
Integrating scFv into xMAP Assays for the Detection of Marine Toxins

 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
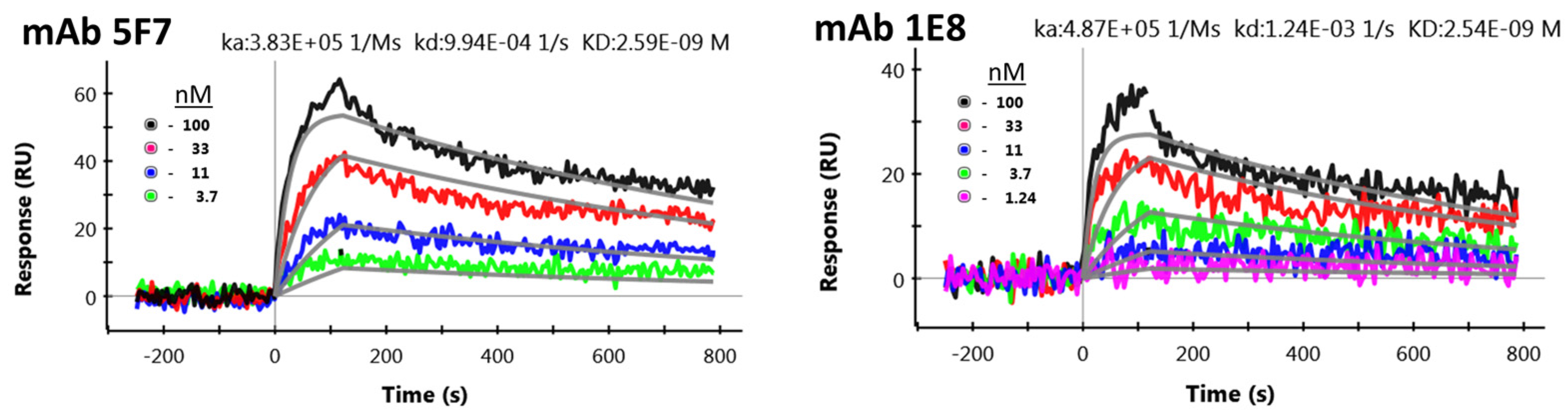
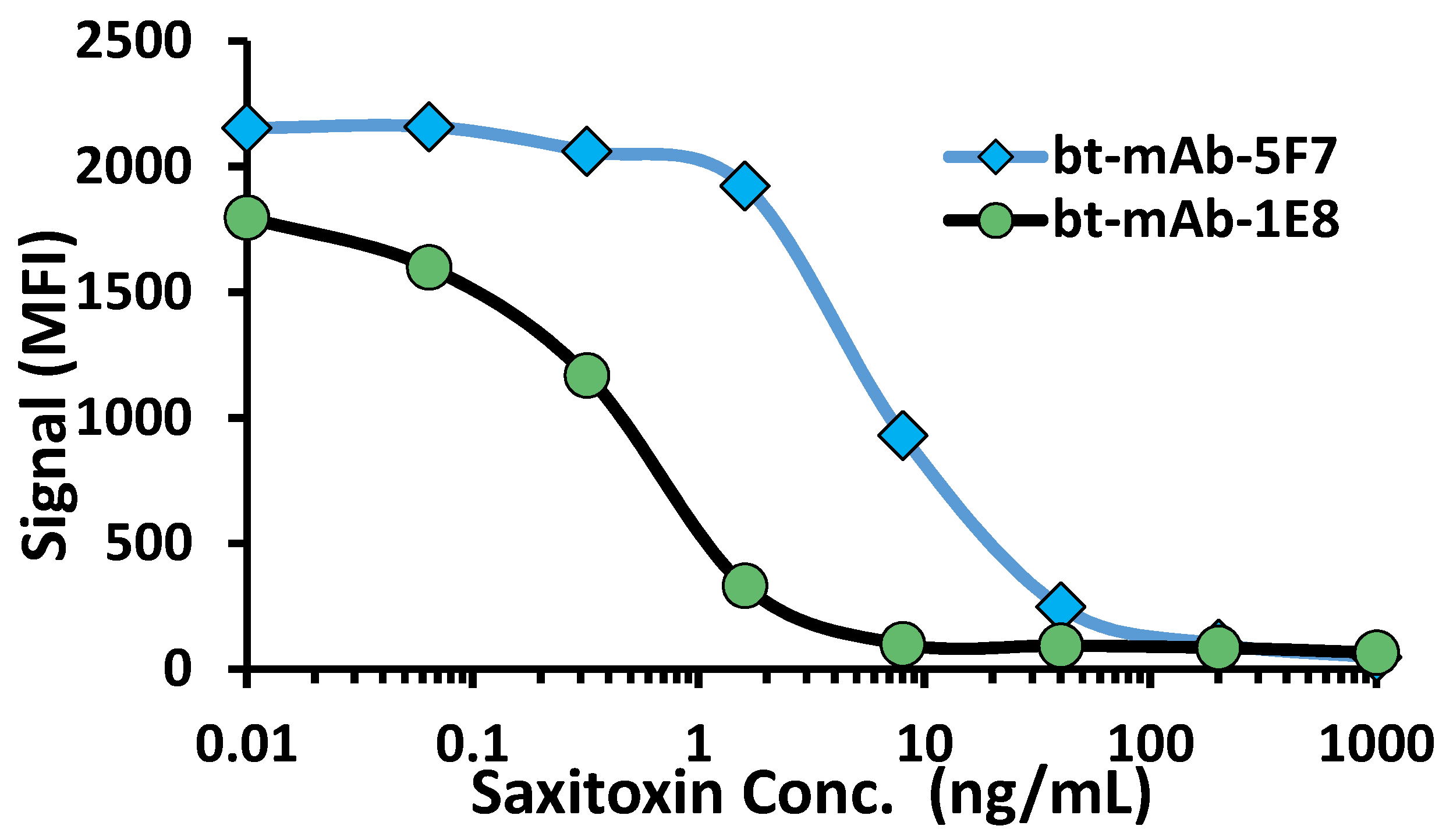
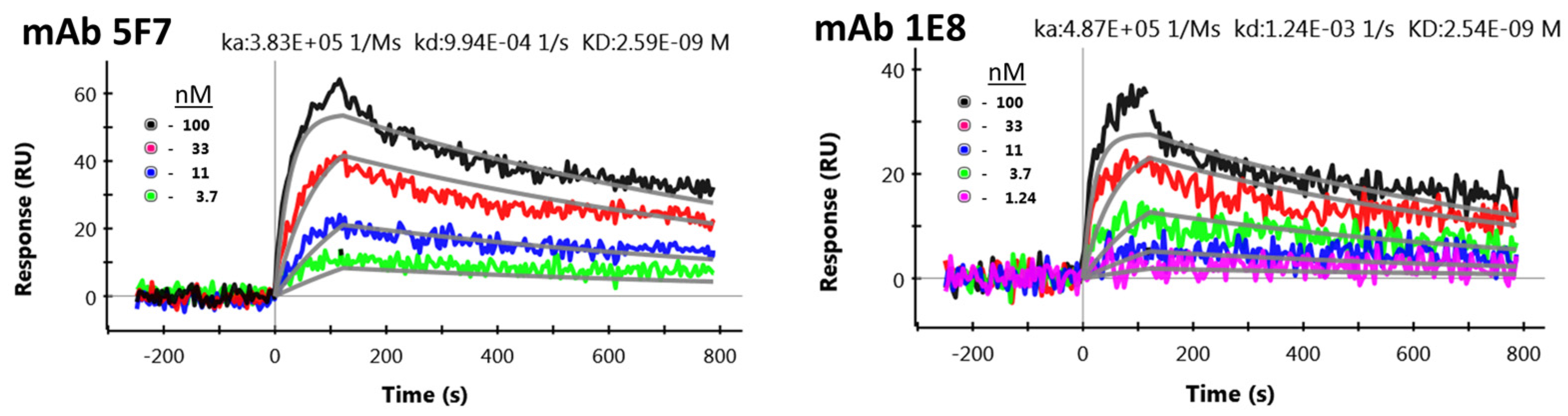
2.1. Sequencing and Evaluation of Anti-STX mAbs for scFv Production
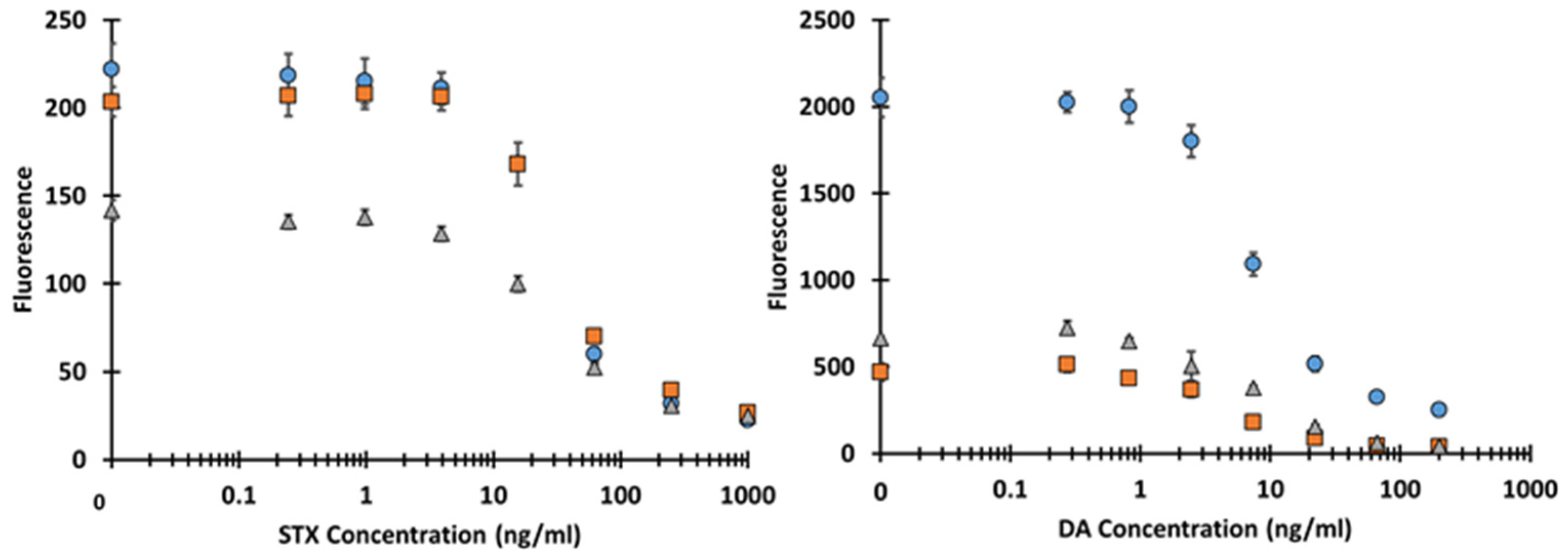
2.2. Production of scFv Targeting STX and DA
2.3. Evaluation of scFv Targeting STX and DA
2.4. Combined STX and DA Assays
3. Conclusions
4. Experimental Section
4.1. Materials
4.2. scFv Construction and Protein Production
4.3. Food Matrices Preparation
4.4. Preparation and Biotinylation of mAbs and ScFv
4.5. Surface Plasmon Resonance Evaluation of Anti-STX mAbs
