
Fibroblast Growth Factor-23—A Potential Uremic Toxin

{kind=link}
Abstract
:1. Introduction
2. FGF23 Properties
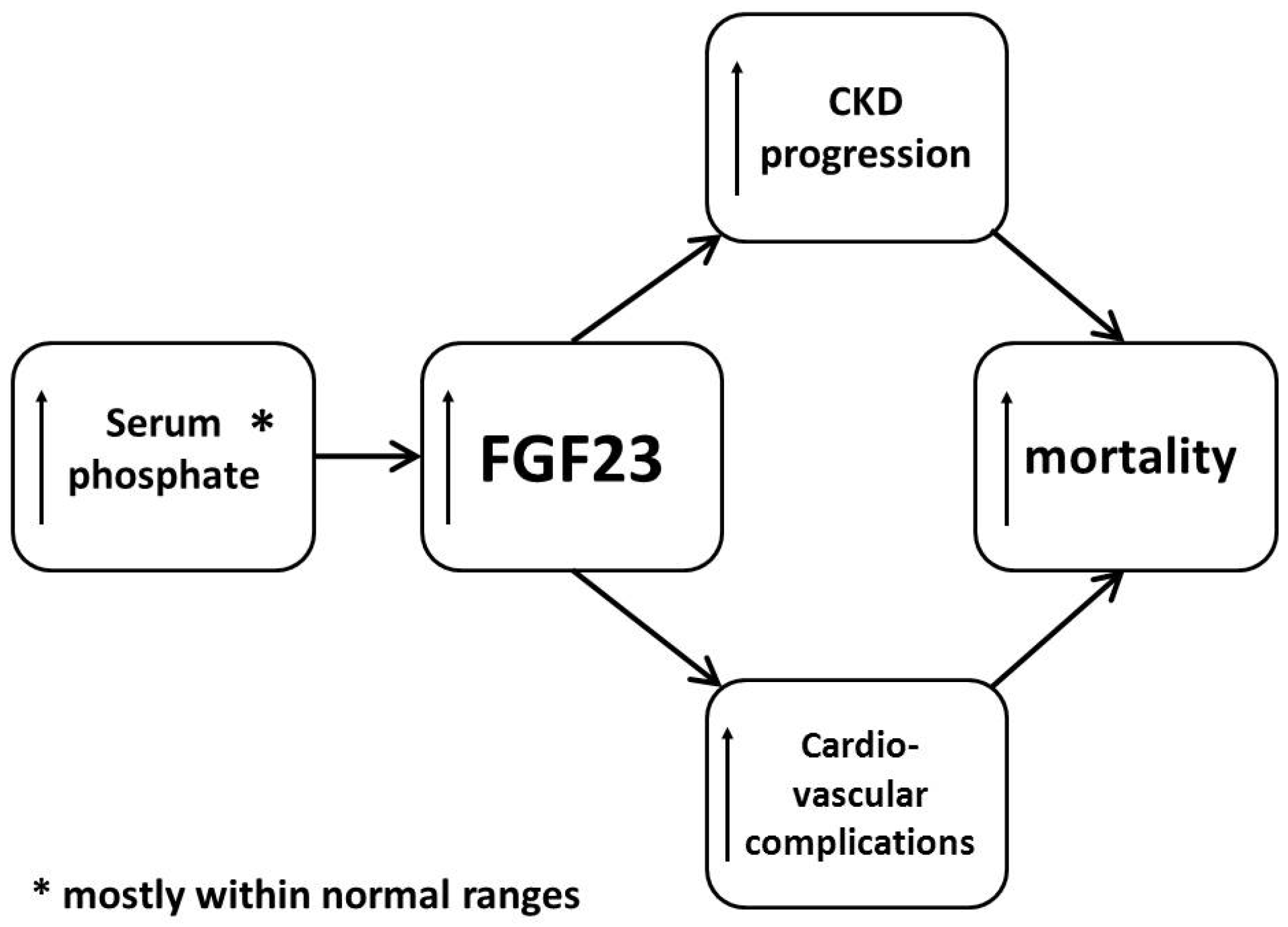
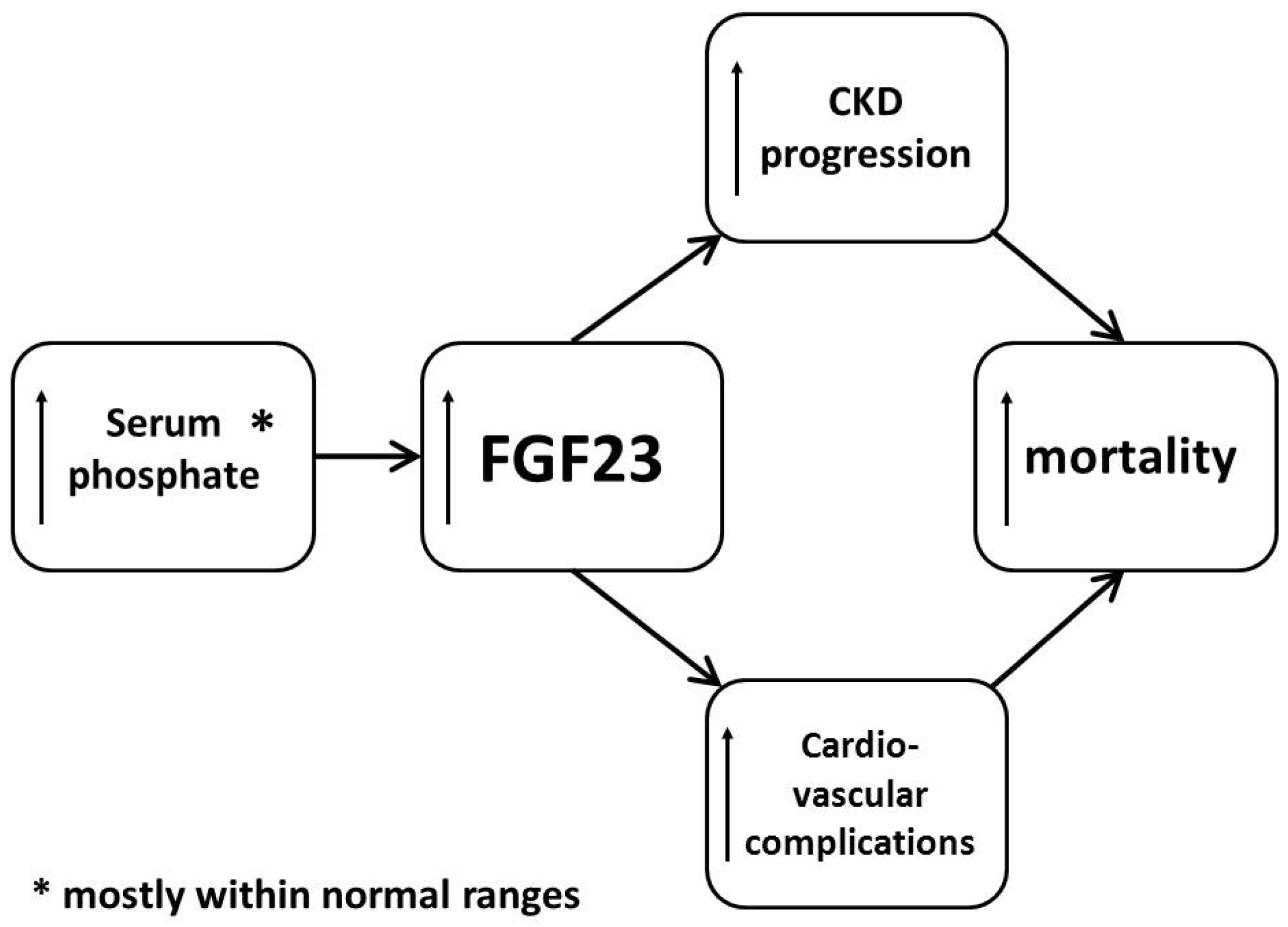
3. Mechanisms of Action and Toxicity
4. Factors Influencing Plasma FGF23 Concentration
Author Contributions
Conflicts of Interest
References
- Meyer, R.A., Jr.; Meyer, M.H.; Gray, R.W. Parabiosis suggests a humoral factor is involved in X-linked hypophosphatemia in mice. J. Bone Miner. Res. 1989, 4, 493–500. [Google Scholar] [CrossRef] [PubMed]
- White, K.E.; Carn, G.; Lorenz-Depiereux, B.; Benet-Pages, A.; Strom, T.M.; Econs, M.J. Autosomal-dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF-23. Kidney Int. 2001, 60, 2079–2086. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Mizutani, S.; Muto, T.; Yoneya, T.; Hino, R.; Takeda, S.; Takeuchi, Y.; Fujita, T.; Fukumoto, S.; Yamashita, T. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc. Natl. Acad. Sci. USA 2001, 98, 6500–6505. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, O.M. Fibroblast growth factor 23, Klotho and disordered mineral metabolism in chronic kidney disease: Unraveling the intricate tapestry of events and implications for therapy. J. Ren. Nutr. 2013, 23, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, S.; Sorenson, A.H.; Austin, A.M.; Mackenzie, D.S.; Fritz, T.A.; Moh, A.; Hui, S.L.; Econs, M.J. Ablation of the Galnt3 gene leads to low-circulating intact fibroblast growth factor 23 (Fgf23) concentrations and hyperphosphatemia despite increased Fgf23 expression. Endocrinology 2009, 150, 2543–2550. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, S.; Li, C.; Gao, T.; Liu, Y.; Rangiani, A.; Sun, Y.; Hao, J.; George, A.; Lu, Y.; et al. Inactivation of a novel FGF23 regulator, FAM20C, leads to hypophosphatemic rickets in mice. PLoS Genet. 2012, 8. [Google Scholar] [CrossRef] [PubMed]
- Farrow, E.G.; Yu, X.; Summers, L.J.; Davis, S.I.; Fleet, J.C.; Allen, M.R.; Robling, A.G.; Stayrook, K.R.; Jideonwo, V.; Magers, M.J.; et al. Iron deficiency drives an autosomal dominant hypophosphatemic rickets (ADHR) phenotype in fibroblast growth factor-23 (Fgf23) knock-in mice. Proc. Natl. Acad. Sci. USA 2011, 108, E1146–E1155. [Google Scholar] [CrossRef] [PubMed]
- Bożentowicz-Wikarek, M.; Owczarek, A.; Kocełak, P.; Olszanecka-Glinianowicz, M.; Więcek, A.; Chudek, J. C-Terminal to intact fibroblast growth factor 23 ratio in relation to estimated glomerular filtration rate in elderly population. Kidney Blood Press. Res. 2016, 41, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Mizutani, S.; Muto, T.; Yoneya, T.; Hino, R.; Takeda, S.; Takeuchi, Y.; Fujita, T.; Fukumoto, S.; Yamashita, T. FGF-23 transgenic mice demonstrate hypophosphatemic rickets with reduced expression of sodium phosphate cotransporter type IIa. Biochem. Biophys. Res. Commun. 