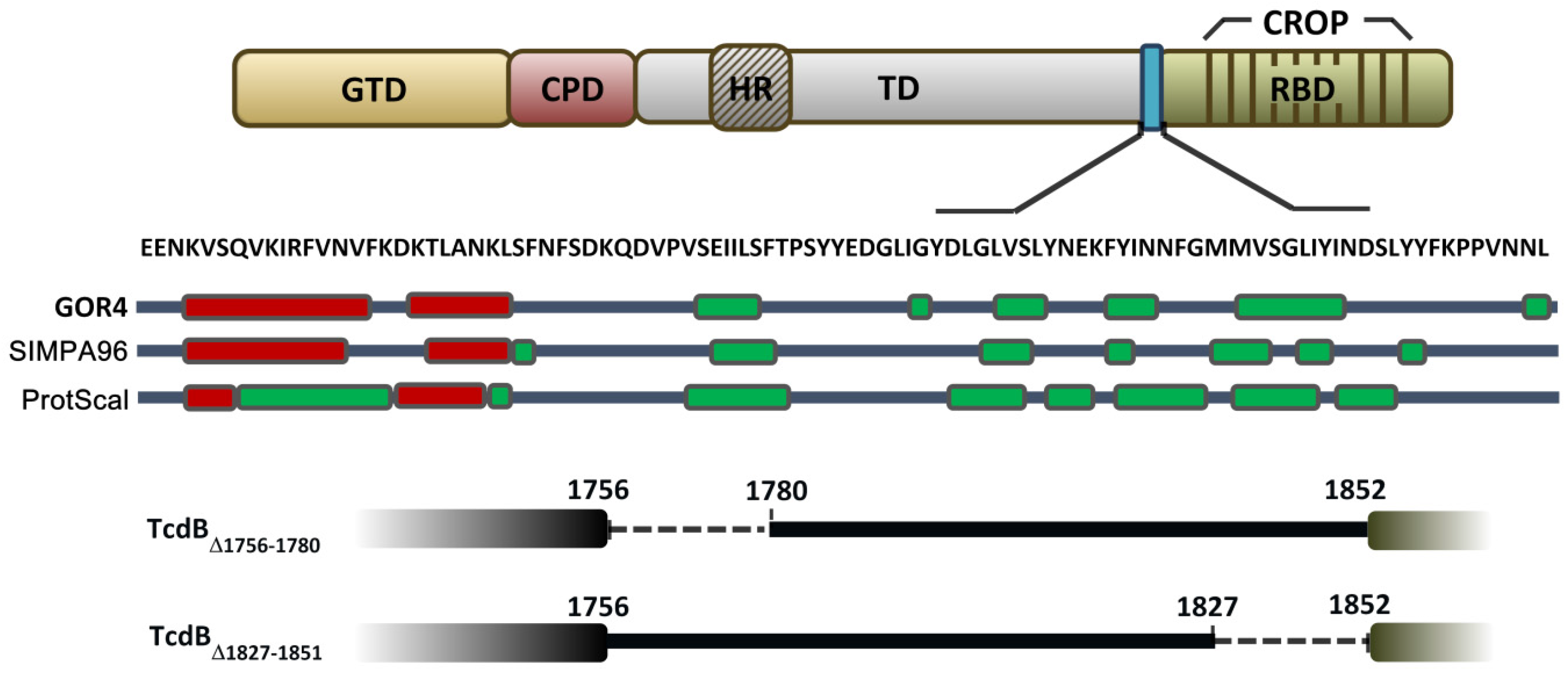
2.1. Prediction of the Secondary Structure of the D97 Segment
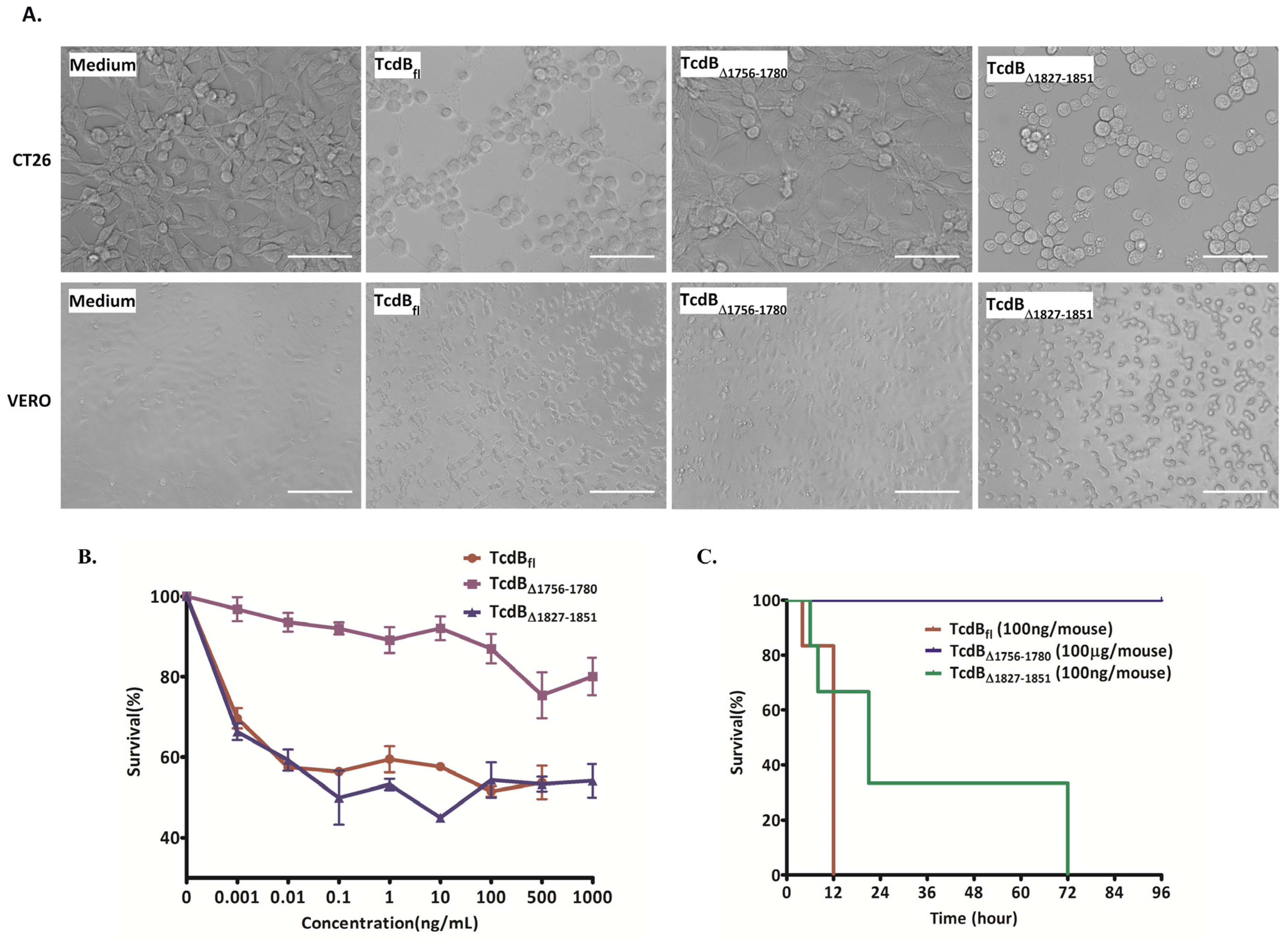
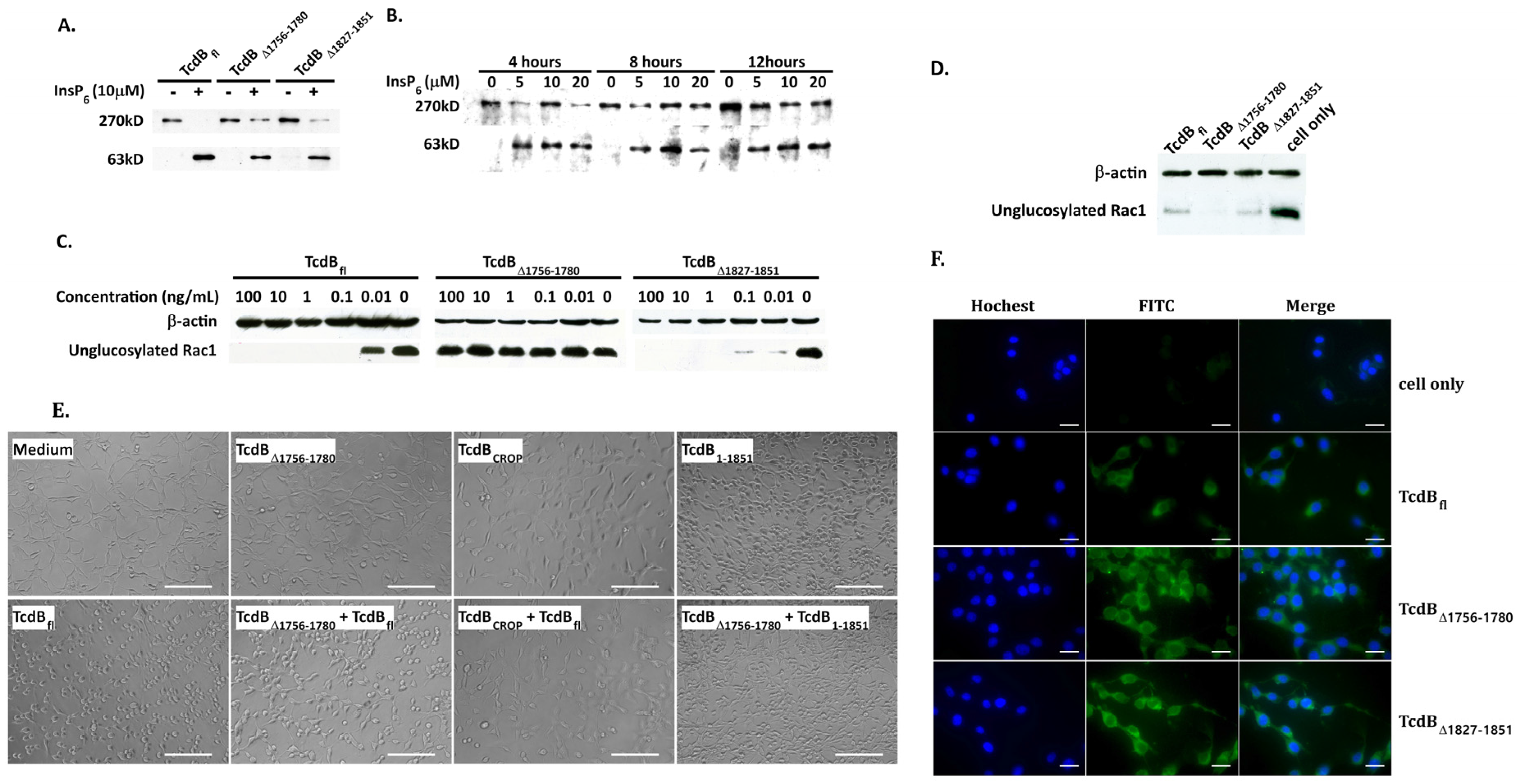
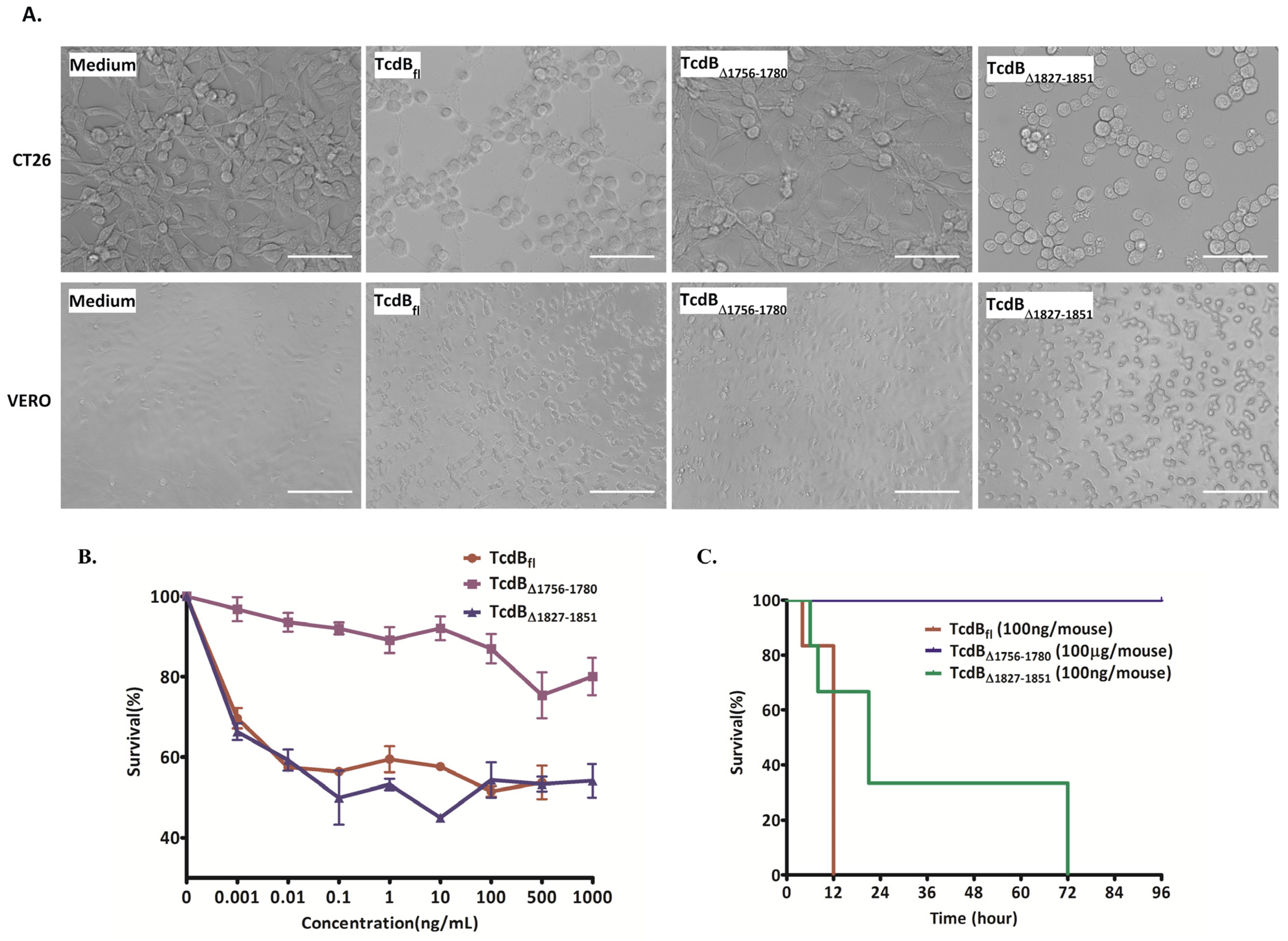
2.2. Cytopathic and Cytotoxic Effects of TcdBΔ1756–1780
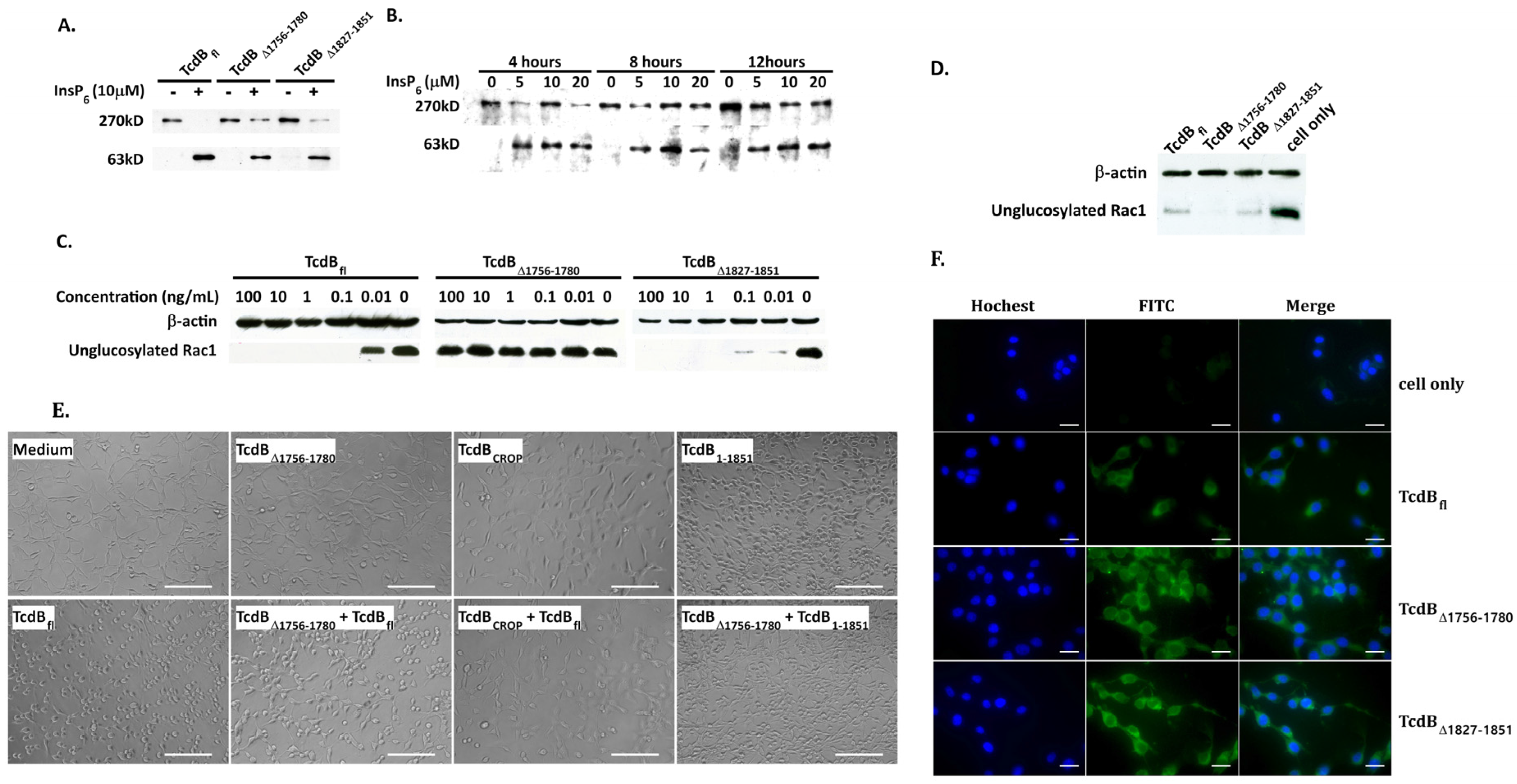
2.3. Analysis of Cysteine Protease, Glucosyltransferase Activity, and Cellular Binding of TcdBΔ1756–1780
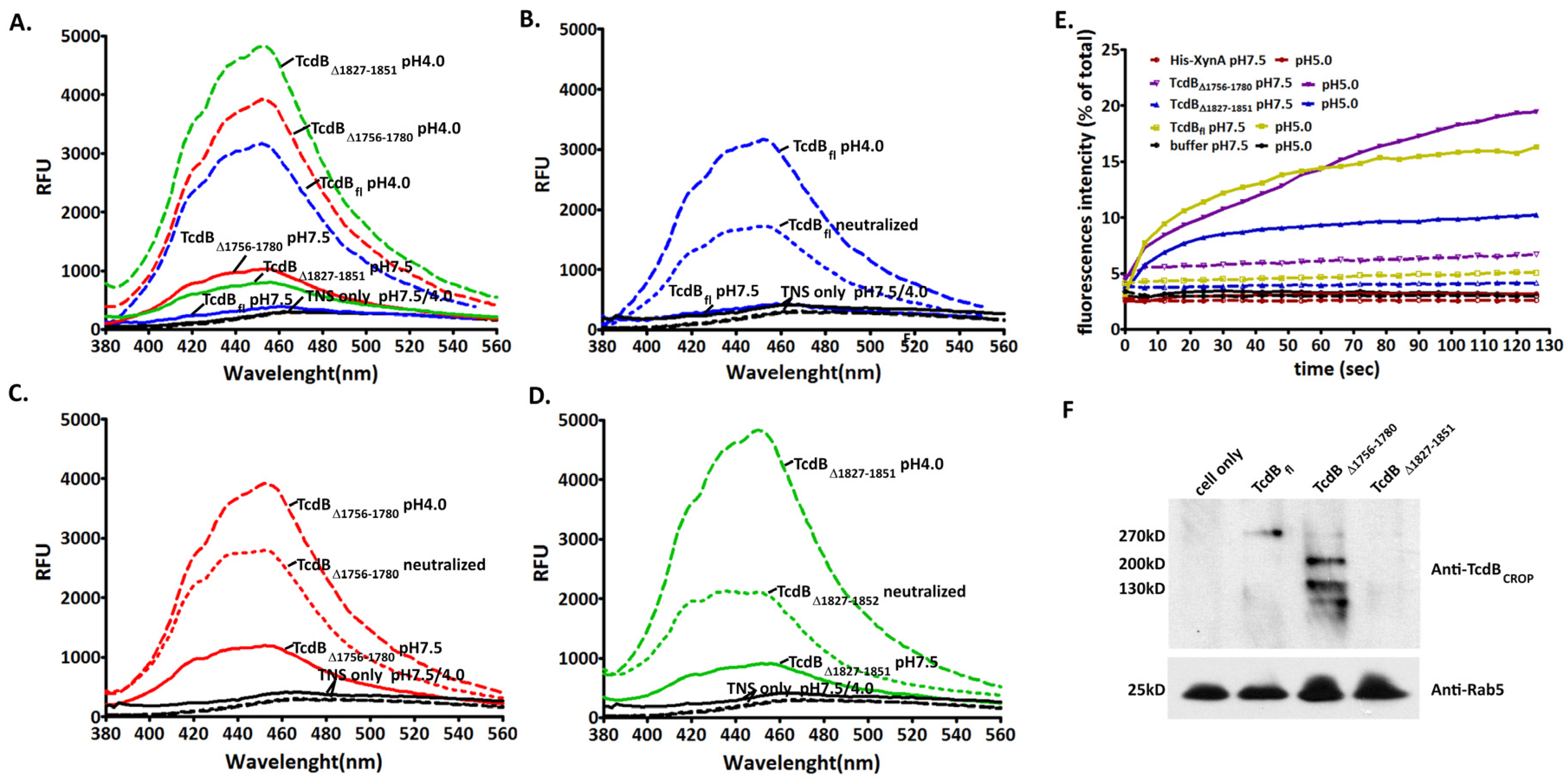
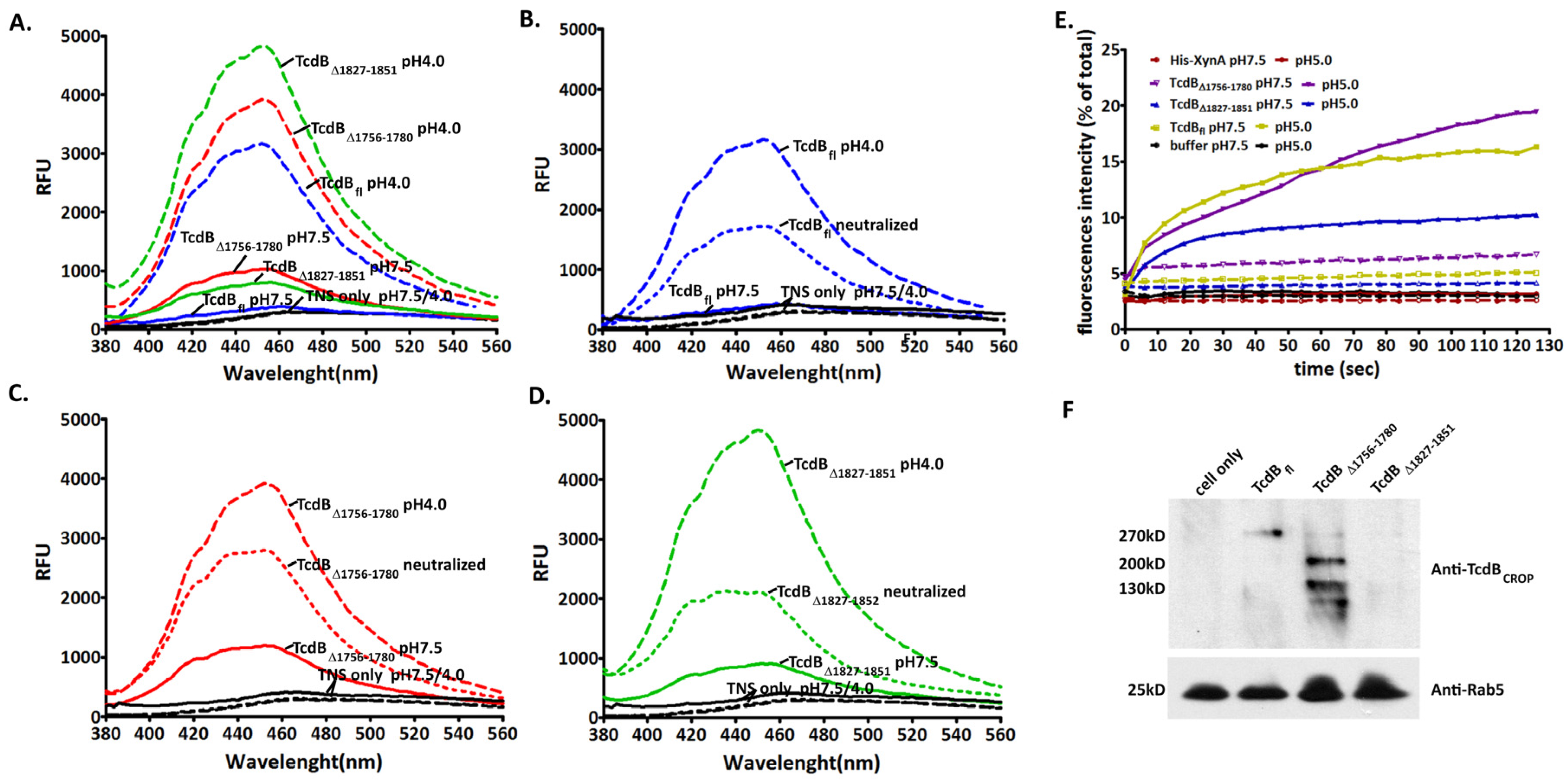
2.4. Conformational Change at Low pH and Pore Formation by TcdB∆1756–1780
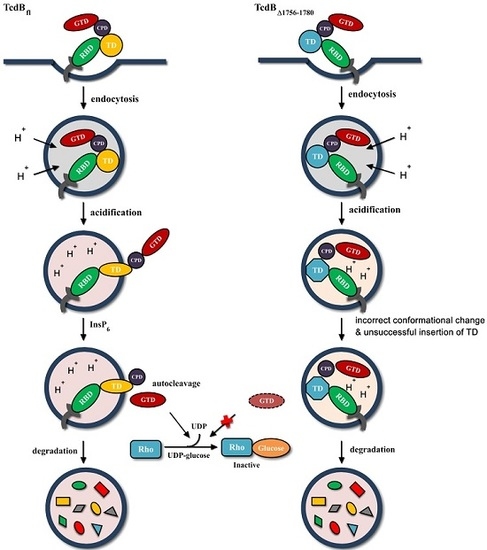
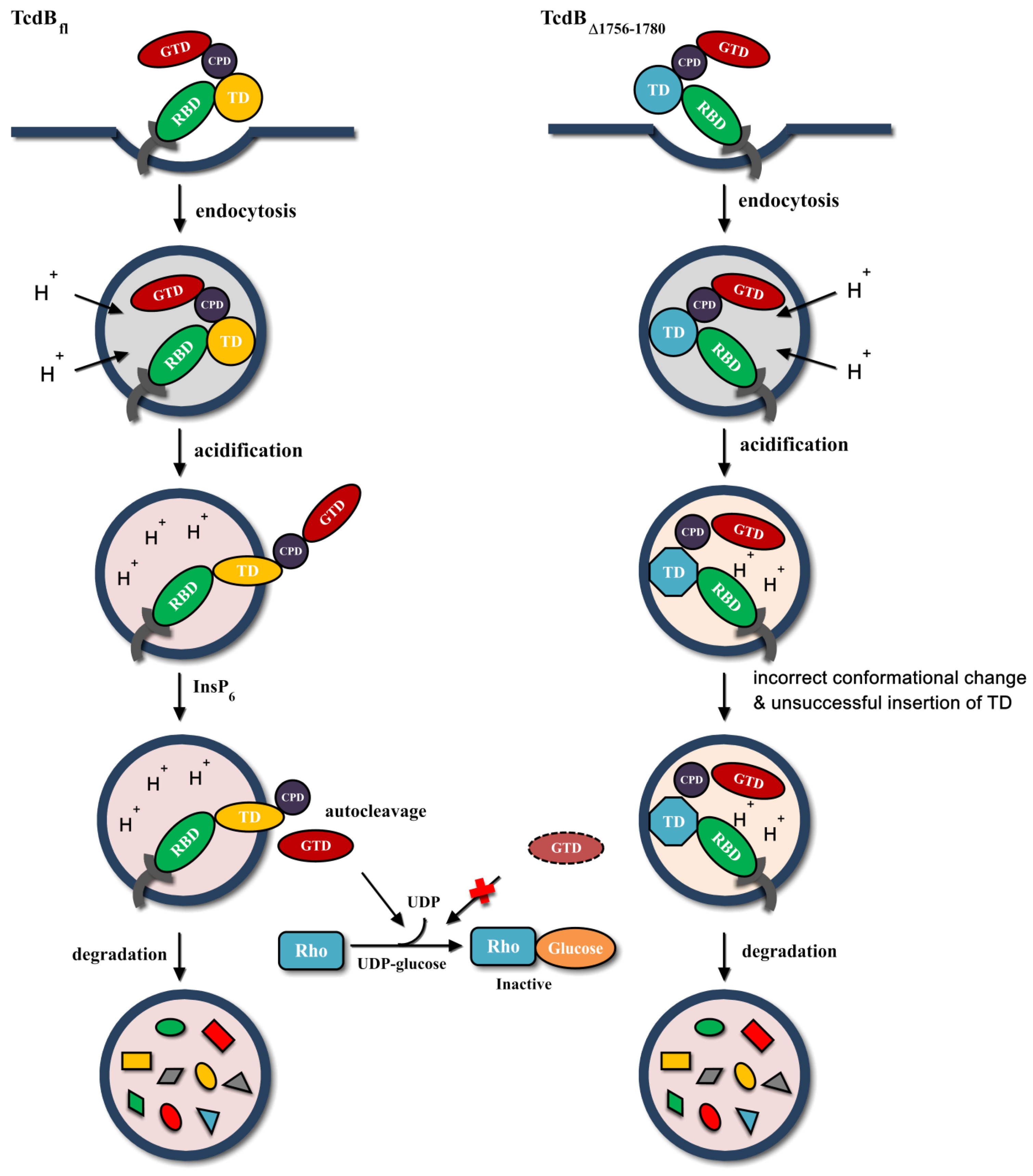
2.5. Behavior in Endosomes
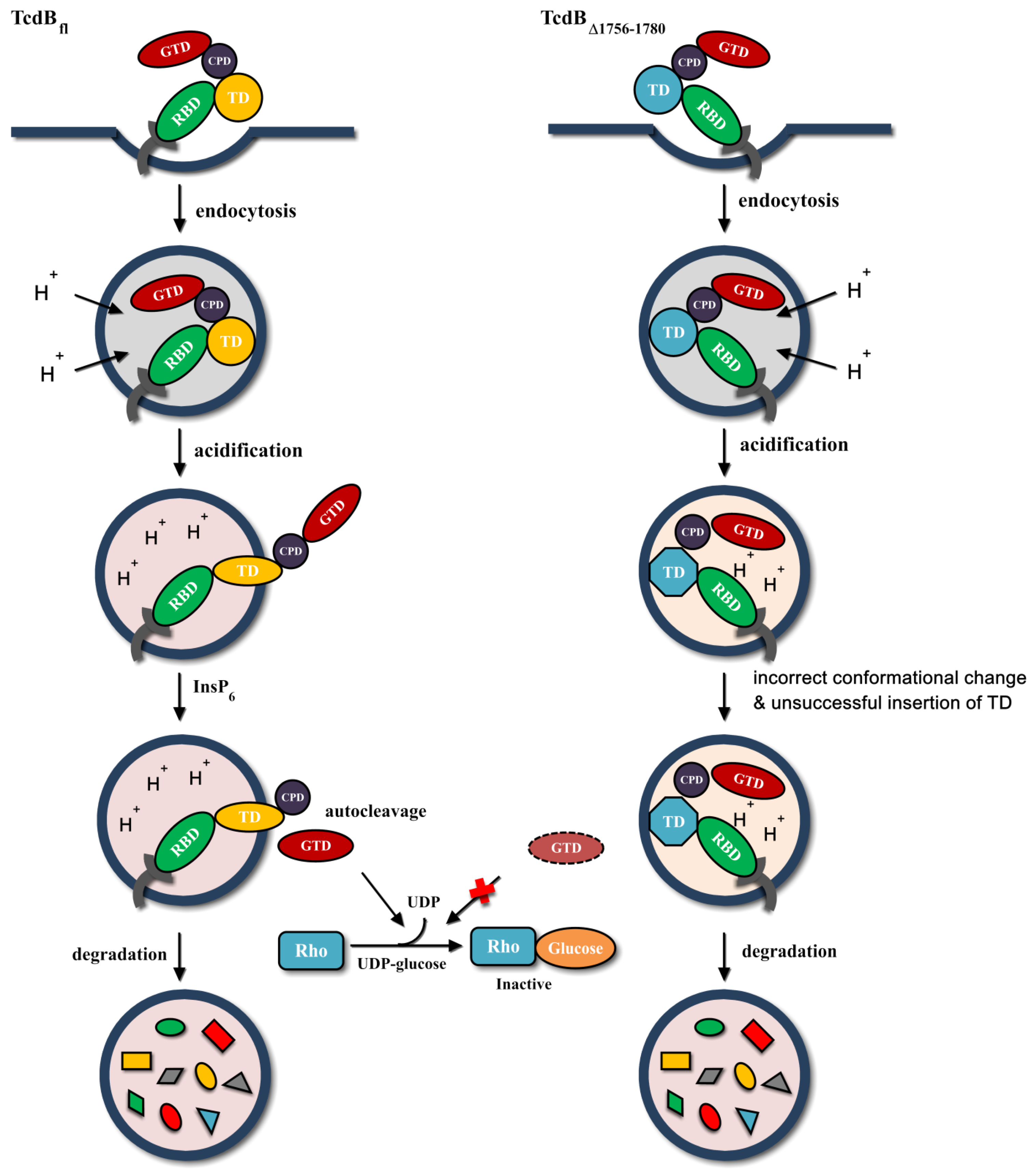
3. Discussion
4. Materials and Methods
4.1. Mammalian Cell Lines
4.2. Bacteria Strains
4.3. Cloning of TcdB Constructs
4.4. Protein Expression and Purification
4.5. Cytotoxic and Cytopathic Effects
4.6. Mouse Systemic Toxin Challenge
4.7. In Vitro Autocleavage Assay
4.8. In Vitro Glucosylation Assay
4.9. Analysis of Cell Surface Binding of the Toxins
4.10. TNS Fluorescence Analysis of Conformational Change of Toxins
