
Tumor Targeting and Drug Delivery by Anthrax Toxin

Abstract
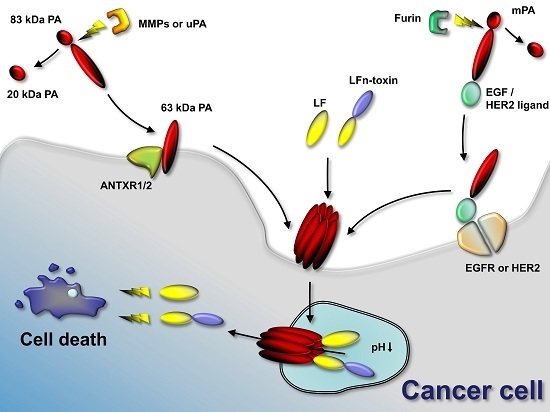
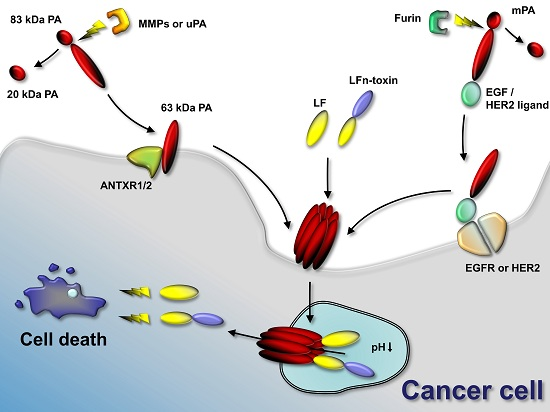
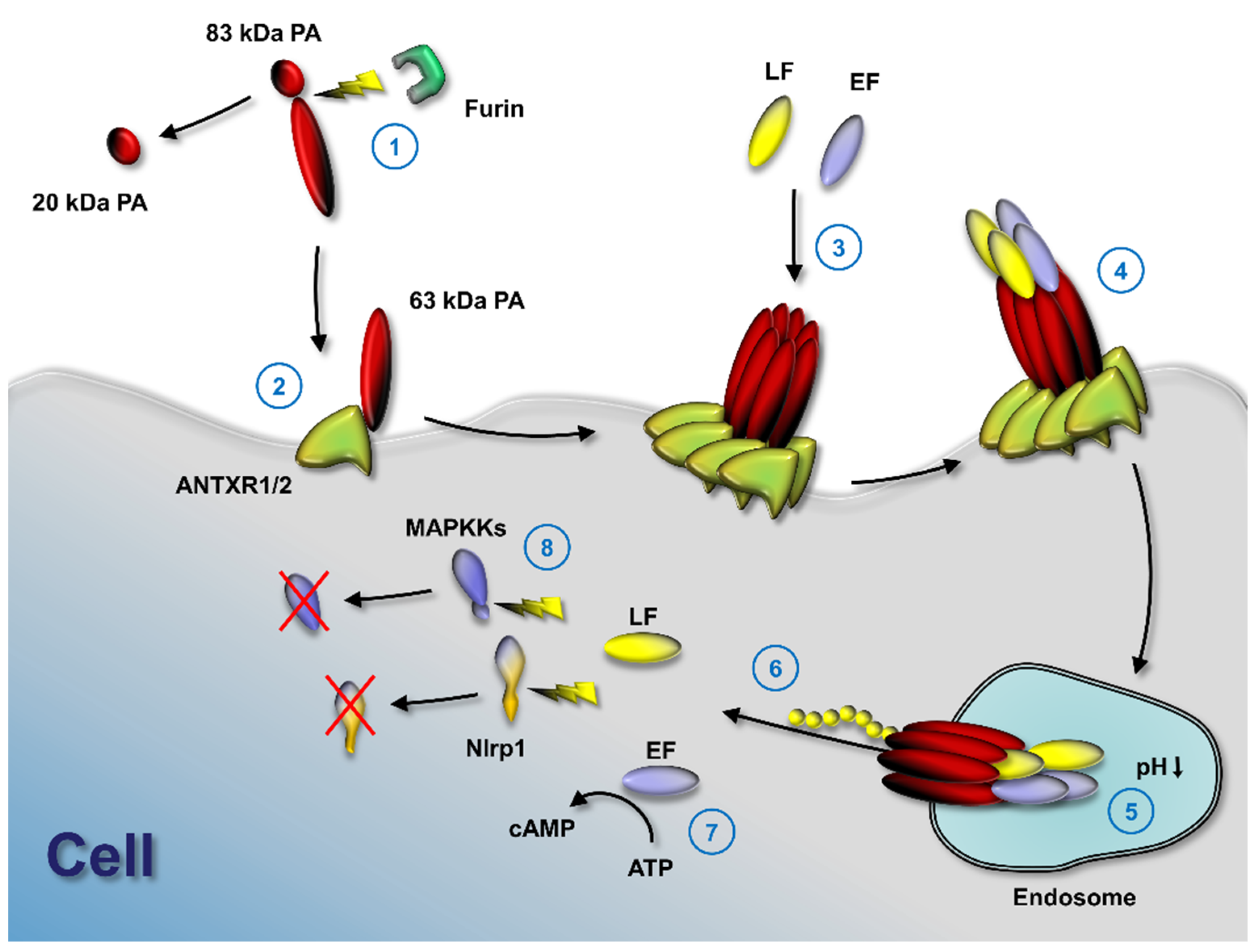
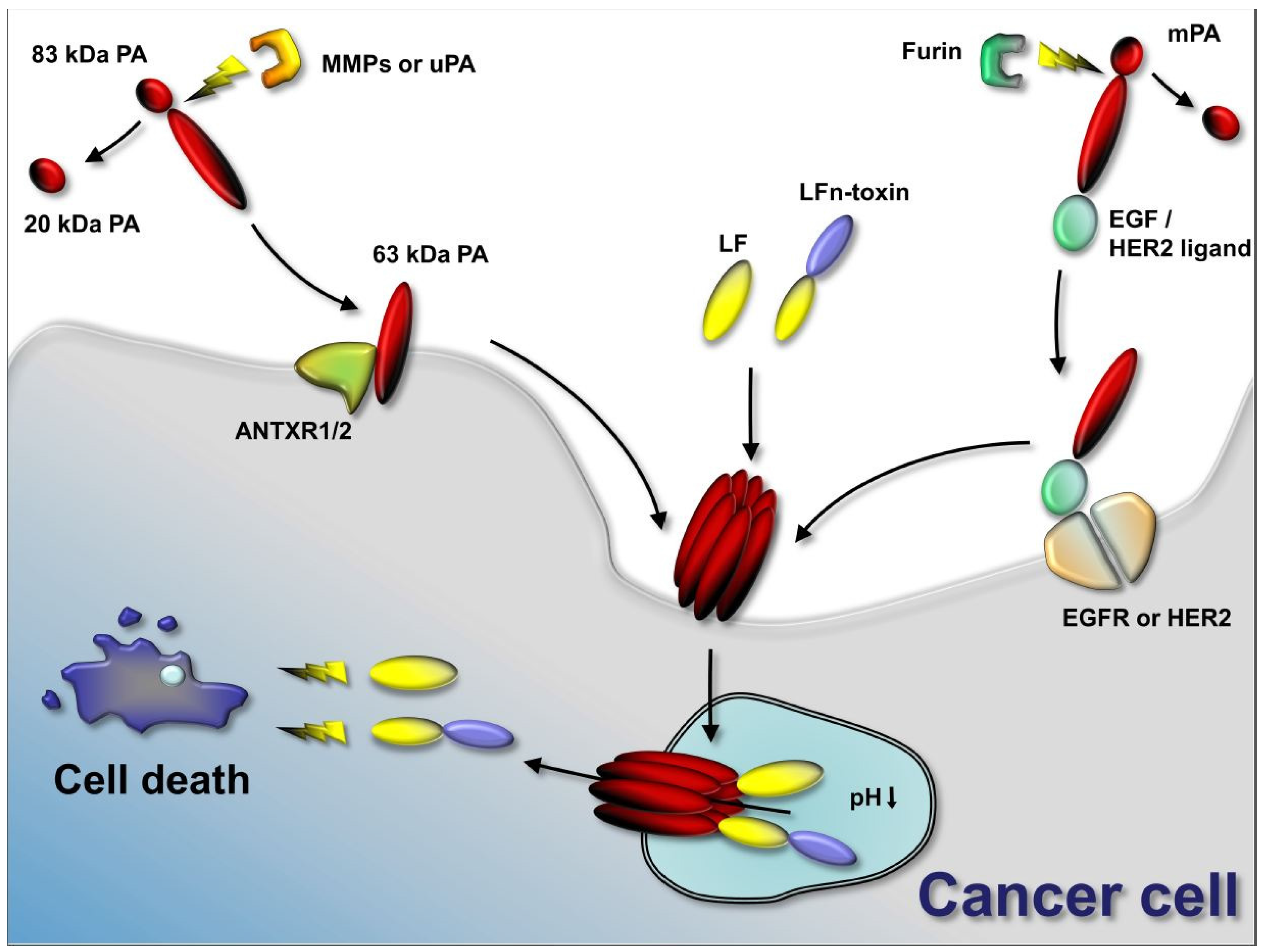
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Tumor-Selective Activation of Protective Antigen and Tumor-Selective Formation of Protective Antigen Octamer
3. Cellular Delivery of Fusion Proteins
4. Retargeting of PA
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AT | anthrax toxin |
| Bcl-XL | B-cell lymphoma-extra large |
| CMG2 or ANTXR2 | capillary morphogenesis gene 2 |
| CdtB | cytolethal distending toxin B |
| DTA | diphtheria toxin A chain |
| EF | edema factor |
| ET | edema toxin |
| LF | lethal factor |
| LFn | LF N-terminal domain |
| LT | lethal toxin |
| MMPs | matrix-metalloproteases |
| MAPKKs | mitogen-activated protein kinase kinases |
| mPA | mutant PA |
| mPA-EGF | mPA genetically fused to human epidermal growth factor |
| mPA-ZHER2 | mPA genetically fused to the affibody ZHER2 |
| PA | protective antigen |
| PA-DK | PA with D512K mutation |
| PA-GN | PA with mutation complementing PA-D512K mutation |
| PE | Pseudomonas exotoxin A |
| PEIII | Pseudomonas exotoxin A catalytic domain |
| PE38 | PE domains II and III |
| Raf-1 | rapidly accelerated fibrosarcoma |
| TEM8 or ANTXR1 | tumor endothelial marker 8 |
| uPa | urokinase plasminogen activator |
References
- Liu, S.; Schubert, R.L.; Bugge, T.H.; Leppla, S.H. Anthrax toxin: Structures, functions and tumour targeting. Expert Opin. Biol. Ther. 2003, 3, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Koehler, T.M. Bacillus anthracis physiology and genetics. Mol. Asp. Med. 2009, 30, 386–396. [Google Scholar] [CrossRef] [PubMed]
- Moayeri, M.; Leppla, S.H. Cellular and systemic effects of anthrax lethal toxin and edema toxin. Mol. Asp. Med. 2009, 30, 439–455. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.; Cho, M.; Lee, H.R.; Cha, K.; Chun, J.H.; Hong, K.J.; Park, J.; Rhie, G.E. Monoclonal antibody against the poly-γ-d-glutamic acid capsule of Bacillus anthracis protects mice from enhanced lethal toxin activity due to capsule and anthrax spore challenge. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 2804–2812. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.; Keppie, J.; Stanley, J.L. The chemical basis of the virulence of Bacillus anthracis. V. The specific toxin produced by B. Anthracis in vivo. Br. J. Exp. Pathol. 1955, 36, 460–472. [Google Scholar] [PubMed]
- Liu, S.; Zhang, Y.; Moayeri, M.; Liu, J.; Crown, D.; Fattah, R.J.; Wein, A.N.; Yu, Z.-X.; Finkel, T.; Leppla, S.H. Key tissue targets responsible for anthrax-toxin-induced lethality. Nature 2013, 501, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Crown, D.; Miller-Randolph, S.; Moayeri, M.; Wang, H.; Hu, H.; Morley, T.; Leppla, S.H. Capillary morphogenesis protein-2 is the major receptor mediating lethality of anthrax toxin in vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 12424–12429. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhang, Y.; Hoover, B.; Leppla, S.H. The receptors that mediate the direct lethality of anthrax toxin. Toxins 2013, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Milne, J.C.; Furlong, D.; Hanna, P.C.; Wall, J.S.; Collier, R.J. Anthrax protective antigen forms oligomers during intoxication of mammalian cells. J. Biol. Chem. 1994, 269, 20607–20612. [Google Scholar] [PubMed]