4.6. Preparation Toxin-Coated MagPlex Microspheres
4.7. Assays
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Landsberg, J.H. The effects of harmful algal blooms on aquatic organisms. Rev. Fish. Sci. 2002, 10, 113–390. [Google Scholar] [CrossRef]
- Botana, A.M.; Otero, P.; Rodriguez, P.; Alfonso, A.; Botana, L.M. Current situation on analysis of marine toxins. Rev. Anal. Chem. 2013, 32, 15–34. [Google Scholar] [CrossRef]
- Zingone, A.; Oksfeldt Enevoldsen, H. The diversity of harmful algal blooms: A challenge for science and management. Ocean Coast. Manag. 2000, 43, 725–748. [Google Scholar] [CrossRef]
- Cusick, K.D.; Sayler, G.S. An overview on the marine neurotoxin, saxitoxin: Genetics, molecular targets, methods of detection and ecological functions. Mar. Drugs 2013, 11, 991–1018. [Google Scholar] [CrossRef] [PubMed]
- Vilarino, N.; Louzao, M.C.; Vieytes, M.R.; Botana, L.M. Biological methods for marine toxin detection. Anal. Bioanal. Chem. 2010, 397, 1673–1681. [Google Scholar] [CrossRef] [PubMed]
- Funk, J.A.; Janech, M.G.; Dillon, J.C.; Bissler, J.J.; Siroky, B.J.; Bell, P.D. Characterization of renal toxicity in mice administered the marine biotoxin domoic acid. J. Am. Soc. Nephrol. 2014, 25, 1187–1197. [Google Scholar] [CrossRef] [PubMed]
- Ansdell, V.E. Food Poisoning From Marine Toxins. Available online: http://wwwnc.cdc.gov/travel/yellowbook/2016/the-pre-travel-consultation/food-poisoning-from-marine-toxins (accessed on 6 March 2016).
