Abstract
Fibroblast growth factor-23 (FGF23) is a circulating member of the FGF family produced mainly by the osteocytes and osteoblasts that can act as a hormone. The main action of FGF23 is to lower phosphatemia via the reduction of urinary phosphate reabsorption and the decrease of 1,25(OH)2-D generation in the kidney. In the course of chronic kidney disease (CKD), plasma FGF23 concentration rises early, most probably to compensate the inability of the deteriorating kidneys to excrete an adequate amount of phosphate. However, this comes at the cost of FGF23-related target organ toxicity. Results of clinical studies suggest that elevated plasma FGF23 concentration is independently associated with the increased risk of CKD progression, occurrence of cardio-vascular complications, and mortality in different stages of CKD. FGF23 also contributes to cardiomyocyte hypertrophy, vascular calcification, and endothelial dysfunction. The impact of FGF23 on heart muscle is not dependent on Klotho, but rather on the PLCγ–calcineurin–NFAT (nuclear factor of activated T-cells) pathway. Among the factors increasing plasma FGF23 concentration, active vitamin D analogues play a significant role. Additionally, inflammation and iron deficiency can contribute to the increase of plasma FGF23. Among the factors decreasing plasma FGF23, dietary phosphate restriction, some intestinal phosphate binders, cinacalcet (and other calcimimetics), and nicotinamide can be enumerated. Anti-FGF23 antibodies have also recently been developed to inhibit the action of FGF23 in target organs. Still, the best way to normalize plasma FGF23 in maintenance hemodialysis patients is restoring kidney function by successful kidney transplantation.
1. Introduction
At the beginning of the 21th century it seemed that the regulation of the calcium–phosphate balance was relatively well understood. The two main “players” involved in the Ca–P homeostasis were thought to be the parathyroid hormone (PTH) and 1,25-dihydroxy-vitamin D3 (1,25(OH)2-D). Then, the discovery of fibroblast growth factor-23 (FGF23) revolutionized our understanding of Ca–P balance regulation, and changed a previous simplistic view to the complex, multi-organ feedback system that acts to maintain the physiological concentrations of calcium and phosphate. The existence and function of FGF23 was firstly hypothesized when Meyers et al. demonstrated that the “phosphate wasting factor” can be transferred from the X-linked hypophosphatemic rickets mice to normal mice in a parabiosis model [1]. Another piece of evidence emerged when a gain of function mutation for the FGF23 gene had been described in patients with autosomal hypophosphatemic rickets [2]. Further confirmation came from the studies documenting the involvement of FGF23 in the etiopathogenesis of Tumor Induced Osteomalacia (TIO) [3].
2. FGF23 Properties
FGF23 is an endocrine-acting 32 kDa protein mostly secreted by osteoblasts and osteocytes. It consists of 251 amino acids, of which 24 undergo limited proteolysis on secretion [3]. The N-terminal end has an FGF homology domain (allowing the binding to the FGF receptor), while the C-terminal end has a unique 72-amino acid sequence which can bind to the FGF23 coreceptor—α-Klotho. The C-terminal domain is also involved in the systemic action of this protein. It was shown that the lack of several amino acids in the C-terminal end makes this protein more soluble—mostly because of lower binding affinity to heparin [4].
It has recently been shown that the amount of FGF23 in circulation is precisely regulated by posttranslational processes. To prevent the intracellular cleavage of FGF23, it has to be O-glycosylated at threonine. This is clinically important, because only the intact and not-cleaved molecule exerts systemic actions; thus, the failure of this glycosylation leads to an FGF23 deficiency-like status [5]. Moreover, the aforementioned O-glycosylation must be counterbalanced by the phosphorylation of serine. The lack of the aforementioned process leads to the increase of circulating intact-FGF23 (iFGF23) concentration, causing a hypophosphatemic rickets-like phenotype [6]. The exact mechanisms of the regulation of posttranslational FGF23 modification are not yet fully understood; they are, however, extremely precise. The excess of C-terminal FGF23 (cFGF23) produced in bone in response to various (e.g., inflammatory) stimuli is counterbalanced by the augmentation of FGF23 cleavage [7]. This phenomenon leads to the stable plasma concentration of the biologically active iFGF23 [7,8]. It is however important to stress that in CKD, the cleavage process seems to be impaired, which may lead to the accumulation of cFGF23 and the increase of the cFGF23/iFGF23 ratio [8].
3. Mechanisms of Action and Toxicity
Clinically, the most important site of FGF23 action (at least under physiological conditions) is the kidney. It has now been clearly documented that plasma FGF23 concentration physiologically rises in order to decrease the excess of serum phosphate [8] (Figure 1).
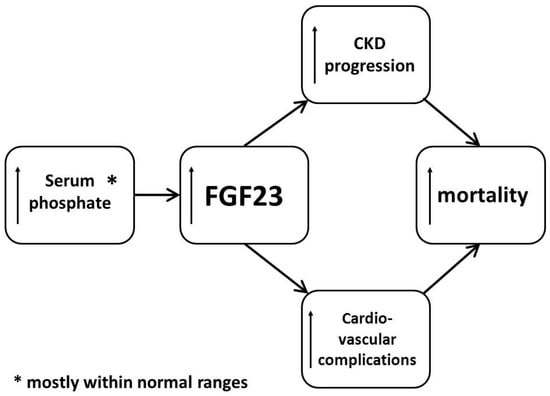
Figure 1.
Brief summary of fibroblast growth factor-23 toxicity in chronic kidney disease. CKD—chronic kidney disease; FGF23—fibroblast growth factor-23.
The FGF23-induced decrease of serum phosphate is exerted mainly through the increase of urinary phosphate excretion. This is due to the suppression of the expression of the NaPi-2a and NaPi-2c sodium-phosphate cotransporters in the apical membrane of the epithelial cells of the renal proximal tubules [9,10].