4.11. Fluorophore Leakage from Lipid Vesicles
4.12. Endosome Isolation
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kelly, C.P.; LaMont, J.T. Clostridium difficile infection. Annu. Rev. Med. 1998, 49, 375–390. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.P.; LaMont, J.T. Clostridium difficile—More difficult than ever. N. Engl. J. Med. 2008, 359, 1932–1940. [Google Scholar] [CrossRef] [PubMed]
- Jank, T.; Aktories, K. Structure and mode of action of clostridial glucosylating toxins: The ABCD model. Trends Microbiol. 2008, 16, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Busch, C.; Hofmann, F.; Selzer, J.; Munro, S.; Jeckel, D.; Aktories, K. A common motif of eukaryotic glycosyltransferases is essential for the enzyme activity of large clostridial cytotoxins. J. Biol. Chem. 1998, 273, 19566–19572. [Google Scholar] [CrossRef] [PubMed]
- Jank, T.; Giesemann, T.; Aktories, K. Clostridium difficile glucosyltransferase toxin B-essential amino acids for substrate binding. J. Biol. Chem. 2007, 282, 35222–35231. [Google Scholar] [CrossRef] [PubMed]
- Egerer, M.; Giesemann, T.; Jank, T.; Satchell, K.J.; Aktories, K. Auto-catalytic cleavage of Clostridium difficile toxins A and B depends on cysteine protease activity. J. Biol. Chem. 2007, 282, 25314–25321. [Google Scholar] [CrossRef] [PubMed]
- Kreimeyer, I.; Euler, F.; Marckscheffel, A.; Tatge, H.; Pich, A.; Olling, A.; Schwarz, J.; Just, I.; Gerhard, R. Autoproteolytic cleavage mediates cytotoxicity of Clostridium difficile toxin A. Naunyn Schmiedebergs Arch. Pharmacol. 2011, 383, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Pruitt, R.N.; Chagot, B.; Cover, M.; Chazin, W.J.; Spiller, B.; Lacy, D.B. Structure-function analysis of inositol hexakisphosphate-induced autoprocessing in Clostridium difficile toxin A. J. Biol. Chem. 2009, 284, 21934–21940. [Google Scholar] [CrossRef] [PubMed]
- Von Eichel-Streiber, C.; Sauerborn, M. Clostridium difficile toxin A carries a C-terminal repetitive structure homologous to the carbohydrate binding region of streptococcal glycosyltransferases. Gene 1990, 96, 107–113. [Google Scholar] [CrossRef]
- Von Eichel-Streiber, C.; Sauerborn, M.; Kuramitsu, H.K. Evidence for a modular structure of the homologous repetitive C-terminal carbohydrate-binding sites of Clostridium difficile toxins and Streptococcus mutans glucosyltransferases. J. Bacteriol. 1992, 174, 6707–6710. [Google Scholar] [PubMed]