2004, 314, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Larsson, T.; Marsell, R.; Schipani, E.; Ohlsson, C.; Ljunggren, O.; Tenenhouse, H.S.; Jüppner, H.; Jonsson, K.B. Transgenic mice expressing fibroblast growth factor 23 under the control of the alpha1(I) collagen promoter exhibit growth retardation, osteomalacia, and disturbed phosphate homeostasis. Endocrinology 2004, 145, 3087–3094. [Google Scholar] [CrossRef] [PubMed]
- Li, S.A.; Watanabe, M.; Yamada, H.; Nagai, A.; Kinuta, M.; Takei, K. Immunohistochemical localization of Klotho protein in brain, kidney, and reproductive organs of mice. Cell Struct. Funct. 2004, 29, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Farrow, E.G.; Davis, S.I.; Summers, L.J.; White, K.E. Initial FGF23-mediated signaling occurs in the distal convoluted tubule. J. Am. Soc. Nephrol. 2009, 20, 955–960. [Google Scholar] [CrossRef] [PubMed]
- Andrukhova, O.; Zeitz, U.; Goetz, R.; Mohammadi, M.; Lanske, B.; Erben, R.G. FGF23 acts directly on renal proximal tubules to induce phosphaturia through activation of the ERK1/2-SGK1 signaling pathway. Bone 2012, 51, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Haussler, M.R.; Whitfield, G.K.; Kaneko, I.; Forster, R.; Saini, R.; Hsieh, J.C.; Haussler, C.A.; Jurutka, P.W. The role of vitamin D in the FGF23, klotho, and phosphate bone-kidney endocrine axis. Rev. Endocr. Metab. Disord. 2012, 13, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Chanakul, A.; Zhang, M.Y.; Louw, A.; Armbrecht, H.J.; Miller, W.L.; Portale, A.A.; Perwad, F. FGF-23 regulates CYP27B1 transcription in the kidney and in extra-renal tissues. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Andrukhova, O.; Smorodchenko, A.; Egerbacher, M.; Streicher, C.; Zeitz, U.; Goetz, R.; Shalhoub, V.; Mohammadi, M.; Pohl, E.E.; Lanske, B.; et al. FGF23 promotes renal calcium reabsorption through the TRPV5 channel. EMBO J. 2014, 33, 229–246. [Google Scholar] [CrossRef] [PubMed]
- Andrukhova, O.; Slavic, S.; Smorodchenko, A.; Zeitz, U.; Shalhoub, V.; Lanske, B.; Pohl, E.E.; Erben, R.G. FGF23 regulates renal sodium handling and blood pressure. EMBO Mol. Med. 2014, 6, 744–759. [Google Scholar] [CrossRef] [PubMed]
- Ben-Dov, I.Z.; Galitzer, H.; Lavi-Moshayoff, V.; Goetz, R.; Kuro-o, M.; Mohammadi, M.; Sirkis, R.; Naveh-Many, T.; Silver, J. The parathyroid is a target organ for FGF23 in rats. J. Clin. Investig. 2007, 117, 4003–4008. [Google Scholar] [CrossRef] [PubMed]
- David, V.; Martin, A.; Isakova, T.; Spaulding, C.; Qi, L.; Ramirez, V.; Zumbrennen-Bullough, K.B.; Sun, C.C.; Lin, H.Y.; Babitt, J.L.; et al. Inflammation and functional iron deficiency regulate fibroblast growth factor 23 production. Kidney Int. 2016, 89, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Di Marco, G.S.; Reuter, S.; Kentrup, D.; Ting, L.; Ting, L.; Grabner, A.; Jacobi, A.M.; Pavenstädt, H.; Baba, H.A.; Tiemann, K.; et al. Cardioprotective effect of calcineurin inhibition in an animal model of renal disease. Eur. Heart J. 2011, 32, 1935–1945. [Google Scholar] [CrossRef] [PubMed]