Still, the exact mechanism of the phosphaturic action of FGF23 has remained elusive for a considerable amount of time. This has arisen from the fact that in the early studies, the expression of Klotho—a mandatory FGF23 co-receptor in the kidney—has been found only in the distal renal tubules [11]. It was therefore not clear how FGF23 could act in the proximal tubules while its mandatory co-receptor was absent there. It was even hypothesized that the distal tubules may secrete some unknown paracrine-acting cytokine that would mediate the action of FGF23 in the proximal tubules in order to decrease urinary phosphate reabsorption [12]. Although this possibility has not been excluded, it has recently been observed that FGF23 can decrease the NaPi-2a expression in the renal epithelial cells in a manner resembling PTH action—through the sodium–hydrogen exchanger regulatory factor (NHERF)-1 [13]. Additionally, the apparent absence of Klotho in the proximal epithelial cells might merely be caused by the specificity of “anti-Klotho” antibodies used in the earlier studies, as Klotho expression in proximal epithelial cells has been confirmed by Andrukova et al. using different types of antibodies [13].
The second pathway in which FGF23 acts to lower serum phosphate concentration is the inhibition of 1,25(OH)2-D synthesis. FGF23 inhibits the synthesis of CYP27B1, which is an enzyme involved in the conversion of 25OH-D into the more active 1,25(OH)2-D. Moreover, FGF23 upregulates the expression of another enzyme: CYP24A1, which is essential in the catabolism of 1,25(OH)2-D, as well as the 25OH-D it stimulates the synthesis of 1,24,25(OH)3-D or 24,25(OH)2-D [14,15].
In addition to the phosphaturic action of FGF23, it seems that this hormone is also involved in renal calcium reabsorption through the regulation of the TRVP5 (transient receptor potential cation channel subfamily V member 5) channel in a Klotho-dependent manner [16]. This is a possible explanation of the fact that the injection of FGF23 does not lead to the development of hypocalcemia, despite the FGF23-related diminution of serum 1,25(OH)2-D concentration [3].
Another important action of FGF23 in the kidney is the conservation of sodium. This may lead to hypervolemia, and therefore, potentially to the development of hypertension [17]. This mechanism is mediated through the regulation of an abundance of NCC (sodium-chloride symporter) in the membrane of distal renal tubules. Interestingly, this effect of FGF23 was abolished by chlorothiazide administration [17].
Another major target organ for FGF23 is the parathyroid gland. FGF23 suppresses the PTH synthesis in the parathyroid gland, probably in a Klotho-dependent manner (the parathyroids are characterized by abundant expression of Klotho) [18]. However, there have been studies conducted with results suggesting otherwise. In Klotho knock-out mice, FGF23 decreased PTH production, probably by PLCγ–calcineurin–NFAT (nuclear factor of activated T-cells) pathway, and cyclosporine-A administration resulted in a lack of FGF23-dependent decrease of PTH production in these animals [19,20].
On the other hand, the excess PTH decreases the production of FGF23 in bone [21]. The mutual interactions between FGF23 and PTH can thus be seen as a typical negative feedback loop.
In CKD (chronic kidney disease) patients, plasma FGF23 concentration raises as early as in stage 2, which is much earlier than when the significant changes of phosphatemia or serum PTH concentration occur. This increase is most likely a counterbalance for the tendency to the higher (but still mostly within the physiological range) serum phosphate concentrations arising from the altered urinary phosphate excretion. Therefore, uremic toxicity of FGF23 seems to be at least partially secondary to the fledgling phosphate accumulation, and phosphate should be regarded as the “primary” uremic toxin. The decreased renal clearance of FGF23 caused by the falling GFR may also contribute to the increase of plasma FGF23 concentrations. An additional mechanism involved in the elevation of plasma FGF23 concentration in CKD is the decrease of the renal expression of Klotho concomitant with the deterioration of kidney function. This causes the renal “resistance” to circulating FGF23 [14,15,16]. An interesting recent finding is the fact that both inflammatory status and/or iron deficiency can be other potent contributors to the increased FGF23 concentration in patients with CKD. It seems that inflammation increases FGF23 release from the bone, both directly and through a mechanism related to the iron-balance with hypoxia induced factor (HIF)-1 as the main orchestrator [19].
Of note, though the plasma concentration of FGF23 is two- to five-fold above the physiological limit in the less-advanced stages of CKD, it can reach even 1000-fold above the upper limit in terminal renal failure [22,23,24,25].
As was mentioned previously, the excess of FGF23 released into the circulation allows for the maintenance of a stable serum phosphate concentration, even in an environment of declining renal function. However, this comes with a cost. Elevated plasma FGF23 concentration is independently associated with an increased risk of CKD progression, cardiovascular complications, and mortality in different stages of CKD [22,26,27,28,29,30,31]. One of the most evident studies was published in 2008. In this study, Gutierrez et al. showed the increased mortality in a large cohort (>10,000 subjects) of end-stage kidney disease (ESKD) patients beginning renal replacement treatment and followed up for one year. Elevated plasma FGF23 concentration (highest vs. lowest quartile) was associated with an almost six-fold higher risk of death [27]. In addition, in patients from the general population with coronary arteries disease, higher plasma concentrations of FGF23 have been associated with higher risk of mortality and CVD events, even after adjusting for traditional cardiovascular risk factors, serum C-reactive protein (CRP) concentrations, or renal function [32]. This was further confirmed in the LURIC (Ludwigshafen Risk and Cardiovascular Health) study, in which in patients undergoing coronary arteriography plasma FGF23 concentrations within the fourth quartile were associated with a 2.5-fold increase in all-cause and CV mortality when compared with patients with plasma FGF23 concentrations within the lowest quartile [33].
In clinical studies, high plasma FGF23 concentrations have also been associated with increased mass of left ventricle and higher prevalence of its hypertrophy, as well as the presence of vascular calcifications and dysfunction of endothelium [34,35,36,37,38,39].