- Yuan, P.; Zhang, H.; Cai, C.; Zhu, S.; Zhou, Y.; Yang, X.; He, R.; Li, C.; Guo, S.; Li, S.; et al. Chondroitin sulfate proteoglycan 4 functions as the cellular receptor for Clostridium difficile toxin B. Cell Res. 2015, 25, 157–168. [Google Scholar] [CrossRef] [PubMed]
- LaFrance, M.E.; Farrow, M.A.; Chandrasekaran, R.; Sheng, J.; Rubin, D.H.; Lacy, D.B. Identification of an epithelial cell receptor responsible for Clostridium difficile TcdB-induced cytotoxicity. Proc. Natl. Acad. Sci. USA 2015, 112, 7073–7078. [Google Scholar] [CrossRef] [PubMed]
- Olling, A.; Goy, S.; Hoffmann, F.; Tatge, H.; Just, I.; Gerhard, R. The repetitive oligopeptide sequences modulate cytopathic potency but are not crucial for cellular uptake of Clostridium difficile toxin A. PLoS ONE 2011, 6, e17623. [Google Scholar] [CrossRef] [PubMed]
- Schorch, B.; Song, S.; van Diemen, F.R.; Bock, H.H.; May, P.; Herz, J.; Brummelkamp, T.R.; Papatheodorou, P.; Aktories, K. LRP1 is a receptor for Clostridium perfringens TpeL toxin indicating a two-receptor model of clostridial glycosylating toxins. Proc. Natl. Acad. Sci. USA 2014, 111, 6431–6436. [Google Scholar] [CrossRef] [PubMed]