- Meir, T.; Durlacher, K.; Pan, Z.; Amir, G.; Richards, W.G.; Silver, J.; Naveh-Many, T. Parathyroid hormone activates the orphan nuclear receptor Nurr1 to induce FGF23 transcription. Kidney Int. 2014, 86, 1106–1115. [Google Scholar] [CrossRef] [PubMed]
- Isakova, T.; Wahl, P.; Vargas, G.S.; Gutiérrez, O.M.; Scialla, J.; Xie, H.; Appleby, D.; Nessel, L.; Bellovich, K.; Chen, J.; et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 2011, 79, 1370–1378. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M. Forging forward with 10 burning questions on FGF23 in kidney disease. J. Am. Soc. Nephrol. 2010, 21, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- Razzaque, M.S. The FGF23-Klotho axis: Endocrine regulation of phosphate homeostasis. Nat. Rev. Endocrinol. 2009, 5, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Urakawa, I.; Isakova, T.; Yamazaki, Y.; Epstein, M.; Wesseling-Perry, K.; Wolf, M.; Salusky, I.B.; Jüppner, H. Circulating fibroblast growth factor 23 in patients with end-stage renal disease treated by peritoneal dialysis is intact and biologically active. J. Clin. Endocrinol. Metab. 2010, 95, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Fliser, D.; Kollerits, B.; Neyer, U.; Ankerst, D.P.; Lhotta, K.; Lingenhel, A.; Ritz, E.; Kronenberg, F.; MMKD Study Group; Kuen, E.; et al. Fibroblast growth factor 23 (FGF23) predicts progression of chronic kidney disease: the Mild to Moderate Kidney Disease (MMKD) Study. J. Am. Soc. Nephrol. 2007, 18, 2600–2608. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, O.M.; Mannstadt, M.; Isakova, T.; Rauh-Hain, J.A.; Tamez, H.; Shah, A.; Smith, K.; Lee, H.; Thadhani, R.; Jüppner, H.; et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N. Engl. J. Med. 2008, 359, 584–592. [Google Scholar] [CrossRef] [PubMed]
- Isakova, T.; Xie, H.; Yang, W.; Xie, D.; Anderson, A.H.; Scialla, J.; Wahl, P.; Gutiérrez, O.M.; Steigerwalt, S.; He, J.; et al. Fibroblast growth factor 23 and risks of mortality and end-stage renal disease in patients with chronic kidney disease. JAMA 2011, 305, 2432–2439. [Google Scholar] [CrossRef] [PubMed]
- Seiler, S.; Reichart, B.; Roth, D.; Seibert, E.; Fliser, D.; Heine, G.H. FGF-23 and future cardiovascular events in patients with chronic kidney disease before initiation of dialysis treatment. Nephrol. Dial. Transplant. 2010, 25, 3983–3989. [Google Scholar] [CrossRef] [PubMed]
- Jean, G.; Terrat, J.C.; Vanel, T.; Hurot, J.M.; Lorriaux, C.; Mayor, B.; Chazot, C. High levels of serum fibroblast growth factor (FGF)-23 are associated with increased mortality in long haemodialysis patients. Nephrol. Dial. Transplant. 2009, 24, 2792–2796. [Google Scholar] [CrossRef] [PubMed]