- Greer, B.; McNamee, S.E.; Boots, B.; Cimarelli, L.; Guillebault, D.; Helmi, K.; Marcheggiani, S.; Panaiotov, S.; Breitenbach, U.; Akçaalan, R.; et al. A validated UPLC–MS/MS method for the surveillance of ten aquatic biotoxins in european brackish and freshwater systems. Harmful Algae 2016, 55, 31–40. [Google Scholar] [CrossRef]
- Muller, C.; Glamuzina, B.; Pozniak, I.; Weber, K.; Cialla, D.; Popp, J.; Pinzaru, S.C. Amnesic shellfish poisoning biotoxin detection in seawater using pure or amino-functionalized Ag nanoparticles and SERS. Talanta 2014, 130, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Olson, T.Y.; Schwartzberg, A.M.; Liu, J.L.; Zhang, J.Z. Raman and surface-enhanced raman detection of domoic acid and saxitoxin. Appl. Spectrosc. 2011, 65, 159–164. [Google Scholar] [CrossRef]
- Banerjee, P.; Kintzios, S.; Prabhakarpandian, B. Biotoxin detection using cell-based sensors. Toxins 2013, 5, 2366–2383. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Fang, J.; Cao, D.; Li, H.; Su, K.; Hu, N.; Wang, P. An improved functional assay for rapid detection of marine toxins, saxitoxin and brevetoxin using a portable cardiomyocyte-based potential biosensor. Biosens. Bioelectron. 2015, 72, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Wu, C.; Wang, Q.; Zhou, J.; Su, K.; Li, H.; Hu, N.; Wang, P. An improved sensitive assay for the detection of PSP toxins with neuroblastoma cell-based impedance biosensor. Biosens. Bioelectron. 2015, 67, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Van Dolan, F.M.; Fire, S.E.; Leighfield, T.A.; Mikulski, C.M.; Doucette, G.J. Determination of paralytic shellfish toxins in shellfish by receptor binding assay: Collaborative study. J. AOAC Int. 2012, 95, 795–812. [Google Scholar] [CrossRef]
- Dietrich, R.; Usleber, E.; Bürk, C.; Märtlbauer, E. Immunochemical approaches to the analysis of paralytic shellfish poisoning toxins. In Immunoassays for Residue Analysis; American Chemical Society: Washington, DC, USA, 1996. [Google Scholar]
- Micheli, L.; Di Stefano, S.; Moscone, D.; Palleschi, G.; Marini, S.; Coletta, M.; Draisci, R.; delli Quadri, F. Production of antibodies and development of highly sensitive formats of enzyme immunoassay for saxitoxin analysis. Anal. Bioanal. Chem. 2014, 373, 678–684. [Google Scholar] [CrossRef] [PubMed]
- Fraga, M.; Vilariño, N.; Louzao, M.C.; Rodríguez, P.; Campbell, K.; Elliott, C.T.; Botana, L.M. Multidetection of paralytic, diarrheic, and amnesic shellfish toxins by an inhibition immunoassay using a microsphere-flow cytometry system. Anal. Chem. 2013, 85, 7794–7802. [Google Scholar] [CrossRef] [PubMed]
- Fonfría, E.S.; Vilariño, N.; Campbell, K.; Elliott, C.; Haughey, S.A.; Ben-Gigirey, B.; Vieites, J.M.; Kawatsu, K.; Botana, L.M. Paralytic shellfish poisoning detection by surface plasmon resonance-based biosensors in shellfish matrixes. Anal. Chem. 2007, 79, 6303–6311. [Google Scholar] [CrossRef] [PubMed]
- Szkola, A.; Linares, E.M.; Worbs, S.; Dorner, B.G.; Dietrich, R.; Martlbauer, E.; Niessner, R.; Seidel, M. Rapid and simultaneous detection of ricin, staphylococcal enterotoxin B and saxitoxin by chemiluminescence-based microarray immunoassay. Analyst 2014, 139, 5885–5892. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.; Huet, A.C.; Charlier, C.; Higgins, C.; Delahaut, P.; Elliott, C.T. Comparison of ELISA and SPR biosensor technology for the detection of paralytic shellfish poisoning toxins. J. Chromatogr. B-Anal. Technol. Biomed. Life Sci. 2009, 877, 4079–4089. [Google Scholar] [CrossRef] [PubMed]