- Singh, Y.; Klimpel, K.R.; Goel, S.; Swain, P.K.; Leppla, S.H. Oligomerization of anthrax toxin protective antigen and binding of lethal factor during endocytic uptake into mammalian cells. Infect. Immun. 1999, 67, 1853–1859. [Google Scholar] [PubMed]
- Feld, G.K.; Brown, M.J.; Krantz, B.A. Ratcheting up protein translocation with anthrax toxin. Protein Sci. 2012, 21, 606–624. [Google Scholar] [CrossRef] [PubMed]
- Gutwein, L.G.; Al-Quran, S.Z.; Fernando, S.; Fletcher, B.S.; Copeland, E.M.; Grobmyer, S.R. Tumor endothelial marker 8 expression in triple-negative breast cancer. Anticancer Res. 2011, 31, 3417–3422. [Google Scholar] [PubMed]
- Maurya, S.K.; Tewari, M.; Kumar, M.; Thakur, M.K.; Shukla, H.S. Expression pattern of tumor endothelial marker 8 protein in gallbladder carcinomas. Asian Pac. J. Cancer Prev. 2011, 12, 507–512. [Google Scholar] [PubMed]
- Singh, A.P.; Bafna, S.; Chaudhary, K.; Venkatraman, G.; Smith, L.; Eudy, J.D.; Johansson, S.L.; Lin, M.-F.; Batra, S.K. Genome-wide expression profiling reveals transcriptomic variation and perturbed gene networks in androgen-dependent and androgen-independent prostate cancer cells. Cancer Lett. 2008, 259, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Peters, D.E.; Zhang, Y.; Molinolo, A.A.; Miller-Randolph, S.; Szabo, R.; Bugge, T.H.; Leppla, S.H.; Liu, S. Capillary morphogenesis protein-2 is required for mouse parturition by maintaining uterine collagen homeostasis. Biochem. Biophys. Res. Commun. 2012, 422, 393–397. [Google Scholar] [CrossRef] [PubMed]
- Gordon, V.M.; Klimpel, K.R.; Arora, N.; Henderson, M.A.; Leppla, S.H. Proteolytic activation of bacterial toxins by eukaryotic cells is performed by furin and by additional cellular proteases. Infect. Immun. 1995, 63, 82–87. [Google Scholar] [PubMed]
- Petosa, C.; Collier, R.J.; Klimpel, K.R.; Leppla, S.H.; Liddington, R.C. Crystal structure of the anthrax toxin protective antigen. Nature 1997, 385, 833–838. [Google Scholar] [CrossRef] [PubMed]
- Kintzer, A.F.; Thoren, K.L.; Sterling, H.J.; Dong, K.C.; Feld, G.K.; Tang, I.I.; Zhang, T.T.; Williams, E.R.; Berger, J.M.; Krantz, B.A. The protective antigen component of anthrax toxin forms functional octameric complexes. J. Mol. Biol. 2009, 392, 614–629. [Google Scholar] [CrossRef] [PubMed]
- Kintzer, A.F.; Sterling, H.J.; Tang, I.I.; Abdul-Gader, A.; Miles, A.J.; Wallace, B.A.; Williams, E.R.; Krantz, B.A. Role of the protective antigen octamer in the molecular mechanism of anthrax lethal toxin stabilization in plasma. J. Mol. Biol. 2010, 399, 741–758. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Milne, J.C.; Collier, R.J. Effect of anthrax toxin’s lethal factor on ion channels formed by the protective antigen. J. Biol. Chem. 1995, 270, 18626–18630. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Pentelute, B.L.; Collier, R.J.; Zhou, Z.H. Atomic structure of anthrax protective antigen pore elucidates toxin translocation. Nature 2015, 521, 545–549. [Google Scholar] [CrossRef] [PubMed]
- Feld, G.K.; Thoren, K.L.; Kintzer, A.F.; Sterling, H.J.; Tang, I.I.; Greenberg, S.G.; Williams, E.R.; Krantz, B.A. Structural basis for the unfolding of anthrax lethal factor by protective antigen oligomers. Nat. Struct. Mol. Biol. 2010, 17, 1383–1390. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.J.; Thoren, K.L.; Krantz, B.A. Charge requirements for proton gradient-driven translocation of anthrax toxin. J. Biol. Chem. 2011, 286, 23189–23199. [Google Scholar] [CrossRef] [PubMed]