- Kendrick, J.; Cheung, A.K.; Kaufman, J.S.; Greene, T.; Roberts, W.L.; Smits, G.; Chonchol, M. FGF-23 associates with death, cardiovascular events, and initiation of chronic dialysis. J. Am. Soc. Nephrol. 2011, 22, 1913–1922. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.D.; Schurgers, L.J.; Brandenburg, V.M.; Christenson, R.H.; Vermeer, C.; Ketteler, M.; Shlipak, M.G.; Whooley, M.A.; Ix, J.H. The associations of fibroblast growth factor 23 and uncarboxylated matrix Gla protein with mortality in coronary artery disease: The Heart and Soul Study. Ann. Intern. Med. 2010, 152, 640–648. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, V.M.; Kleber, M.E.; Vervloet, M.G.; Tomaschitz, A.; Pilz, S.; Stojakovic, T.; Delgado, G.; Grammer, T.B.; Marx, N.; März, W.; et al. Fibroblast growth factor 23 (FGF23) and mortality: The Ludwigshafen Risk and Cardiovascular Health Study. Atherosclerosis 2014, 237, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Faul, C.; Amaral, A.P.; Oskouei, B.; Hu, M.C.; Sloan, A.; Isakova, T.; Gutiérrez, O.M.; Aguillon-Prada, R.; Lincoln, J.; Hare, J.M.; et al. FGF23 induces left ventricular hypertrophy. J. Clin. Investig. 2011, 121, 4393–4408. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, O.M.; Januzzi, J.L.; Isakova, T.; Laliberte, K.; Smith, K.; Collerone, G.; Sarwar, A.; Hoffmann, U.; Coglianese, E.; Christenson, R.; et al. Fibroblast growth factor 23 and left ventricular hypertrophy in chronic kidney disease. Circulation 2009, 119, 2545–2552. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.A.; Larsson, A.; Melhus, H.; Lind, L.; Larsson, T.E. Serum intact FGF23 associate with left ventricular mass, hypertrophy and geometry in an elderly population. Atherosclerosis 2009, 207, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Kanbay, M.; Nicoleta, M.; Selcoki, Y.; Ikizek, M.; Aydin, M.; Eryonucu, B.; Duranay, M.; Akcay, A.; Armutcu, F.; Covic, A. Fibroblast growth factor 23 and fetuin A are independent predictors for the coronary artery disease extent in mild chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2010, 5, 1780–1786. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.A.; Hansen, T.; Johansson, L.; Ahlström, H.; Larsson, A.; Lind, L.; Larsson, T.E. Relationship between circulating FGF23 and total body atherosclerosis in the community. Nephrol. Dial. Transplant. 2009, 24, 3125–3131. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.A.; Larsson, A.; Lind, L.; Larsson, T.E. Circulating fibroblast growth factor-23 is associated with vascular dysfunction in the community. Atherosclerosis 2009, 205, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Grabner, A.; Amaral, A.P.; Schramm, K.; Singh, S.; Sloan, A.; Yanucil, C.; Li, J.; Shehadeh, L.A.; Hare, J.M.; David, V.; et al. Activation of cardiac fibroblast growth factor receptor 4 causes left ventricular hypertrophy. Cell Metab. 2015, 22, 1020–1032. [Google Scholar] [CrossRef] [PubMed]