The results obtained in the aforementioned studies may arise from a direct action of FGF23 on the cardiomyocytes, as this hormone has been shown to directly induce cardiomyocyte hypertrophy in both in vivo and in vitro studies [34].
Interestingly, the hypertrophic action of FGF23 on cardiomyocytes is not Klotho-dependent. It was shown by Faul et al. [34] that Klotho is virtually absent in cardiomyocytes. Nevertheless, when treated with pan-FGFR inhibitor (PD 173074), the mouse cardiomyocytes did not develop hypertrophy, regardless of the high plasma FGF23 concentration. This gives a strong assumption for the conclusion that, in fact, FGF23-induced cardiomyocyte hypertrophy is FGFR but not Klotho dependent. What is more, these authors have shown that, although FGF23 exerts its action on cardiomyocytes through FGFR, it uses a distinct specific pathway different than other FGFs. In a series of experiments, Grabner et al. recently identified FGFR4 as the target receptor for FGF23 binding in the myocardium [40]. The results of both of the aforementioned studies strongly suggest that the FGF23 signaling (after binding with FGFR4) is through the PLCγ–calcineurin–NFAT pathway rather than via the MAPK cascade (activated in a hypertrophic action of, e.g., FGF2—a classical protein stimulation of cardiac hypertrophy). This is interesting because of the possibility of attenuating the FGF23 related cardiomyocyte hypertrophy with cyclosporine-A, for example, which has already been documented by the authors in an experimental model [20]. This set of observations was then further confirmed by Touchberry et al., who showed that FGF23 significantly increases the size of adult cardiomyocyte cells [41]. Moreover, the very low expression of Klotho in the heart muscle has been confirmed in this study. It also seems likely that the final link in the pathway of FGF23’s influence on cardiomyocytes is the increase of Ca2+ influx to the cell, resulting in contractility changes. This action can be abolished by verapamil administration [41].
Finally, an interesting observation by Rossiant et al. must be mentioned. They have convincingly reported an involvement of FGF23 in an impairment of neutrophil recruitment and host defense in the experimental model of CKD. Briefly, as expected, CKD animals have been more prone to infection. In the experiments, the neutralization of FGF23 at different levels (e.g., with anti-FGF23 antibodies or FGF23 receptor blockade) tended to restore leukocyte recruitment into the inflammatory tissue, which was initially impaired by CKD, whereas the administration of FGF23 to control animals had the opposite effect [42].
4. Factors Influencing Plasma FGF23 Concentration
Results of clinical studies conducted so far have shown that increased plasma FGF23 concentration is associated with severe adverse outcomes in CKD patients. This gives a strong rationale for the attempts to lower the plasma FGF23 concentrations in patients with CKD.
As elevated serum phosphate concentrations lead to an increase of FGF23 secretion, the most obvious approach would be to limit the intestinal phosphate delivery. The results of a small study with the application of a low-phosphate (vegetarian) diet seem to support this thesis [42]. Similar results have been obtained in patients without kidney disease [43]. The effects of a low phosphate/low protein diet on plasma concentrations of FGF23 were further confirmed by other authors [44,45,46].
Studies evaluating the effects of the administration of phosphate binders on plasma FGF23 concentrations yielded conflicting results. The use of non-calcium-based phosphate binders generally led to an approximately 30% decrease of plasma FGF23 [47,48,49]. This effect was not seen after the administration of calcium-based binders [49,50], probably because calcium is regarded as a secondary factor stimulating FGF23 synthesis [50].
Another potential method of reducing intestinal phosphate absorption is the blocking of NPT2b (sodium dependent phosphate transporter 2b). NPT2b is an intestinal phosphate transporter whose expression can be blocked by nicotinamide [51,52,53], causing the decrease of the intestinal phosphate absorption in experimental models [52] and in CKD patients [52,54]. It was shown that nicotinamide administration leads to an 11% decrease of plasma FGF23 concentration compared to placebo [55] in patients with CKD stage 3.
Vitamin D and its active metabolites are also known to modify the plasma concentrations of FGF23. As far as native vitamin D is concerned, the data presented so far is conflicting. In a study by Turner et al. [56] and Burnett-Bowie et al. [57], ergocalciferol treatment in patients with hypovitaminosis D and normal kidney function led to a considerable increase in plasma FGF23 concentration. Conversely, Uzum et al. showed that in vitamin D-deficient women with preserved renal function, cholecalciferol loading with a subsequent maintenance vitamin D3 dose led to a decrease in plasma FGF23 concentration (which was already low at baseline) [58].
The data concerning active vitamin D and its analogues is more straight-forward. It has been well documented that 1,25(OH)2-D increases plasma FGF23 concentrations in both rodents [59] and humans with preserved renal function [60], and the so-called active vitamin D analogues (e.g., doxecalciferol) increase the plasma concentrations of FGF23 in peritoneal dialysis patients with secondary hyperparathyroidism (sHPT) [61]. Moreover, in hemodialysis patients with sHPT, both alfacalcidol and paricalcitol have been documented to increase plasma FGF23 roughly 2–3-fold with a return to baseline values after the washout period [62]. This could give a strong rationale to not overuse active vitamin D analogues in the treatment of CKD-MBD in patients with renal failure; however, as has already been mentioned, there is some experimental data suggesting that Klotho restoration in the vasculature obtained by VDR activation with calcitriol led to the FGF23 mediated inhibition of calcification [63].
On the other hand, treatment with cinacalcet has been associated with the decrease of plasma FGF23 concentrations [64,65,66]. However, whether this is a direct influence of cinacalcet or a secondary effect of cinacalcet-related serum phosphate concentration decrease has not been univocally elucidated. Interestingly, it has already been shown that in healthy males, a single intravenous dose of a novel peptide activating the CaSR—velcaletide decreases plasma FGF23 concentration along with the serum PTH, not significantly influencing the serum phosphate concentrations [67].