- Tamayo, A.G.; Slater, L.; Taylor-Parker, J.; Bharti, A.; Harrison, R.; Hung, D.T.; Murphy, J.R. GRP78(BiP) facilitates the cytosolic delivery of anthrax lethal factor (LF) in vivo and functions as an unfoldase in vitro. Mol. Microbiol. 2011, 81, 1390–1401. [Google Scholar] [CrossRef] [PubMed]
- Dmochewitz, L.; Lillich, M.; Kaiser, E.; Jennings, L.D.; Lang, A.E.; Buchner, J.; Fischer, G.; Aktories, K.; Collier, R.J.; Barth, H. Role of CypA and Hsp90 in membrane translocation mediated by anthrax protective antigen. Cell. Microbiol. 2011, 13, 359–373. [Google Scholar] [CrossRef] [PubMed]
- Leppla, S.H. Anthrax toxin edema factor: A bacterial adenylate cyclase that increases cyclic AMP concentrations of eukaryotic cells. Proc. Natl. Acad. Sci. USA 1982, 79, 3162–3166. [Google Scholar] [CrossRef] [PubMed]
- Firoved, A.M.; Miller, G.F.; Moayeri, M.; Kakkar, R.; Shen, Y.; Wiggins, J.F.; McNally, E.M.; Tang, W.-J.; Leppla, S.H. Bacillus anthracis edema toxin causes extensive tissue lesions and rapid lethality in mice. Am. J. Pathol. 2005, 167, 1309–1320. [Google Scholar] [CrossRef]
- Gnade, B.T.; Moen, S.T.; Chopra, A.K.; Peterson, J.W.; Yeager, L.A. Emergence of anthrax edema toxin as a master manipulator of macrophage and B cell functions. Toxins 2010, 2, 1881–1897. [Google Scholar] [CrossRef] [PubMed]
- Chou, P.-J.J.; Newton, C.A.; Perkins, I.; Friedman, H.; Klein, T.W. Suppression of dendritic cell activation by anthrax lethal toxin and edema toxin depends on multiple factors including cell source, stimulus used, and function tested. DNA Cell Biol. 2008, 27, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Paccani, S.R.; Tonello, F.; Ghittoni, R.; Natale, M.; Muraro, L.; D’Elios, M.M.; Tang, W.-J.; Montecucco, C.; Baldari, C.T. Anthrax toxins suppress T lymphocyte activation by disrupting antigen receptor signaling. J. Exp. Med. 2005, 201, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Comer, J.E.; Galindo, C.L.; Zhang, F.; Wenglikowski, A.M.; Bush, K.L.; Garner, H.R.; Peterson, J.W.; Chopra, A.K. Murine macrophage transcriptional and functional responses to Bacillus anthracis edema toxin. Microb. Pathog. 2006, 41, 96–110. [Google Scholar] [CrossRef] [PubMed]
- Crawford, M.A.; Aylott, C.V.; Bourdeau, R.W.; Bokoch, G.M. Bacillus anthracis toxins inhibit human neutrophil NADPH oxidase activity. J. Immunol. 2006, 176, 7557–7565. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Doebele, R.C.; Lingen, M.W.; Quilliam, L.A.; Tang, W.-J.; Rosner, M.R. Anthrax edema toxin inhibits endothelial cell chemotaxis via Epac and Rap1. J. Biol. Chem. 2007, 282, 19781–19787. [Google Scholar] [CrossRef] [PubMed]
- Duesbery, N.S.; Webb, C.P.; Leppla, S.H.; Gordon, V.M.; Klimpel, K.R.; Copeland, T.D.; Ahn, N.G.; Oskarsson, M.K.; Fukasawa, K.; Paull, K.D.; et al. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science 1998, 280, 734–737. [Google Scholar] [CrossRef] [PubMed]
- Levinsohn, J.L.; Newman, Z.L.; Hellmich, K.A.; Fattah, R.; Getz, M.A.; Liu, S.; Sastalla, I.; Leppla, S.H.; Moayeri, M. Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PLoS Pathog. 2012, 8. [Google Scholar] [CrossRef] [PubMed]
- Vitale, G.; Pellizzari, R.; Recchi, C.; Napolitani, G.; Mock, M.; Montecucco, C. Anthrax lethal factor cleaves the N-terminus of MAPKKs and induces tyrosine/threonine phosphorylation of MAPKs in cultured macrophages. Biochem. Biophys. Res. Commun. 1998, 248, 706–711. [Google Scholar] [CrossRef] [PubMed]