- Touchberry, C.D.; Green, T.M.; Tchikrizov, V.; Mannix, J.E.; Mao, T.F.; Carney, B.W.; Girgis, M.; Vincent, R.J.; Wetmore, L.A.; Dawn, B.; et al. FGF23 is a novel regulator of intracellular calcium and cardiac contractility in addition to cardiac hypertrophy. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E863–E873. [Google Scholar] [CrossRef] [PubMed]
- Rossaint, J.; Oehmichen, J.; Van Aken, H.; Reuter, S.; Pavenstädt, H.J.; Meersch, M.; Unruh, M.; Zarbock, A. FGF23 signaling impairs neutrophil recruitment and host defense during CKD. J. Clin. Investig. 2016, 126, 962–974. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.L.; Bonjour, J.P.; Rizzoli, R. Fibroblast growth factor-23 relationship to dietary phosphate and renal phosphate handling in healthy young men. J. Clin. Endocrinol. Metab. 2005, 90, 1519–1524. [Google Scholar] [CrossRef] [PubMed]
- Moe, S.M.; Zidehsarai, M.P.; Chambers, M.A.; Jackman, L.A.; Radcliffe, J.S.; Trevino, L.L.; Donahue, S.E.; Asplin, J.R. Vegetarian compared with meat dietary protein source and phosphorus homeostasis in chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2011, 6, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Di Iorio, B.; Di Micco, L.; Torraca, S.; Sirico, M.L.; Russo, L.; Pota, A.; Mirenghi, F.; Russo, D. Acute effects of very-low-protein diet on FGF23 levels: A randomized study. Clin. J. Am. Soc. Nephrol. 2012, 7, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Sigrist, M.; Tang, M.; Beaulieu, M.; Espino-Hernandez, G.; Er, L.; Djurdjev, O.; Levin, A. Responsiveness of FGF-23 and mineral metabolism to altered dietary phosphate intake in chronic kidney disease (CKD): Results of a randomized trial. Nephrol. Dial. Transplant. 2013, 28, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, R.B.; Cancela, A.L.; Graciolli, F.G.; Dos Reis, L.M.; Draibe, S.A.; Cuppari, L.; Carvalho, A.B.; Jorgetti, V.; Canziani, M.E.; Moysés, R.M. Early control of PTH and FGF23 in normophosphatemic CKD patients: A new target in CKD-MBD therapy? Clin. J. Am. Soc. Nephrol. 2010, 5, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Isakova, T.; Barchi-Chung, A.; Enfield, G.; Smith, K.; Vargas, G.; Houston, J.; Xie, H.; Wahl, P.; Schiavenato, E.; Dosch, A.; et al. Effects of dietary phosphate restriction and phosphate binders on FGF23 levels in CKD. Clin. J. Am. Soc. Nephrol. 2013, 8, 1009–1018. [Google Scholar] [CrossRef] [PubMed]
- Isakova, T.; Ix, J.H.; Sprague, S.M.; Raphael, K.L.; Fried, L.; Gassman, J.J.; Raj, D.; Cheung, A.K.; Kusek, J.W.; Flessner, M.F.; et al. Rationale and approaches to phosphate and fibroblast growth factor 23 reduction in CKD. J. Am. Soc. Nephrol. 2015, 26, 2328–2339. [Google Scholar] [CrossRef] [PubMed]