Another approach is the development of anti-FGF23 antibodies. It was recently documented [68] that the administration of these antibodies in animals on a high phosphate diet somewhat reversed the CKD-related 1,25(-OH)2D and calcium concentration decrease, and ameliorated the severity of secondary hyperparathyroidism with the improvement of bone histomorphometry. However, this came at the cost of higher serum phosphate concentrations, with concomitant aggravated calcification of the aorta and increased incidence of death in animals treated with anti-FGF23 antibodies compared with controls. This is an interesting finding, especially in the light of results obtained by Lim et al. [63], which showed that FGF23 may in fact have some anti-calcifying properties. Nevertheless, in the first clinical studies involving patients with hypophosphatemic rickets/osteomalacia, the use of anti-FGF23 antibodies gave promising results [68,69,70].
As was mentioned previously, there are lines of evidence emerging that suggest a role of inflammation and/or iron deficiency in the increased production of FGF23 in bone. Nevertheless, the exact underlying mechanisms remain somewhat elusive. It seems that a main player is the hypoxia-inducible factor-1a (HIF-1a), which is involved in the regulation of FGF23 cleavage processes. Besides HIF-1a, the direct inflammatory signals (e.g., interleukin-1) also stimulate the FGF23 production [19,71]. Interestingly, it seems that the acute inflammation and functional iron deficiency lead only to an increase of cFGF23, while the plasma concentration of iFGF23 remains stable. This is probably due to the augmentation of FGF23 cleavage, and results in normal concentrations of the biologically active form—iFGF23 [7,71,72]. However, this is only true in subjects with preserved renal function. In animals with CKD, IL-1β injection resulted in an increase of both cFGF23 and iFGF23 concentrations [72]. In addition, chronic inflammation can lead to elevated plasma concentrations of iFGF23, which is probably caused by the alteration of FGF23 production and cleavage balance [72].
However tempting the theory is, no studies have been conducted so far implying that the limitation of inflammation or the correction of iron deficiency could decrease the elevated plasma FGF23 concentrations in CKD. Conversely, the injection of iron polymaltose caused an increase in plasma FGF23 concentration, concomitantly with the decrease of serum phosphate concentration in patients with iron deficiency and preserved kidney function [73].
Finally, it should be noted that successful kidney transplantation usually tends to lower the markedly increased FGF23 concentrations so often seen in advanced stages of CKD [74,75,76,77]. The results of some clinical studies suggest that despite the rapid decline in plasma FGF23 concentration after kidney transplantation, it is still slightly but significantly higher than in the general population [77]. This could be attributed to the common use of phosphate supplementation or active vitamin D treatment of the post-transplant hypophosphatemia [74,78], but may also be related to the observations that calcineurin inhibitors and sirolimus in fact stimulate FGF23 production [75,76]. On the other hand, no differences in the plasma concentration of FGF23 between the post-transplant and CKD population with equal eGFR have been found [74].
In conclusion: Plasma FGF23 concentration rises early in the course of chronic kidney disease, most probably to compensate the inability of the deteriorating kidneys to excrete an adequate amount of phosphate. In addition, the results of clinical studies suggest that elevated plasma FGF23 concentration is independently associated with an increased risk of CKD progression, occurrence of cardio-vascular complications, and mortality in different stages of CKD. These facts give a strong rationale for regarding FGF23 as a uremic toxin.
Author Contributions
Each of the authors has contributed equally in the writing of this manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Meyer, R.A., Jr.; Meyer, M.H.; Gray, R.W. Parabiosis suggests a humoral factor is involved in X-linked hypophosphatemia in mice. J. Bone Miner. Res. 1989, 4, 493–500. [Google Scholar] [CrossRef] [PubMed]
- White, K.E.; Carn, G.; Lorenz-Depiereux, B.; Benet-Pages, A.; Strom, T.M.; Econs, M.J. Autosomal-dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF-23. Kidney Int. 2001, 60, 2079–2086. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Mizutani, S.; Muto, T.; Yoneya, T.; Hino, R.; Takeda, S.; Takeuchi, Y.; Fujita, T.; Fukumoto, S.; Yamashita, T. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc. Natl. Acad. Sci. USA 2001, 98, 6500–6505. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, O.M. Fibroblast growth factor 23, Klotho and disordered mineral metabolism in chronic kidney disease: Unraveling the intricate tapestry of events and implications for therapy. J. Ren. Nutr. 2013, 23, 250–254. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, S.; Sorenson, A.H.; Austin, A.M.; Mackenzie, D.S.; Fritz, T.A.; Moh, A.; Hui, S.L.; Econs, M.J. Ablation of the Galnt3 gene leads to low-circulating intact fibroblast growth factor 23 (Fgf23) concentrations and hyperphosphatemia despite increased Fgf23 expression. Endocrinology 2009, 150, 2543–2550. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, S.; Li, C.; Gao, T.; Liu, Y.; Rangiani, A.; Sun, Y.; Hao, J.; George, A.; Lu, Y.; et al. Inactivation of a novel FGF23 regulator, FAM20C, leads to hypophosphatemic rickets in mice. PLoS Genet. 2012, 8. [Google Scholar] [CrossRef] [PubMed]