- Barth, H.; Pfeifer, G.; Hofmann, F.; Maier, E.; Benz, R.; Aktories, K. Low pH-induced formation of ion channels by Clostridium difficile toxin B in target cells. J. Biol. Chem. 2001, 276, 10670–10676. [Google Scholar] [CrossRef] [PubMed]
- Genisyuerek, S.; Papatheodorou, P.; Guttenberg, G.; Schubert, R.; Benz, R.; Aktories, K. Structural determinants for membrane insertion, pore formation and translocation of Clostridium difficile toxin B. Mol. Microbiol. 2011, 79, 1643–1654. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, G.; Schirmer, J.; Leemhuis, J.; Busch, C.; Meyer, D.K.; Aktories, K.; Barth, H. Cellular uptake of Clostridium difficile toxin B. Translocation of the N-terminal catalytic domain into the cytosol of eukaryotic cells. J. Biol. Chem. 2003, 278, 44535–44541. [Google Scholar] [CrossRef] [PubMed]
- Qa’Dan, M.; Spyres, L.M.; Ballard, J.D. pH-induced conformational changes in Clostridium difficile toxin B. Infect. Immun. 2000, 68, 2470–2474. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, M.O.; Jank, T.; Aktories, K.; Schulz, G.E. Conformational changes and reaction of clostridial glycosylating toxins. J. Mol. Biol. 2008, 377, 1346–1356. [Google Scholar] [CrossRef] [PubMed]
- Rupnik, M.; Pabst, S.; von Eichel-Streiber, C.; Urlaub, H.; Soling, H.D. Characterization of the cleavage site and function of resulting cleavage fragments after limited proteolysis of Clostridium difficile toxin B (TcdB) by host cells. Microbiology 2005, 151, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Giesemann, T.; Jank, T.; Gerhard, R.; Maier, E.; Just, I.; Benz, R.; Aktories, K. Cholesterol-dependent pore formation of Clostridium difficile toxin A. J. Biol. Chem. 2006, 281, 10808–10815. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Park, M.; Tam, J.; Auger, A.; Beihartz, G.L.; Lacy, D.B.; Melnyk, R.A. Translocation domain mutations affecting cellular toxicity identify the Clostridium difficile toxin B pore. Proc. Natl. Acad. Sci. USA 2014, 111, 3721–3726. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Shi, L.; Li, S.; Yang, Z.; Standley, C.; ZhuGe, R.; Savidge, T.; Wang, X.; Feng, H. A segment of 97 amino acids within the translocation domain of Clostridium difficile toxin B is essential for toxicity. PLoS ONE 2013, 8, e58634. [Google Scholar] [CrossRef] [PubMed]
- Gilleron, J.; Zeigerer, A.; Marsico, G.; Galvez, T.; Zerial, M. Key role of Rab5: From endosome biogenesis to liver metabolism. Med. Sci. 2012, 28, 1041–1044. [Google Scholar]
- Woodman, P.G. Biogenesis of the sorting endosome: The role of Rab5. Traffic 2000, 1, 695–701. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Sun, X.; Wang, J.; Wang, X.; Zhang, Q.; Tzipori, S.; Feng, H. Antibody-enhanced, Fc gamma receptor-mediated endocytosis of Clostridium difficile toxin A. Infect. Immun. 2009, 77, 2294–2303. [Google Scholar] [CrossRef] [PubMed]
- Egerer, M.; Giesemann, T.; Herrmann, C.; Aktories, K. Autocatalytic processing of Clostridium difficile toxin B. Binding of inositol hexakisphosphate. J. Biol. Chem. 2009, 284, 3389–3395. [Google Scholar] [CrossRef] [PubMed]
- Torchilin, V.; Weissig, V. Liposomes, a Practical Approach; Oxford University Press: Oxford, UK, 2003. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Α-helix (%) | β-sheet (%) | Random Coil (%) | Total (%) |