- Dubois, M.; Demoulin, L.; Charlier, C.; Singh, G.; Godefroy, S.B.; Campbell, K.; Elliott, C.T.; Delahaut, P. Development of ELISAs for detecting domoic acid, okadaic acid, and saxitoxin and their applicability for the detection of marine toxins in samples collected in Belgium. Food Addit. Contam. A Chem. Anal. Control Expo. Risk Assess. 2010, 27, 859–868. [Google Scholar] [CrossRef] [PubMed]
- McNamee, S.E.; Elliott, C.T.; Delahaut, P.; Campbell, K. Multiplex biotoxin surface plasmon resonance method for marine biotoxins in algal and seawater samples. Environ. Sci. Pollut. Res. 2013, 20, 6794–6807. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.; McNamee, S.E.; Huet, A.C.; Delahaut, P.; Vilarino, N.; Botana, L.M.; Poli, M.; Elliott, C.T. Evolving to the optoelectronic mouse for phycotoxin analysis in shellfish. Anal. Bioanal. Chem. 2014, 406, 6867–6881. [Google Scholar] [CrossRef] [PubMed]
- Haughey, S.A.; Campbell, K.; Yakes, B.J.; Prezioso, S.M.; DeGrasse, S.L.; Kawatsu, K.; Elliott, C.T. Comparison of biosensor platforms for surface plasmon resonance based detection of paralytic shellfish toxins. Talanta 2011, 85, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Rawn, D.F.K.; Niedzwiadek, B.; Campbell, K.; Higgins, H.C.; Elliott, C.T. Evaluation of surface plasmon resonance relative to high pressure liquid chromatography for the determination of paralytic shellfish toxins. J. Agric. Food Chem. 2009, 57, 10022–10031. [Google Scholar] [CrossRef] [PubMed]
- McNamee, S.E.; Elliott, C.T.; Greer, B.; Lochhead, M.; Campbell, K. Development of a planar waveguide microarray for the monitoring and early detection of five harmful algal toxins in water and cultures. Environ. Sci. Technol. 2014, 48, 13340–13349. [Google Scholar] [CrossRef] [PubMed]
- Szkola, A.; Campbell, K.; Elliott, C.T.; Niessner, R.; Seidel, M. Automated, high performance, flow-through chemiluminescence microarray for the multiplexed detection of phycotoxins. Anal. Chim. Acta 2013, 787, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Fraga, M.; Vilarino, N.; Carmen Louzao, M.; Rodriguez, L.P.; Alfonso, A.; Campbell, K.; Elliott, C.T.; Taylor, P.; Ramos, V.; Vasconcelos, V.; et al. Multi-detection method for five common microalgal toxins based on the use of microspheres coupled to a flow-cytometry system. Anal. Chim. Acta 2014, 850, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Fraga, M.; Vilarino, N.; Louzao, M.C.; Campbell, K.; Elliott, C.T.; Kawatsu, K.; Vieytes, M.R.; Botana, L.M. Detection of paralytic shellfish toxins by a solid-phase inhibition immunoassay using a microsphere-flow cytometry system. Anal. Chem. 2012, 84, 4350–4356. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-H.; Choi, S.-J. Immunoassay of paralytic shellfish toxins by moving magnetic particles in a stationary liquid-phase lab-on-a-chip. Biosens. Bioelectron. 2015, 66, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Jawaid, W.; Campbell, K.; Melville, K.; Holmes, S.J.; Rice, J.; Elliott, C.T. Development and validation of a novel lateral flow immunoassay (LFIA) for the rapid screening of paralytic shellfish toxins (PSTs) from shellfish extracts. Anal. Chem. 2015, 87, 5324–5332. [Google Scholar] [CrossRef] [PubMed]