- Farrow, E.G.; Yu, X.; Summers, L.J.; Davis, S.I.; Fleet, J.C.; Allen, M.R.; Robling, A.G.; Stayrook, K.R.; Jideonwo, V.; Magers, M.J.; et al. Iron deficiency drives an autosomal dominant hypophosphatemic rickets (ADHR) phenotype in fibroblast growth factor-23 (Fgf23) knock-in mice. Proc. Natl. Acad. Sci. USA 2011, 108, E1146–E1155. [Google Scholar] [CrossRef] [PubMed]
- Bożentowicz-Wikarek, M.; Owczarek, A.; Kocełak, P.; Olszanecka-Glinianowicz, M.; Więcek, A.; Chudek, J. C-Terminal to intact fibroblast growth factor 23 ratio in relation to estimated glomerular filtration rate in elderly population. Kidney Blood Press. Res. 2016, 41, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Mizutani, S.; Muto, T.; Yoneya, T.; Hino, R.; Takeda, S.; Takeuchi, Y.; Fujita, T.; Fukumoto, S.; Yamashita, T. FGF-23 transgenic mice demonstrate hypophosphatemic rickets with reduced expression of sodium phosphate cotransporter type IIa. Biochem. Biophys. Res. Commun. 2004, 314, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Larsson, T.; Marsell, R.; Schipani, E.; Ohlsson, C.; Ljunggren, O.; Tenenhouse, H.S.; Jüppner, H.; Jonsson, K.B. Transgenic mice expressing fibroblast growth factor 23 under the control of the alpha1(I) collagen promoter exhibit growth retardation, osteomalacia, and disturbed phosphate homeostasis. Endocrinology 2004, 145, 3087–3094. [Google Scholar] [CrossRef] [PubMed]
- Li, S.A.; Watanabe, M.; Yamada, H.; Nagai, A.; Kinuta, M.; Takei, K. Immunohistochemical localization of Klotho protein in brain, kidney, and reproductive organs of mice. Cell Struct. Funct. 2004, 29, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Farrow, E.G.; Davis, S.I.; Summers, L.J.; White, K.E. Initial FGF23-mediated signaling occurs in the distal convoluted tubule. J. Am. Soc. Nephrol. 2009, 20, 955–960. [Google Scholar] [CrossRef] [PubMed]
- Andrukhova, O.; Zeitz, U.; Goetz, R.; Mohammadi, M.; Lanske, B.; Erben, R.G. FGF23 acts directly on renal proximal tubules to induce phosphaturia through activation of the ERK1/2-SGK1 signaling pathway. Bone 2012, 51, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Haussler, M.R.; Whitfield, G.K.; Kaneko, I.; Forster, R.; Saini, R.; Hsieh, J.C.; Haussler, C.A.; Jurutka, P.W. The role of vitamin D in the FGF23, klotho, and phosphate bone-kidney endocrine axis. Rev. Endocr. Metab. Disord. 2012, 13, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Chanakul, A.; Zhang, M.Y.; Louw, A.; Armbrecht, H.J.; Miller, W.L.; Portale, A.A.; Perwad, F. FGF-23 regulates CYP27B1 transcription in the kidney and in extra-renal tissues. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Andrukhova, O.; Smorodchenko, A.; Egerbacher, M.; Streicher, C.; Zeitz, U.; Goetz, R.; Shalhoub, V.; Mohammadi, M.; Pohl, E.E.; Lanske, B.; et al. FGF23 promotes renal calcium reabsorption through the TRPV5 channel. EMBO J. 2014, 33, 229–246. [Google Scholar] [CrossRef] [PubMed]
- Andrukhova, O.; Slavic, S.; Smorodchenko, A.; Zeitz, U.; Shalhoub, V.; Lanske, B.; Pohl, E.E.; Erben, R.G. FGF23 regulates renal sodium handling and blood pressure. EMBO Mol. Med. 2014, 6, 744–759. [Google Scholar] [CrossRef] [PubMed]
- Ben-Dov, I.Z.; Galitzer, H.; Lavi-Moshayoff, V.; Goetz, R.; Kuro-o, M.; Mohammadi, M.; Sirkis, R.; Naveh-Many, T.; Silver, J. The parathyroid is a target organ for FGF23 in rats. J. Clin. Investig. 2007, 117, 4003–4008. [Google Scholar] [CrossRef] [PubMed]
- David, V.; Martin, A.; Isakova, T.; Spaulding, C.; Qi, L.; Ramirez, V.; Zumbrennen-Bullough, K.B.; Sun, C.C.; Lin, H.Y.; Babitt, J.L.; et al. Inflammation and functional iron deficiency regulate fibroblast growth factor 23 production. Kidney Int. 2016, 89, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Di Marco, G.S.; Reuter, S.; Kentrup, D.; Ting, L.; Ting, L.; Grabner, A.; Jacobi, A.M.; Pavenstädt, H.; Baba, H.A.; Tiemann, K.; et al. Cardioprotective effect of calcineurin inhibition in an animal model of renal disease. Eur. Heart J. 2011, 32, 1935–1945. [Google Scholar] [CrossRef] [PubMed]
- Meir, T.; Durlacher, K.; Pan, Z.; Amir, G.; Richards, W.G.; Silver, J.; Naveh-Many, T. Parathyroid hormone activates the orphan nuclear receptor Nurr1 to induce FGF23 transcription. Kidney Int. 2014, 86, 1106–1115. [Google Scholar] [CrossRef] [PubMed]