- Vitale, G.; Bernardi, L.; Napolitani, G.; Mock, M.; Montecucco, C. Susceptibility of mitogen-activated protein kinase kinase family members to proteolysis by anthrax lethal factor. Biochem. J. 2000, 352, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Pellizzari, R.; Guidi-Rontani, C.; Vitale, G.; Mock, M.; Montecucco, C. Anthrax lethal factor cleaves MKK3 in macrophages and inhibits the LPS/IFNgamma-induced release of NO and TNFalpha. FEBS Lett. 1999, 462, 199–204. [Google Scholar] [CrossRef]
- Liu, S.; Bugge, T.H.; Leppla, S.H. Targeting of tumor cells by cell surface urokinase plasminogen activator-dependent anthrax toxin. J. Biol. Chem. 2001, 276, 17976–17984. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Netzel-Arnett, S.; Birkedal-Hansen, H.; Leppla, S.H. Tumor cell-selective cytotoxicity of matrix metalloproteinase-activated anthrax toxin. Cancer Res. 2000, 60, 6061–6067. [Google Scholar] [PubMed]
- Choi, K.Y.; Swierczewska, M.; Lee, S.; Chen, X. Protease-activated drug development. Theranostics 2012, 2, 156–178. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Redeye, V.; Kuremsky, J.G.; Kuhnen, M.; Molinolo, A.; Bugge, T.H.; Leppla, S.H. Intermolecular complementation achieves high-specificity tumor targeting by anthrax toxin. Nat. Biotechnol. 2005, 23, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Phillips, D.D.; Fattah, R.J.; Crown, D.; Zhang, Y.; Liu, S.; Moayeri, M.; Fischer, E.R.; Hansen, B.T.; Ghirlando, R.; Nestorovich, E.M.; et al. Engineering anthrax toxin variants that exclusively form octamers and their application to targeting tumors. J. Biol. Chem. 2013, 288, 9058–9065. [Google Scholar] [CrossRef] [PubMed]
- Mogridge, J.; Cunningham, K.; Lacy, D.B.; Mourez, M.; Collier, R.J. The lethal and edema factors of anthrax toxin bind only to oligomeric forms of the protective antigen. Proc. Natl. Acad. Sci. USA 2002, 99, 7045–7048. [Google Scholar] [CrossRef] [PubMed]
- Arora, N.; Klimpel, K.R.; Singh, Y.; Leppla, S.H. Fusions of anthrax toxin lethal factor to the ADP-ribosylation domain of Pseudomonas exotoxin A are potent cytotoxins which are translocated to the cytosol of mammalian cells. J. Biol. Chem. 1992, 267, 15542–15548. [Google Scholar] [PubMed]
- Arora, N.; Leppla, S.H. Residues 1-254 of anthrax toxin lethal factor are sufficient to cause cellular uptake of fused polypeptides. J. Biol. Chem. 1993, 268, 3334–3341. [Google Scholar] [PubMed]
- Sharma, O.; Collier, R.J. Polylysine-mediated translocation of the diphtheria toxin catalytic domain through the anthrax protective antigen pore. Biochemistry 2014, 53, 6934–6940. [Google Scholar] [CrossRef] [PubMed]
- Blanke, S.R.; Milne, J.C.; Benson, E.L.; Collier, R.J. Fused polycationic peptide mediates delivery of diphtheria toxin A chain to the cytosol in the presence of anthrax protective antigen. Proc. Natl. Acad. Sci. USA 1996, 93, 8437–8442. [Google Scholar] [CrossRef] [PubMed]
- Milne, J.C.; Blanke, S.R.; Hanna, P.C.; Collier, R.J. Protective antigen-binding domain of anthrax lethal factor mediates translocation of a heterologous protein fused to its amino- or carboxy-terminus. Mol. Microbiol. 1995, 15, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Arora, N.; Leppla, S.H. Fusions of anthrax toxin lethal factor with shiga toxin and diphtheria toxin enzymatic domains are toxic to mammalian cells. Infect. Immun. 1994, 62, 4955–4961. [Google Scholar] [PubMed]