|---|---|---|---|---|
| rTcdBfl | 6.5 | 52.6 | 40.8 | 99 |
| TcdBΔ1756-1780 | 5.8 | 53.5 | 40.9 | 100.2 |
| TcdBΔ1827-1851 | 6.1 | 53.1 | 40.8 | 100 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, S.; Wang, H.; Gu, H.; Sun, C.; Li, S.; Feng, H.; Wang, J. Identification of an Essential Region for Translocation of Clostridium difficile Toxin B. Toxins 2016, 8, 241. https://doi.org/10.3390/toxins8080241
Chen S, Wang H, Gu H, Sun C, Li S, Feng H, Wang J. Identification of an Essential Region for Translocation of Clostridium difficile Toxin B. Toxins. 2016; 8(8):241. https://doi.org/10.3390/toxins8080241
Chicago/Turabian StyleChen, Shuyi, Haiying Wang, Huawei Gu, Chunli Sun, Shan Li, Hanping Feng, and Jufang Wang. 2016. "Identification of an Essential Region for Translocation of Clostridium difficile Toxin B" Toxins 8, no. 8: 241. https://doi.org/10.3390/toxins8080241
APA StyleChen, S., Wang, H., Gu, H., Sun, C., Li, S., Feng, H., & Wang, J. (2016). Identification of an Essential Region for Translocation of Clostridium difficile Toxin B. Toxins, 8(8), 241. https://doi.org/10.3390/toxins8080241