- Isakova, T.; Wahl, P.; Vargas, G.S.; Gutiérrez, O.M.; Scialla, J.; Xie, H.; Appleby, D.; Nessel, L.; Bellovich, K.; Chen, J.; et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 2011, 79, 1370–1378. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M. Forging forward with 10 burning questions on FGF23 in kidney disease. J. Am. Soc. Nephrol. 2010, 21, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- Razzaque, M.S. The FGF23-Klotho axis: Endocrine regulation of phosphate homeostasis. Nat. Rev. Endocrinol. 2009, 5, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Urakawa, I.; Isakova, T.; Yamazaki, Y.; Epstein, M.; Wesseling-Perry, K.; Wolf, M.; Salusky, I.B.; Jüppner, H. Circulating fibroblast growth factor 23 in patients with end-stage renal disease treated by peritoneal dialysis is intact and biologically active. J. Clin. Endocrinol. Metab. 2010, 95, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Fliser, D.; Kollerits, B.; Neyer, U.; Ankerst, D.P.; Lhotta, K.; Lingenhel, A.; Ritz, E.; Kronenberg, F.; MMKD Study Group; Kuen, E.; et al. Fibroblast growth factor 23 (FGF23) predicts progression of chronic kidney disease: the Mild to Moderate Kidney Disease (MMKD) Study. J. Am. Soc. Nephrol. 2007, 18, 2600–2608. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, O.M.; Mannstadt, M.; Isakova, T.; Rauh-Hain, J.A.; Tamez, H.; Shah, A.; Smith, K.; Lee, H.; Thadhani, R.; Jüppner, H.; et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N. Engl. J. Med. 2008, 359, 584–592. [Google Scholar] [CrossRef] [PubMed]
- Isakova, T.; Xie, H.; Yang, W.; Xie, D.; Anderson, A.H.; Scialla, J.; Wahl, P.; Gutiérrez, O.M.; Steigerwalt, S.; He, J.; et al. Fibroblast growth factor 23 and risks of mortality and end-stage renal disease in patients with chronic kidney disease. JAMA 2011, 305, 2432–2439. [Google Scholar] [CrossRef] [PubMed]
- Seiler, S.; Reichart, B.; Roth, D.; Seibert, E.; Fliser, D.; Heine, G.H. FGF-23 and future cardiovascular events in patients with chronic kidney disease before initiation of dialysis treatment. Nephrol. Dial. Transplant. 2010, 25, 3983–3989. [Google Scholar] [CrossRef] [PubMed]
- Jean, G.; Terrat, J.C.; Vanel, T.; Hurot, J.M.; Lorriaux, C.; Mayor, B.; Chazot, C. High levels of serum fibroblast growth factor (FGF)-23 are associated with increased mortality in long haemodialysis patients. Nephrol. Dial. Transplant. 2009, 24, 2792–2796. [Google Scholar] [CrossRef] [PubMed]
- Kendrick, J.; Cheung, A.K.; Kaufman, J.S.; Greene, T.; Roberts, W.L.; Smits, G.; Chonchol, M. FGF-23 associates with death, cardiovascular events, and initiation of chronic dialysis. J. Am. Soc. Nephrol. 2011, 22, 1913–1922. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.D.; Schurgers, L.J.; Brandenburg, V.M.; Christenson, R.H.; Vermeer, C.; Ketteler, M.; Shlipak, M.G.; Whooley, M.A.; Ix, J.H. The associations of fibroblast growth factor 23 and uncarboxylated matrix Gla protein with mortality in coronary artery disease: The Heart and Soul Study. Ann. Intern. Med. 2010, 152, 640–648. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, V.M.; Kleber, M.E.; Vervloet, M.G.; Tomaschitz, A.; Pilz, S.; Stojakovic, T.; Delgado, G.; Grammer, T.B.; Marx, N.; März, W.; et al. Fibroblast growth factor 23 (FGF23) and mortality: The Ludwigshafen Risk and Cardiovascular Health Study. Atherosclerosis 2014, 237, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Faul, C.; Amaral, A.P.; Oskouei, B.; Hu, M.C.; Sloan, A.; Isakova, T.; Gutiérrez, O.M.; Aguillon-Prada, R.; Lincoln, J.; Hare, J.M.; et al. FGF23 induces left ventricular hypertrophy. J. Clin. Investig. 2011, 121, 4393–4408. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, O.M.; Januzzi, J.L.; Isakova, T.; Laliberte, K.; Smith, K.; Collerone, G.; Sarwar, A.; Hoffmann, U.; Coglianese, E.; Christenson, R.; et al. Fibroblast growth factor 23 and left ventricular hypertrophy in chronic kidney disease. Circulation 2009, 119, 2545–2552. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.A.; Larsson, A.; Melhus, H.; Lind, L.; Larsson, T.E. Serum intact FGF23 associate with left ventricular mass, hypertrophy and geometry in an elderly population. Atherosclerosis 2009, 207, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Kanbay, M.; Nicoleta, M.; Selcoki, Y.; Ikizek, M.; Aydin, M.; Eryonucu, B.; Duranay, M.; Akcay, A.; Armutcu, F.; Covic, A. Fibroblast growth factor 23 and fetuin A are independent predictors for the coronary artery disease extent in mild chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2010, 5, 1780–1786. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.A.; Hansen, T.; Johansson, L.; Ahlström, H.; Larsson, A.; Lind, L.; Larsson, T.E. Relationship between circulating FGF23 and total body atherosclerosis in the community. Nephrol. Dial. Transplant. 2009, 24, 3125–3131. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.A.; Larsson, A.; Lind, L.; Larsson, T.E. Circulating fibroblast growth factor-23 is associated with vascular dysfunction in the community. Atherosclerosis 2009, 205, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Grabner, A.; Amaral, A.P.; Schramm, K.; Singh, S.; Sloan, A.; Yanucil, C.; Li, J.; Shehadeh, L.A.; Hare, J.M.; David, V.; et al. Activation of cardiac fibroblast growth factor receptor 4 causes left ventricular hypertrophy. Cell Metab. 2015, 22, 1020–1032. [Google Scholar] [CrossRef] [PubMed]
- Touchberry, C.D.; Green, T.M.; Tchikrizov, V.; Mannix, J.E.; Mao, T.F.; Carney, B.W.; Girgis, M.; Vincent, R.J.; Wetmore, L.A.; Dawn, B.; et al. FGF23 is a novel regulator of intracellular calcium and cardiac contractility in addition to cardiac hypertrophy. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E863–E873. [Google Scholar] [CrossRef] [PubMed]