- Taft, S.C.; Weiss, A.A. Toxicity of anthrax toxin is influenced by receptor expression. Clin. Vaccine Immunol. 2008, 15, 1330–1336. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.S.; Christensen, K.A.; Birsner, A.E.; Short, S.M.; Wigelsworth, D.J.; Collier, R.J.; D’Amato, R.J. Mutant anthrax toxin B moiety (protective antigen) inhibits angiogenesis and tumor growth. Cancer Res. 2007, 67, 9980–9985. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.W.; Buzza, M.S.; Driesbaugh, K.H.; Liu, S.; Fortenberry, Y.M.; Leppla, S.H.; Antalis, T.M. Targeting the membrane-anchored serine protease testisin with a novel engineered anthrax toxin prodrug to kill tumor cells and reduce tumor burden. Oncotarget 2015, 6, 33534–33553. [Google Scholar] [PubMed]
- Varughese, M.; Chi, A.; Teixeira, A.V.; Nicholls, P.J.; Keith, J.M.; Leppla, S.H. Internalization of a Bacillus anthracis protective antigen-c-Myc fusion protein mediated by cell surface anti-c-Myc antibodies. Mol. Med. 1998, 4, 87–95. [Google Scholar] [PubMed]
- Liu, S.; Aaronson, H.; Mitola, D.J.; Leppla, S.H.; Bugge, T.H. Potent antitumor activity of a urokinase-activated engineered anthrax toxin. Proc. Natl. Acad. Sci. USA 2003, 100, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Rønø, B.; Rømer, J.; Liu, S.; Bugge, T.H.; Leppla, S.H.; Kristjansen, P.E.G. Antitumor efficacy of a urokinase activation-dependent anthrax toxin. Mol. Cancer Ther. 2006, 5, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Abi-Habib, R.J.; Singh, R.; Liu, S.; Bugge, T.H.; Leppla, S.H.; Frankel, A.E. A urokinase-activated recombinant anthrax toxin is selectively cytotoxic to many human tumor cell types. Mol. Cancer Ther. 2006, 5, 2556–2562. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Ortiz, J.; Liu, S.; Bugge, T.H.; Singh, R.; Leppla, S.H.; Frankel, A.E. Systematic urokinase-activated anthrax toxin therapy produces regressions of subcutaneous human non-small cell lung tumor in athymic nude mice. Cancer Res. 2007, 67, 3329–3336. [Google Scholar] [CrossRef] [PubMed]
- Schafer, J.M.; Peters, D.E.; Morley, T.; Liu, S.; Molinolo, A.A.; Leppla, S.H.; Bugge, T.H. Efficient targeting of head and neck squamous cell carcinoma by systemic administration of a dual uPA and MMP-activated engineered anthrax toxin. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Alfano, R.W.; Leppla, S.H.; Liu, S.; Bugge, T.H.; Herlyn, M.; Smalley, K.S.; Bromberg-White, J.L.; Duesbery, N.S.; Frankel, A.E. Cytotoxicity of the matrix metalloproteinase-activated anthrax lethal toxin is dependent on gelatinase expression and B-RAF status in human melanoma cells. Mol. Cancer Ther. 2008, 7, 1218–1226. [Google Scholar] [CrossRef] [PubMed]
- Abi-Habib, R.J.; Urieto, J.O.; Liu, S.; Leppla, S.H.; Duesbery, N.S.; Frankel, A.E. BRAF status and mitogen-activated protein/extracellular signal-regulated kinase kinase 1/2 activity indicate sensitivity of melanoma cells to anthrax lethal toxin. Mol. Cancer Ther. 2005, 4, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Abi-Habib, R.J.; Singh, R.; Leppla, S.H.; Greene, J.J.; Ding, Y.; Berghuis, B.; Duesbery, N.S.; Frankel, A.E. Systemic anthrax lethal toxin therapy produces regressions of subcutaneous human melanoma tumors in athymic nude mice. Clin. Cancer Res. 2006, 12, 7437–7443. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wang, H.; Currie, B.M.; Molinolo, A.; Leung, H.J.; Moayeri, M.; Basile, J.R.; Alfano, R.W.; Gutkind, J.S.; Frankel, A.E.; et al. Matrix metalloproteinase-activated anthrax lethal toxin demonstrates high potency in targeting tumor vasculature. J. Biol. Chem. 2008, 283, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Ding, Y.; Luo, W.-M.; Bender, S.; Qian, C.-N.; Kort, E.; Zhang, Z.-F.; VandenBeldt, K.; Duesbery, N.S.; Resau, J.H.; et al. Inhibition of MAPK kinase signaling pathways suppressed renal cell carcinoma growth and angiogenesis in vivo. Cancer Res. 2008, 68, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Boguslawski, E.A.; Berghuis, B.D.; Young, J.J.; Zhang, Z.; Hardy, K.; Furge, K.; Kort, E.; Frankel, A.E.; Hay, R.V.; et al. Mitogen-activated protein kinase kinase signaling promotes growth and vascularization of fibrosarcoma. Mol. Cancer Ther. 2008, 7, 648–658. [Google Scholar] [CrossRef] [PubMed]
- Rouleau, C.; Menon, K.; Boutin, P.; Guyre, C.; Yoshida, H.; Kataoka, S.; Perricone, M.; Shankara, S.; Frankel, A.E.; Duesbery, N.S.; et al. The Systemic Administration of Lethal Toxin Achieves a Growth delay of Human Melanoma and Neuroblastoma Xenografts: Assessment of Receptor Contribution. Int. J. Oncol. 2008, 32, 739–748. [Google Scholar] [PubMed]
- Zhuo, W.; Tao, G.; Zhang, L.; Chen, Z. Vector-mediated selective expression of lethal factor, a toxic element of Bacillus anthracis, damages A549 cells via inhibition of MAPK and AKT pathways. Int. J. Med. Sci. 2013, 10, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Wein, A.N.; Liu, S.; Zhang, Y.; McKenzie, A.T.; Leppla, S.H. Tumor therapy with a urokinase plasminogen activator-activated anthrax lethal toxin alone and in combination with paclitaxel. Investig. New Drugs 2013, 31, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Goldufsky, J.; Wood, S.; Hajihossainlou, B.; Rehman, T.; Majdobeh, O.; Kaufman, H.L.; Ruby, C.E.; Shafikhani, S.H. Pseudomonas aeruginosa exotoxin T induces potent cytotoxicity against a variety of murine and human cancer cell lines. J. Med. Microbiol. 2015, 64, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Mattoo, A.R.; FitzGerald, D.J. Combination treatments with ABT-263 and an immunotoxin produce synergistic killing of ABT-263-resistant small cell lung cancer cell lines. Int. J. Cancer 2013, 132, 978–987. [Google Scholar] [CrossRef] [PubMed]
- Mazor, R.; Vassall, A.N.; Eberle, J.A.; Beers, R.; Weldon, J.E.; Venzon, D.J.; Tsang, K.Y.; Benhar, I.; Pastan, I. Identification and elimination of an immunodominant T-cell epitope in recombinant immunotoxins based on Pseudomonas exotoxin A. Proc. Natl. Acad. Sci. USA 2012, 109, E3597–E3603. [Google Scholar] [CrossRef] [PubMed]
- Onda, M.; Beers, R.; Xiang, L.; Nagata, S.; Wang, Q.-C.; Pastan, I. An immunotoxin with greatly reduced immunogenicity by identification and removal of B cell epitopes. Proc. Natl. Acad. Sci. USA 2008, 105, 11311–11316. [Google Scholar] [CrossRef] [PubMed]
- Mossoba, M.E.; Onda, M.; Taylor, J.; Massey, P.R.; Treadwell, S.; Sharon, E.; Hassan, R.; Pastan, I.; Fowler, D.H. Pentostatin plus cyclophosphamide safely and effectively prevents immunotoxin immunogenicity in murine hosts. Clin. Cancer Res. 2011, 17, 3697–3705. [Google Scholar] [CrossRef] [PubMed]
- Hassan, R.; Miller, A.C.; Sharon, E.; Thomas, A.; Reynolds, J.C.; Ling, A.; Kreitman, R.J.; Miettinen, M.M.; Steinberg, S.M.; Fowler, D.H.; et al. Major cancer regressions in mesothelioma after treatment with an anti-mesothelin immunotoxin and immune suppression. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.K.; Moayeri, M.; Crown, D.; Fattah, R.J.; Leppla, S.H. Role of N-terminal amino acids in the potency of anthrax lethal factor. PLoS ONE 2008, 3. [Google Scholar] [CrossRef] [PubMed]