- Jawaid, W.; Meneely, J.; Campbell, K.; Hooper, M.; Melville, K.; Holmes, S.; Rice, J.; Elliott, C. Development and validation of the first high performance-lateral flow immunoassay (HP-LFIA) for the rapid screening of domoic acid from shellfish extracts. Talanta 2013, 116, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Turner, A.D.; Tarnovius, S.; Johnson, S.; Higman, W.A.; Algoet, M. Testing and application of a refined rapid detection method for paralytic shellfish poisoning toxins in UK shellfish. Toxicon 2015, 100, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.; Rawn, D.F.K.; Niedzwiadek, B.; Elliott, C.T. Paralytic shellfish poisoning (PSP) toxin binders for optical biosensor technology: Problems and possibilities for the future: A review. Food Addit. Contam. A Chem. Anal. Control Expo. Risk Assess. 2011, 28, 711–725. [Google Scholar] [CrossRef] [PubMed]
- Bradbury, A.; Pluckthun, A. Standardize antibodies used in research. Nature 2015, 518, 27–29. [Google Scholar] [CrossRef] [PubMed]
- Fodey, T.; Leonard, P.; O’Mahony, J.; O’Kennedy, R.; Danaher, M. Developments in the production of biological and synthetic binders for immunoassay and sensor-based detection of small molecules. TrAC Trends Anal. Chem. 2011, 30, 254–269. [Google Scholar] [CrossRef]
- Shaw, I.; O’Reilly, A.; Charleton, M.; Kane, M. Development of a high-affinity anti-domoic acid sheep scFv and its use in detection of the toxin in shellfish. Anal. Chem. 2008, 80, 3205–3212. [Google Scholar] [CrossRef] [PubMed]
- Kavanagh, O.; Elliott, C.T.; Campbell, K. Progress in the development of immunoanalytical methods incorporating recombinant antibodies to small molecular weight biotoxins. Anal. Bioanal. Chem. 2015, 407, 2749–2770. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.L.; Zabetakis, D.; Walper, S.A.; Goldman, E.R.; Anderson, G.P. Bioconjugates of rhizavidin with single domain antibodies as bifunctional immunoreagents. J. Immunol. Methods 2014, 411, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.L.; Zabetakis, D.; Lee, A.B.; Goldman, E.R.; Anderson, G.P. Single domain antibody–alkaline phosphatase fusion proteins for antigen detection—Analysis of affinity and thermal stability of single domain antibody. J. Immunol. Methods 2013, 393, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, Z.A.; Yeap, S.K.; Ali, A.M.; Ho, W.Y.; Alitheen, N.B.M.; Hamid, M. scFv antibody: Principles and clinical application. Clin. Dev. Immunol. 2012, 15, 125–130. [Google Scholar] [CrossRef] [PubMed]
- McConnell, A.D.; Spasojevich, V.; Macomber, J.L.; Krapf, I.P.; Chen, A.; Sheffer, J.C.; Berkebile, A.; Horlick, R.A.; Neben, S.; King, D.J.; et al. An integrated approach to extreme thermostabilization and affinity maturation of an antibody. Protein Eng. Des. Sel. 2013, 26, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Hayhurst, A.; Harris, W.J. Escherichia coli Skp chaperone coexpression improves solubility and phage display of single-chain antibody fragments. Protein Expr. Purif. 1999, 15, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Hayhurst, A. Improved expression characteristics of single-chain Fv fragments when fused downstream of the Escherichia coli maltose-binding protein or upstream of a single immunoglobulin-constant domain. Protein Expr. Purif. 