- Rossaint, J.; Oehmichen, J.; Van Aken, H.; Reuter, S.; Pavenstädt, H.J.; Meersch, M.; Unruh, M.; Zarbock, A. FGF23 signaling impairs neutrophil recruitment and host defense during CKD. J. Clin. Investig. 2016, 126, 962–974. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.L.; Bonjour, J.P.; Rizzoli, R. Fibroblast growth factor-23 relationship to dietary phosphate and renal phosphate handling in healthy young men. J. Clin. Endocrinol. Metab. 2005, 90, 1519–1524. [Google Scholar] [CrossRef] [PubMed]
- Moe, S.M.; Zidehsarai, M.P.; Chambers, M.A.; Jackman, L.A.; Radcliffe, J.S.; Trevino, L.L.; Donahue, S.E.; Asplin, J.R. Vegetarian compared with meat dietary protein source and phosphorus homeostasis in chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2011, 6, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Di Iorio, B.; Di Micco, L.; Torraca, S.; Sirico, M.L.; Russo, L.; Pota, A.; Mirenghi, F.; Russo, D. Acute effects of very-low-protein diet on FGF23 levels: A randomized study. Clin. J. Am. Soc. Nephrol. 2012, 7, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Sigrist, M.; Tang, M.; Beaulieu, M.; Espino-Hernandez, G.; Er, L.; Djurdjev, O.; Levin, A. Responsiveness of FGF-23 and mineral metabolism to altered dietary phosphate intake in chronic kidney disease (CKD): Results of a randomized trial. Nephrol. Dial. Transplant. 2013, 28, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, R.B.; Cancela, A.L.; Graciolli, F.G.; Dos Reis, L.M.; Draibe, S.A.; Cuppari, L.; Carvalho, A.B.; Jorgetti, V.; Canziani, M.E.; Moysés, R.M. Early control of PTH and FGF23 in normophosphatemic CKD patients: A new target in CKD-MBD therapy? Clin. J. Am. Soc. Nephrol. 2010, 5, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Isakova, T.; Barchi-Chung, A.; Enfield, G.; Smith, K.; Vargas, G.; Houston, J.; Xie, H.; Wahl, P.; Schiavenato, E.; Dosch, A.; et al. Effects of dietary phosphate restriction and phosphate binders on FGF23 levels in CKD. Clin. J. Am. Soc. Nephrol. 2013, 8, 1009–1018. [Google Scholar] [CrossRef] [PubMed]
- Isakova, T.; Ix, J.H.; Sprague, S.M.; Raphael, K.L.; Fried, L.; Gassman, J.J.; Raj, D.; Cheung, A.K.; Kusek, J.W.; Flessner, M.F.; et al. Rationale and approaches to phosphate and fibroblast growth factor 23 reduction in CKD. J. Am. Soc. Nephrol. 2015, 26, 2328–2339. [Google Scholar] [CrossRef] [PubMed]
- Jono, S.; McKee, M.D.; Murry, C.E.; Shioi, A.; Nishizawa, Y.; Mori, K.; Morii, H.; Giachelli, C.M. Phosphate regulation of vascular smooth muscle cell calcification. Circ. Res. 2000, 87, E10–E17. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Ortiz, M.E.; Lopez, I.; Muñoz-Castañeda, J.R.; Martinez-Moreno, J.M.; Ramírez, A.P.; Pineda, C.; Canalejo, A.; Jaeger, P.; Aguilera-Tejero, E.; Rodriguez, M.; et al. Calcium deficiency reduces circulating levels of FGF23. J. Am. Soc. Nephrol. 2012, 23, 1190–1197. [Google Scholar] [CrossRef] [PubMed]
- Sabbagh, Y.; O’Brien, S.P.; Song, W.; Boulanger, J.H.; Stockmann, A.; Arbeeny, C.; Schiavi, S.C. Intestinal npt2b plays a major role in phosphate absorption and homeostasis. J. Am. Soc. Nephrol. 2009, 20, 2348–2358. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.C.; Young, D.O.; Huang, Y.; Delmez, J.A.; Coyne, D.W. A randomized, double-blind, placebo-controlled trial of niacinamide for reduction of phosphorus in hemodialysis patients. Clin. J. Am. Soc. Nephrol. 2008, 3, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Tanaka, A.; Nakamura, T.; Fukuwatari, T.; Shibata, K.; Shimada, N.; Ebihara, I.; Koide, H. Nicotinamide suppresses hyperphosphatemia in hemodialysis patients. Kidney Int. 2004, 65, 1099–1104. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.; Steffes, M.; Bostom, A.; Ix, J.H. Effect of niacin on FGF23 concentration in chronic kidney disease. Am. J. Nephrol. 2014, 39, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Turner, C.; Dalton, N.; Inaoui, R.; Fogelman, I.; Fraser, W.D.; Hampson, G. Effect of a 300 000-IU loading dose of ergocalciferol (vitamin D2) on circulating 1,25(OH)2-vitamin D and fibroblast growth factor-23 (FGF-23) in vitamin D insufficiency. J. Clin. Endocrinol. Metab. 2013, 98, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Burnett-Bowie, S.A.; Leder, B.Z.; Henao, M.P.; Baldwin, C.M.; Hayden, D.L.; Finkelstein, J.S. Randomized trial assessing the effects of ergocalciferol administration on circulating FGF23. Clin. J. Am. Soc. Nephrol. 2012, 7, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Uzum, A.K.; Salman, S.; Telci, A.; Boztepe, H.; Tanakol, R.; Alagol, F.; Ozbey, N.C. Effects of vitamin D replacement therapy on serum FGF23 concentrations in vitamin D-deficient women in short term. Eur. J. Endocrinol. 2010, 163, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Saito, H.; Maeda, A.; Ohtomo, S.; Hirata, M.; Kusano, K.; Kato, S.; Ogata, E.; Segawa, H.; Miyamoto, K.; Fukushima, N. Circulating FGF-23 is regulated by 1α,25-dihydroxyvitamin D3 and phosphorus in vivo. J. Biol. Chem. 2005, 280, 2543–2549. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.T.; Lindsay, J.R.; Jain, A.; Kelly, M.H.; Cutler, C.M.; Weinstein, L.S.; Liu, J.; Fedarko, N.S.; Winer, K.K. Fibroblast growth factor-23 is regulated by 1α,25-dihydroxyvitamin D. J. Bone Miner. Res. 2005, 20, 1944–1950. [Google Scholar] [CrossRef] [PubMed]