- Wesche, J.; Elliott, J.L.; Falnes, P.O.; Olsnes, S.; Collier, R.J. Characterization of membrane translocation by anthrax protective antigen. Biochemistry 1998, 37, 15737–15746. [Google Scholar] [CrossRef] [PubMed]
- Bachran, C.; Morley, T.; Abdelazim, S.; Fattah, R.J.; Liu, S.; Leppla, S.H. Anthrax toxin-mediated delivery of the Pseudomonas exotoxin A enzymatic domain to the cytosol of tumor cells via cleavable ubiquitin fusions. mBio 2013, 4, e00201–e00213. [Google Scholar] [CrossRef] [PubMed]
- McCluskey, A.J.; Olive, A.J.; Starnbach, M.N.; Collier, R.J. Targeting HER2-positive cancer cells with receptor-redirected anthrax protective antigen. Mol. Oncol. 2013, 7, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Bachran, C.; Hasikova, R.; Leysath, C.E.; Sastalla, I.; Zhang, Y.; Fattah, R.J.; Liu, S.; Leppla, S.H. Cytolethal distending toxin B as a cell-killing component of tumor-targeted anthrax toxin fusion proteins. Cell Death Dis. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Rabideau, A.E.; Liao, X.; Akçay, G.; Pentelute, B.L. Translocation of Non-Canonical Polypeptides into Cells Using Protective Antigen. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef]
- Liao, X.; Rabideau, A.E.; Pentelute, B.L. Delivery of antibody mimics into mammalian cells via anthrax toxin protective antigen. Chembiochem 2014, 15, 2458–2466. [Google Scholar] [PubMed]
- Arora, N.; Williamson, L.C.; Leppla, S.H.; Halpern, J.L. Cytotoxic effects of a chimeric protein consisting of Tetanus toxin light chain and anthrax toxin lethal factor in non-neuronal cells. J. Biol. Chem. 1994, 269, 26165–26171. [Google Scholar] [PubMed]
- Goletz, T.J.; Klimpel, K.R.; Arora, N.; Leppla, S.H.; Keith, J.M.; Berzofsky, J.A. Targeting HIV proteins to the major histocompatibility complex class I processing pathway with a novel gp120-anthrax toxin fusion protein. Proc. Natl. Acad. Sci. USA 1997, 94, 12059–12064. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.H.; Collier, R.J.; Youle, R.J. Inhibition of axotomy-induced neuronal apoptosis by extracellular delivery of a Bcl-XL fusion protein. J. Biol. Chem. 2001, 276, 46326–46332. [Google Scholar] [CrossRef] [PubMed]
- Von Moltke, J.; Trinidad, N.J.; Moayeri, M.; Kintzer, A.F.; Wang, S.B.; van Rooijen, N.; Brown, C.R.; Krantz, B.A.; Leppla, S.H.; Gronert, K.; et al. Rapid induction of inflammatory lipid mediators by the inflammasome in vivo. Nature 2012, 490, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Hobson, J.P.; Liu, S.; Rønø, B.; Leppla, S.H.; Bugge, T.H. Imaging specific cell-surface proteolytic activity in single living cells. Nat. Methods 2006, 3, 259–261. [Google Scholar] [CrossRef] [PubMed]
- Hobson, J.P.; Liu, S.; Leppla, S.H.; Bugge, T.H. Imaging specific cell surface protease activity in living cells using reengineered bacterial cytotoxins. Methods Mol. Biol. 2009, 539, 115–129. [Google Scholar] [PubMed]
- Zhu, P.J.; Hobson, J.P.; Southall, N.; Qiu, C.; Thomas, C.J.; Lu, J.; Inglese, J.; Zheng, W.; Leppla, S.H.; Bugge, T.H.; et al. Quantitative high-throughput screening identifies inhibitors of anthrax-induced cell death. Bioorg. Med. Chem. 2009, 17, 5139–5145. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Leppla, S.H. Anthrax toxin uptake by primary immune cells as determined with a lethal factor-beta-lactamase fusion protein. PLoS ONE 2009, 4. [Google Scholar] [CrossRef] [PubMed]