- Jono, S.; McKee, M.D.; Murry, C.E.; Shioi, A.; Nishizawa, Y.; Mori, K.; Morii, H.; Giachelli, C.M. Phosphate regulation of vascular smooth muscle cell calcification. Circ. Res. 2000, 87, E10–E17. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Ortiz, M.E.; Lopez, I.; Muñoz-Castañeda, J.R.; Martinez-Moreno, J.M.; Ramírez, A.P.; Pineda, C.; Canalejo, A.; Jaeger, P.; Aguilera-Tejero, E.; Rodriguez, M.; et al. Calcium deficiency reduces circulating levels of FGF23. J. Am. Soc. Nephrol. 2012, 23, 1190–1197. [Google Scholar] [CrossRef] [PubMed]
- Sabbagh, Y.; O’Brien, S.P.; Song, W.; Boulanger, J.H.; Stockmann, A.; Arbeeny, C.; Schiavi, S.C. Intestinal npt2b plays a major role in phosphate absorption and homeostasis. J. Am. Soc. Nephrol. 2009, 20, 2348–2358. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.C.; Young, D.O.; Huang, Y.; Delmez, J.A.; Coyne, D.W. A randomized, double-blind, placebo-controlled trial of niacinamide for reduction of phosphorus in hemodialysis patients. Clin. J. Am. Soc. Nephrol. 2008, 3, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Tanaka, A.; Nakamura, T.; Fukuwatari, T.; Shibata, K.; Shimada, N.; Ebihara, I.; Koide, H. Nicotinamide suppresses hyperphosphatemia in hemodialysis patients. Kidney Int. 2004, 65, 1099–1104. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.; Steffes, M.; Bostom, A.; Ix, J.H. Effect of niacin on FGF23 concentration in chronic kidney disease. Am. J. Nephrol. 2014, 39, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Turner, C.; Dalton, N.; Inaoui, R.; Fogelman, I.; Fraser, W.D.; Hampson, G. Effect of a 300 000-IU loading dose of ergocalciferol (vitamin D2) on circulating 1,25(OH)2-vitamin D and fibroblast growth factor-23 (FGF-23) in vitamin D insufficiency. J. Clin. Endocrinol. Metab. 2013, 98, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Burnett-Bowie, S.A.; Leder, B.Z.; Henao, M.P.; Baldwin, C.M.; Hayden, D.L.; Finkelstein, J.S. Randomized trial assessing the effects of ergocalciferol administration on circulating FGF23. Clin. J. Am. Soc. Nephrol. 2012, 7, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Uzum, A.K.; Salman, S.; Telci, A.; Boztepe, H.; Tanakol, R.; Alagol, F.; Ozbey, N.C. Effects of vitamin D replacement therapy on serum FGF23 concentrations in vitamin D-deficient women in short term. Eur. J. Endocrinol. 2010, 163, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Saito, H.; Maeda, A.; Ohtomo, S.; Hirata, M.; Kusano, K.; Kato, S.; Ogata, E.; Segawa, H.; Miyamoto, K.; Fukushima, N. Circulating FGF-23 is regulated by 1α,25-dihydroxyvitamin D3 and phosphorus in vivo. J. Biol. Chem. 2005, 280, 2543–2549. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.T.; Lindsay, J.R.; Jain, A.; Kelly, M.H.; Cutler, C.M.; Weinstein, L.S.; Liu, J.; Fedarko, N.S.; Winer, K.K. Fibroblast growth factor-23 is regulated by 1α,25-dihydroxyvitamin D. J. Bone Miner. Res. 2005, 20, 1944–1950. [Google Scholar] [CrossRef] [PubMed]