- Wesseling-Perry, K.; Pereira, R.C.; Sahney, S.; Gales, B.; Wang, H.J.; Elashoff, R.; Jüppner, H.; Salusky, I.B. Calcitriol and doxercalciferol are equivalent in controlling bone turnover, suppressing parathyroid hormone, and increasing fibroblast growth factor-23 in secondary hyperparathyroidism. Kidney Int. 2011, 79, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Hansen, D.; Rasmussen, K.; Pedersen, S.M.; Rasmussen, L.M.; Brandi, L. Changes in fibroblast growth factor 23 during treatment of secondary hyperparathyroidism with alfacalcidol or paricalcitol. Nephrol. Dial. Transplant. 2012, 27, 2263–2269. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.; Lu, T.S.; Molostvov, G.; Lee, C.; Lam, F.T.; Zehnder, D.; Hsiao, L.L. Vascular Klotho deficiency potentiates the development of human artery calcification and mediates resistance to fibroblast growth factor 23. Circulation 2012, 125, 2243–2255. [Google Scholar] [CrossRef] [PubMed]
- Wetmore, J.B.; Liu, S.; Krebill, R.; Menard, R.; Quarles, L.D. Effects of cinacalcet and concurrent low-dose vitamin D on FGF23 levels in ESRD. Clin. J. Am. Soc. Nephrol. 2010, 5, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, M.; Komaba, H.; Nakanishi, S.; Fujimori, A.; Fukagawa, M. Cinacalcet treatment and serum FGF23 levels in haemodialysis patients with secondary hyperparathyroidism. Nephrol. Dial. Transplant. 2012, 27, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Kuczera, P.; Adamczak, M.; Wiecek, A. Cinacalcet treatment decreases plasma fibroblast growth factor 23 concentration in haemodialysed patients with chronic kidney disease and secondary hyperparathyroidism. Clin. Endocrinol. (Oxf.) 2014, 80, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.J.; Bell, G.; Pickthorn, K.; Huang, S.; Vick, A.; Hodsman, P.; Peacock, M. Velcalcetide (AMG 416), a novel peptide agonist of the calcium-sensing receptor, reduces serum parathyroid hormone and FGF23 levels in healthy male subjects. Nephrol. Dial. Transplant. 2014, 29, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Shalhoub, V.; Shatzen, E.M.; Ward, S.C.; Davis, J.; Stevens, J.; Bi, V.; Renshaw, L.; Hawkins, N.; Wang, W.; Chen, C.; et al. FGF23 neutralization improves chronic kidney disease-associated hyperparathyroidism yet increases mortality. J. Clin. Investig. 2012, 122, 2543–2553. [Google Scholar] [CrossRef] [PubMed]
- Aono, Y.; Yamazaki, Y.; Yasutake, J.; Kawata, T.; Hasegawa, H.; Urakawa, I.; Fujita, T.; Wada, M.; Yamashita, T.; Fukumoto, S.; et al. Therapeutic effects of anti-FGF23 antibodies in hypophosphatemic rickets/osteomalacia. J. Bone Miner. Res. 2009, 24, 1879–1888. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Imel, E.A.; Ruppe, M.D.; Weber, T.J.; Klausner, M.A.; Ito, T.; Vergeire, M.; Humphrey, J.; Glorieux, F.H.; Portale, A.A.; et al. Pharmacokinetics and pharmacodynamics of a human monoclonal anti-FGF23 antibody (KRN23) in the first multiple ascending dose trial treating adults with X-linked hypophosphatemia. J. Clin. Pharmacol. 2016, 56, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.; Koch, T.A.; Bregman, D.B. Effects of iron deficiency anemia and its treatment on fibroblast growth factor 23 and phosphate homeostasis in women. J. Bone Miner. Res. 2013, 28, 1793–1803. [Google Scholar] [CrossRef] [PubMed]
- Olauson, H.; Lindberg, K.; Amin, R.; Sato, T.; Jia, T.; Goetz, R.; Mohammadi, M.; Andersson, G.; Lanske, B.; Larsson, T.E. Parathyroid-specific deletion of Klotho unravels a novel calcineurin-dependent FGF23 signaling pathway that regulates PTH secretion. PLoS Genet. 2013, 9. [Google Scholar] [CrossRef] [PubMed]
- Schouten, B.J.; Hunt, P.J.; Livesey, J.H.; Frampton, C.M.; Soule, S.G. FGF23 elevation and hypophosphatemia after intravenous iron polymaltose: A prospective study. J. Clin. Endocrinol. Metab. 2009, 94, 2332–2337. [Google Scholar] [CrossRef] [PubMed]
- Coskun, Y.; Paydas, S.; Balal, M.; Soyupak, S.; Kara, E. Bone disease and serum fibroblast growth factor-23 levels in renal transplant recipients. Transplant Proc. 2016, 48, 2040–2045. [Google Scholar] [CrossRef] [PubMed]
- Economidou, D.; Dovas, S.; Papagianni, A.; Pateinakis, P.; Memmos, D. FGF-23 levels before and after renal transplantation. J. Transplant. 2009. [Google Scholar] [CrossRef] [PubMed]
- Krocker, D.; Perka, C.; Tuischer, J.; Funk, J.; Tohtz, S.; Buttgereit, F.; Matziolis, G. Effects of tacrolimus, cyclosporin A and sirolimus on MG63 cells. Transpl. Int. 2006, 19, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Bleskestad, I.H.; Thorsen, I.S.; Jonsson, G.; Skadberg, Ø.; Bergrem, H.; Gøransson, L.G. Soluble Klotho and intact fibroblast growth factor 23 in long-term kidney transplant patients. Eur. J. Endocrinol. 2015, 172, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Bhan, I.; Shah, A.; Holmes, J.; Isakova, T.; Gutierrez, O.; Burnett, S.M.; Jüppner, H.; Wolf, M. Post-transplant hypophosphatemia: Tertiary ‘Hyper-Phosphatoninism’? Kidney Int. 2006, 70, 1486–1494. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).