- Ballard, J.D.; Collier, R.J.; Starnbach, M.N. Anthrax toxin-mediated delivery of a cytotoxic T-cell epitope in vivo. Proc. Natl. Acad. Sci. USA 1996, 93, 12531–12534. [Google Scholar] [CrossRef] [PubMed]
- Ballard, J.D.; Collier, R.J.; Starnbach, M.N. Anthrax toxin as a molecular tool for stimulation of cytotoxic T lymphocytes: Disulfide-linked epitopes, multiple injections, and role of CD4(+) cells. Infect. Immun. 1998, 66, 4696–4699. [Google Scholar] [PubMed]
- Ballard, J.D.; Doling, A.M.; Beauregard, K.; Collier, R.J.; Starnbach, M.N. Anthrax toxin-mediated delivery in vivo and in vitro of a cytotoxic T-lymphocyte epitope from ovalbumin. Infect. Immun. 1998, 66, 615–619. [Google Scholar] [PubMed]
- Doling, A.M.; Ballard, J.D.; Shen, H.; Krishna, K.M.; Ahmed, R.; Collier, R.J.; Starnbach, M.N. Cytotoxic T-lymphocyte epitopes fused to anthrax toxin induce protective antiviral immunity. Infect. Immun. 1999, 67, 3290–3296. [Google Scholar] [PubMed]
- Chen, K.-H.; Liu, S.; Bankston, L.A.; Liddington, R.C.; Leppla, S.H. Selection of anthrax toxin protective antigen variants that discriminate between the cellular receptors TEM8 and CMG2 and achieve targeting of tumor cells. J. Biol. Chem. 2007, 282, 9834–9845. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.H.; Liu, S.; Leysath, C.E.; Miller-Randolph, S.; Zhang, Y.; Fattah, R.; Bugge, T.H.; Leppla, S.H. Anthrax toxin protective antigen variants that selectively utilize either the CMG2 or TEM8 receptors for cellular uptake and tumor targeting. J. Biol. Chem. 2016. in revision. [Google Scholar]
- Mechaly, A.; McCluskey, A.J.; Collier, R.J. Changing the receptor specificity of anthrax toxin. mBio 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Rosovitz, M.J.; Schuck, P.; Varughese, M.; Chopra, A.P.; Mehra, V.; Singh, Y.; McGinnis, L.M.; Leppla, S.H. Alanine-scanning mutations in domain 4 of anthrax toxin protective antigen reveal residues important for Binding to the Cellular Receptor and to a Neutralizing Monoclonal Antibody. J. Biol. Chem. 2003, 278, 30936–30944. [Google Scholar] [CrossRef] [PubMed]
- Orlova, A.; Magnusson, M.; Eriksson, T.L.J.; Nilsson, M.; Larsson, B.; Höidén-Guthenberg, I.; Widström, C.; Carlsson, J.; Tolmachev, V.; Ståhl, S.; et al. Tumor imaging using a picomolar affinity HER2 binding affibody molecule. Cancer Res. 2006, 66, 4339–4348. [Google Scholar] [CrossRef] [PubMed]
- McCluskey, A.J.; Collier, R.J. Receptor-directed chimeric toxins created by sortase-mediated protein fusion. Mol. Cancer Ther. 2013, 12, 2273–2281. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the U.S. Department of National Institutes of Health ; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bachran, C.; Leppla, S.H. Tumor Targeting and Drug Delivery by Anthrax Toxin. Toxins 2016, 8, 197. https://doi.org/10.3390/toxins8070197
Bachran C, Leppla SH. Tumor Targeting and Drug Delivery by Anthrax Toxin. Toxins. 2016; 8(7):197. https://doi.org/10.3390/toxins8070197
Chicago/Turabian StyleBachran, Christopher, and Stephen H. Leppla. 2016. "Tumor Targeting and Drug Delivery by Anthrax Toxin" Toxins 8, no. 7: 197. https://doi.org/10.3390/toxins8070197
APA StyleBachran, C., & Leppla, S. H. (2016). Tumor Targeting and Drug Delivery by Anthrax Toxin. Toxins, 8(7), 197. https://doi.org/10.3390/toxins8070197