- Wesseling-Perry, K.; Pereira, R.C.; Sahney, S.; Gales, B.; Wang, H.J.; Elashoff, R.; Jüppner, H.; Salusky, I.B. Calcitriol and doxercalciferol are equivalent in controlling bone turnover, suppressing parathyroid hormone, and increasing fibroblast growth factor-23 in secondary hyperparathyroidism. Kidney Int. 2011, 79, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.; Rasmussen, K.; Pedersen, S.M.; Rasmussen, L.M.; Brandi, L. Changes in fibroblast growth factor 23 during treatment of secondary hyperparathyroidism with alfacalcidol or paricalcitol. Nephrol. Dial. Transplant. 2012, 27, 2263–2269. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.; Lu, T.S.; Molostvov, G.; Lee, C.; Lam, F.T.; Zehnder, D.; Hsiao, L.L. Vascular Klotho deficiency potentiates the development of human artery calcification and mediates resistance to fibroblast growth factor 23. Circulation 2012, 125, 2243–2255. [Google Scholar] [CrossRef] [PubMed]
- Wetmore, J.B.; Liu, S.; Krebill, R.; Menard, R.; Quarles, L.D. Effects of cinacalcet and concurrent low-dose vitamin D on FGF23 levels in ESRD. Clin. J. Am. Soc. Nephrol. 2010, 5, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, M.; Komaba, H.; Nakanishi, S.; Fujimori, A.; Fukagawa, M. Cinacalcet treatment and serum FGF23 levels in haemodialysis patients with secondary hyperparathyroidism. Nephrol. Dial. Transplant. 2012, 27, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Kuczera, P.; Adamczak, M.; Wiecek, A. Cinacalcet treatment decreases plasma fibroblast growth factor 23 concentration in haemodialysed patients with chronic kidney disease and secondary hyperparathyroidism. Clin. Endocrinol. (Oxf.) 2014, 80, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.J.; Bell, G.; Pickthorn, K.; Huang, S.; Vick, A.; Hodsman, P.; Peacock, M. Velcalcetide (AMG 416), a novel peptide agonist of the calcium-sensing receptor, reduces serum parathyroid hormone and FGF23 levels in healthy male subjects. Nephrol. Dial. Transplant. 2014, 29, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Shalhoub, V.; Shatzen, E.M.; Ward, S.C.; Davis, J.; Stevens, J.; Bi, V.; Renshaw, L.; Hawkins, N.; Wang, W.; Chen, C.; et al. FGF23 neutralization improves chronic kidney disease-associated hyperparathyroidism yet increases mortality. J. Clin. Investig. 2012, 122, 2543–2553. [Google Scholar] [CrossRef] [PubMed]
- Aono, Y.; Yamazaki, Y.; Yasutake, J.; Kawata, T.; Hasegawa, H.; Urakawa, I.; Fujita, T.; Wada, M.; Yamashita, T.; Fukumoto, S.; et al. Therapeutic effects of anti-FGF23 antibodies in hypophosphatemic rickets/osteomalacia. J. Bone Miner. Res. 2009, 24, 1879–1888. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Imel, E.A.; Ruppe, M.D.; Weber, T.J.; Klausner, M.A.; Ito, T.; Vergeire, M.; Humphrey, J.; Glorieux, F.H.; Portale, A.A.; et al. Pharmacokinetics and pharmacodynamics of a human monoclonal anti-FGF23 antibody (KRN23) in the first multiple ascending dose trial treating adults with X-linked hypophosphatemia. J. Clin. Pharmacol. 