2000, 18, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Conway, J.O.; Sherwood, L.J.; Collazo, M.T.; Garza, J.A.; Hayhurst, A. Llama single domain antibodies specific for the 7 botulinum neurotoxin serotypes as heptaplex immunoreagents. PLoS ONE 2010, 5, e8818. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.P.; Liu, J.L.; Hale, M.L.; Bernstein, R.D.; Moore, M.; Swain, M.D.; Goldman, E.R. Development of antiricin single domain antibodies toward detection and therapeutic reagents. Anal. Chem. 2008, 80, 9604–9611. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.; Haughey, S.A.; van den Top, H.; van Egmond, H.; Vilarino, N.; Botana, L.M.; Elliott, C.T. Single laboratory validation of a surface plasmon resonance biosensor screening method for paralytic shellfish poisoning toxins. Anal. Chem. 2010, 82, 2977–2988. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.L.; Zabetakis, D.; Acevedo-Velez, G.; Goldman, E.R.; Anderson, G.P. Comparison of an antibody and its recombinant derivative for the detection of the small molecule explosive 2,4,6-trinitrotoluene. Anal. Chim. Acta 2013, 759, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Anderson, G.P.; Ortiz-Vera, Y.A.; Hayhurst, A.; Czarnecki, J.; Dabbs, J.; Vo, B.; Goldman, E.R. Evaluation of llama anti-botulinum toxin heavy chain antibody. Botulinum J. 2008, 1, 100–115. [Google Scholar] [CrossRef]
- Anderson, G.P.; Taitt, C.R. Suspension microarray immunoassay signal amplification using multilayer formation. Sens. Lett. 2008, 6, 213–218. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| STX | DA | |||||
|---|---|---|---|---|---|---|
| Buffer | Oyster | Scallops | Buffer | Oyster | Scallops | |
| Min (%) | 3.89 ± 0.92 | 2.88 ± 0.87 | 2.37 ± 1.13 | 3.67 ± 1.09 | 4.90 ± 2.31 | 3.43 ± 3.00 |
| Max (%) | 89.18 ± 1.37 | 85.48 ± 1.31 | 84.65 ± 1.86 | 87.24 ± 1.36 | 92.08 ± 2.44 | 97.44 ± 4.33 |
| IC10 (ng/mL) | 8.08 | 9.55 | 4.43 | 1.85 | 1.05 | 0.94 |
| IC50 (ng/mL) | 25.57 ± 1.56 | 31.91 ± 1.75 | 27.38 ± 2.29 | 6.91 ± 0.36 | 4.57 ± 0.47 | 6.93 ± 1.12 |
| IC90 (ng/mL) | 93.98 | 106.85 | 169.10 | 25.75 | 19.96 | 51.06 |
| STX | DA | |||
|---|---|---|---|---|
| % of Buffer Signal * | Signal/Noise (Max/Min) | % of Buffer Signal * | Signal/Noise (Max/Min) | |
| Buffer | 100 | 7.7 ± 3.4 | 100 | 8.8 ± 3.6 |
| Oyster | 80 ± 22 | 8.3 ± 2.2 | 23 ± 3 | 12.9 ± 3.2 |
| Bay Scallops | 51 ± 11 | 6.3 ± 1.9 | 50 ± 23 | 27 ± 11 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shriver-Lake, L.C.; Liu, J.L.; Brozozog Lee, P.A.; Goldman, E.R.; Dietrich, R.; Märtlbauer, E.; Anderson, G.P. Integrating scFv into xMAP Assays for the Detection of Marine Toxins. Toxins 2016, 8, 346. https://doi.org/10.3390/toxins8110346
Shriver-Lake LC, Liu JL, Brozozog Lee PA, Goldman ER, Dietrich R, Märtlbauer E, Anderson GP. Integrating scFv into xMAP Assays for the Detection of Marine Toxins. Toxins. 2016; 8(11):346. https://doi.org/10.3390/toxins8110346
Chicago/Turabian StyleShriver-Lake, Lisa C., Jinny L. Liu, P. Audrey Brozozog Lee, Ellen R. Goldman, Richard Dietrich, Erwin Märtlbauer, and George P. Anderson. 2016. "Integrating scFv into xMAP Assays for the Detection of Marine Toxins" Toxins 8, no. 11: 346. https://doi.org/10.3390/toxins8110346
APA StyleShriver-Lake, L. C., Liu, J. L., Brozozog Lee, P. A., Goldman, E. R., Dietrich, R., Märtlbauer, E., & Anderson, G. P. (2016). Integrating scFv into xMAP Assays for the Detection of Marine Toxins. Toxins, 8(11), 346. https://doi.org/10.3390/toxins8110346