2016, 56, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.; Koch, T.A.; Bregman, D.B. Effects of iron deficiency anemia and its treatment on fibroblast growth factor 23 and phosphate homeostasis in women. J. Bone Miner. Res. 2013, 28, 1793–1803. [Google Scholar] [CrossRef] [PubMed]
- Olauson, H.; Lindberg, K.; Amin, R.; Sato, T.; Jia, T.; Goetz, R.; Mohammadi, M.; Andersson, G.; Lanske, B.; Larsson, T.E. Parathyroid-specific deletion of Klotho unravels a novel calcineurin-dependent FGF23 signaling pathway that regulates PTH secretion. PLoS Genet. 2013, 9. [Google Scholar] [CrossRef] [PubMed]
- Schouten, B.J.; Hunt, P.J.; Livesey, J.H.; Frampton, C.M.; Soule, S.G. FGF23 elevation and hypophosphatemia after intravenous iron polymaltose: A prospective study. J. Clin. Endocrinol. Metab. 2009, 94, 2332–2337. [Google Scholar] [CrossRef] [PubMed]
- Coskun, Y.; Paydas, S.; Balal, M.; Soyupak, S.; Kara, E. Bone disease and serum fibroblast growth factor-23 levels in renal transplant recipients. Transplant Proc. 2016, 48, 2040–2045. [Google Scholar] [CrossRef] [PubMed]
- Economidou, D.; Dovas, S.; Papagianni, A.; Pateinakis, P.; Memmos, D. FGF-23 levels before and after renal transplantation. J. Transplant. 2009. [Google Scholar] [CrossRef] [PubMed]
- Krocker, D.; Perka, C.; Tuischer, J.; Funk, J.; Tohtz, S.; Buttgereit, F.; Matziolis, G. Effects of tacrolimus, cyclosporin A and sirolimus on MG63 cells. Transpl. Int. 2006, 19, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Bleskestad, I.H.; Thorsen, I.S.; Jonsson, G.; Skadberg, Ø.; Bergrem, H.; Gøransson, L.G. Soluble Klotho and intact fibroblast growth factor 23 in long-term kidney transplant patients. Eur. J. Endocrinol. 2015, 172, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Bhan, I.; Shah, A.; Holmes, J.; Isakova, T.; Gutierrez, O.; Burnett, S.M.; Jüppner, H.; Wolf, M. Post-transplant hypophosphatemia: Tertiary ‘Hyper-Phosphatoninism’? Kidney Int. 2006, 70, 1486–1494. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuczera, P.; Adamczak, M.; Wiecek, A. Fibroblast Growth Factor-23—A Potential Uremic Toxin. Toxins 2016, 8, 369. https://doi.org/10.3390/toxins8120369
Kuczera P, Adamczak M, Wiecek A. Fibroblast Growth Factor-23—A Potential Uremic Toxin. Toxins. 2016; 8(12):369. https://doi.org/10.3390/toxins8120369
Chicago/Turabian StyleKuczera, Piotr, Marcin Adamczak, and Andrzej Wiecek. 2016. "Fibroblast Growth Factor-23—A Potential Uremic Toxin" Toxins 8, no. 12: 369. https://doi.org/10.3390/toxins8120369
APA StyleKuczera, P., Adamczak, M., & Wiecek, A. (2016). Fibroblast Growth Factor-23—A Potential Uremic Toxin. Toxins, 8(12), 369. https://doi.org/10.3390/toxins8120369