
Crystal Structure of Ribosome-Inactivating Protein Ricin A Chain in Complex with the C-Terminal Peptide of the Ribosomal Stalk Protein P2


Abstract
:1. Introduction
2. Results
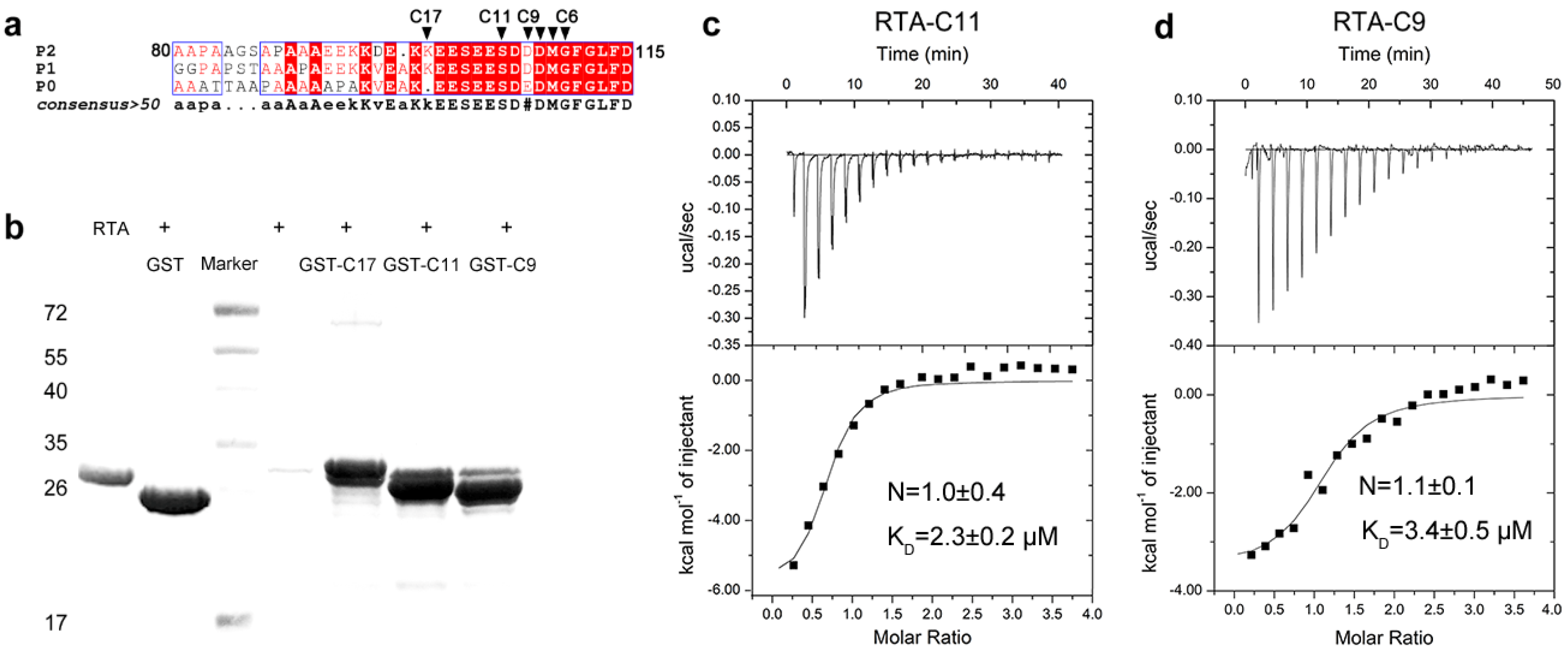
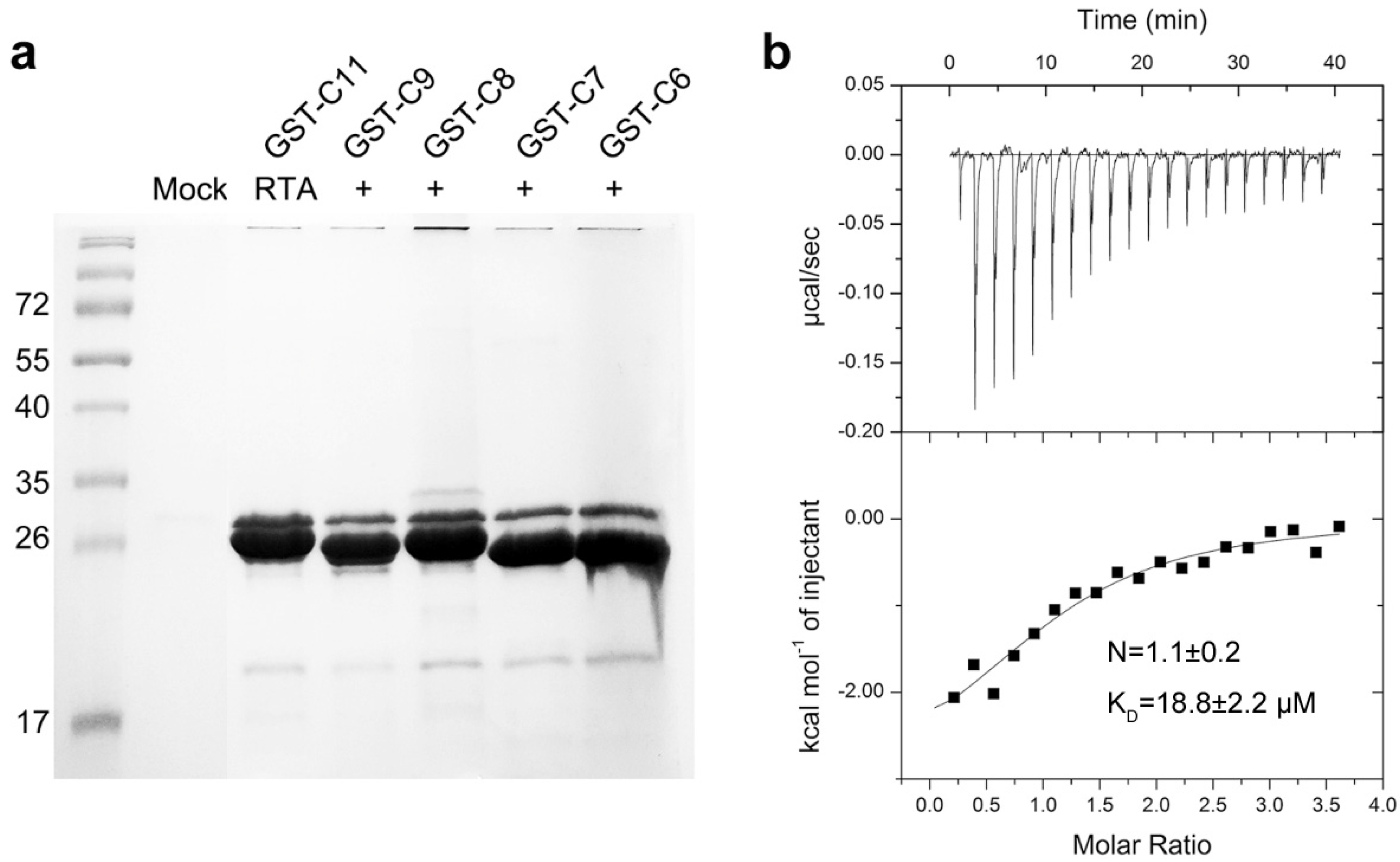
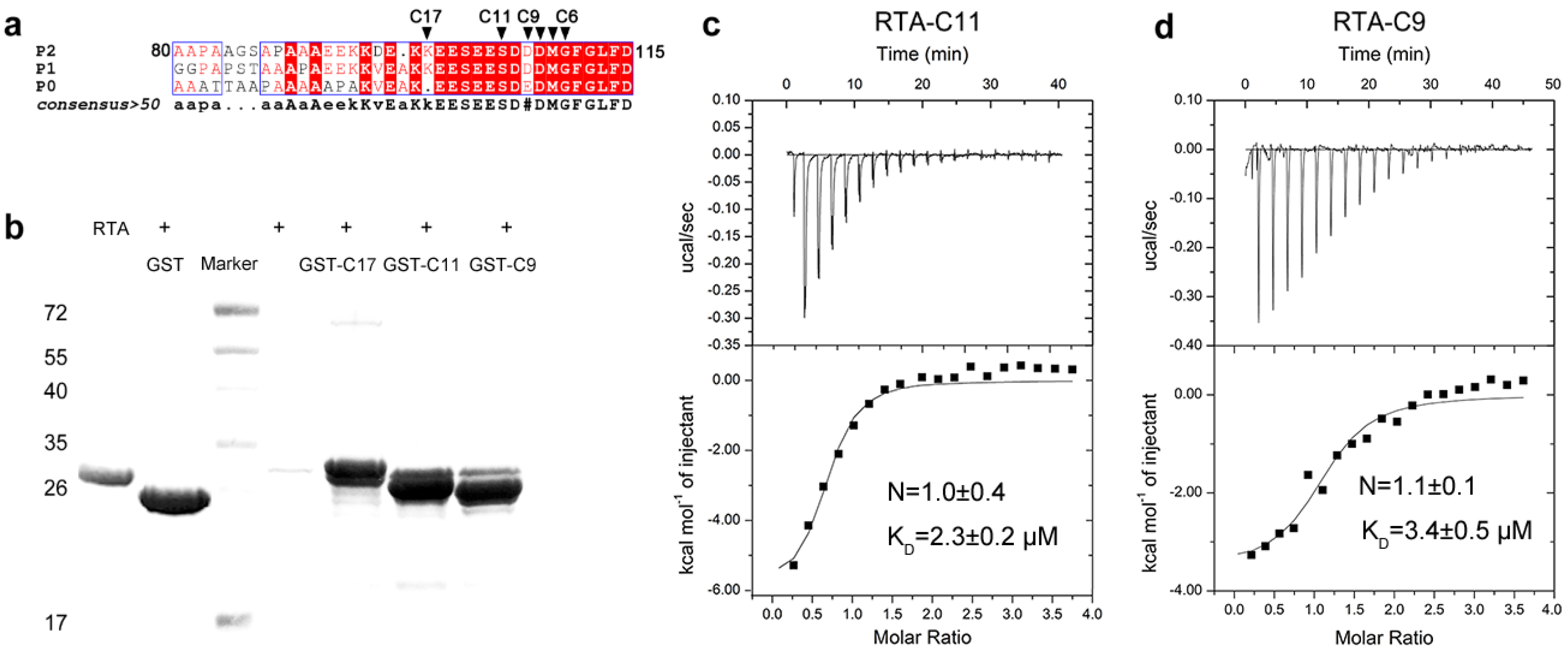
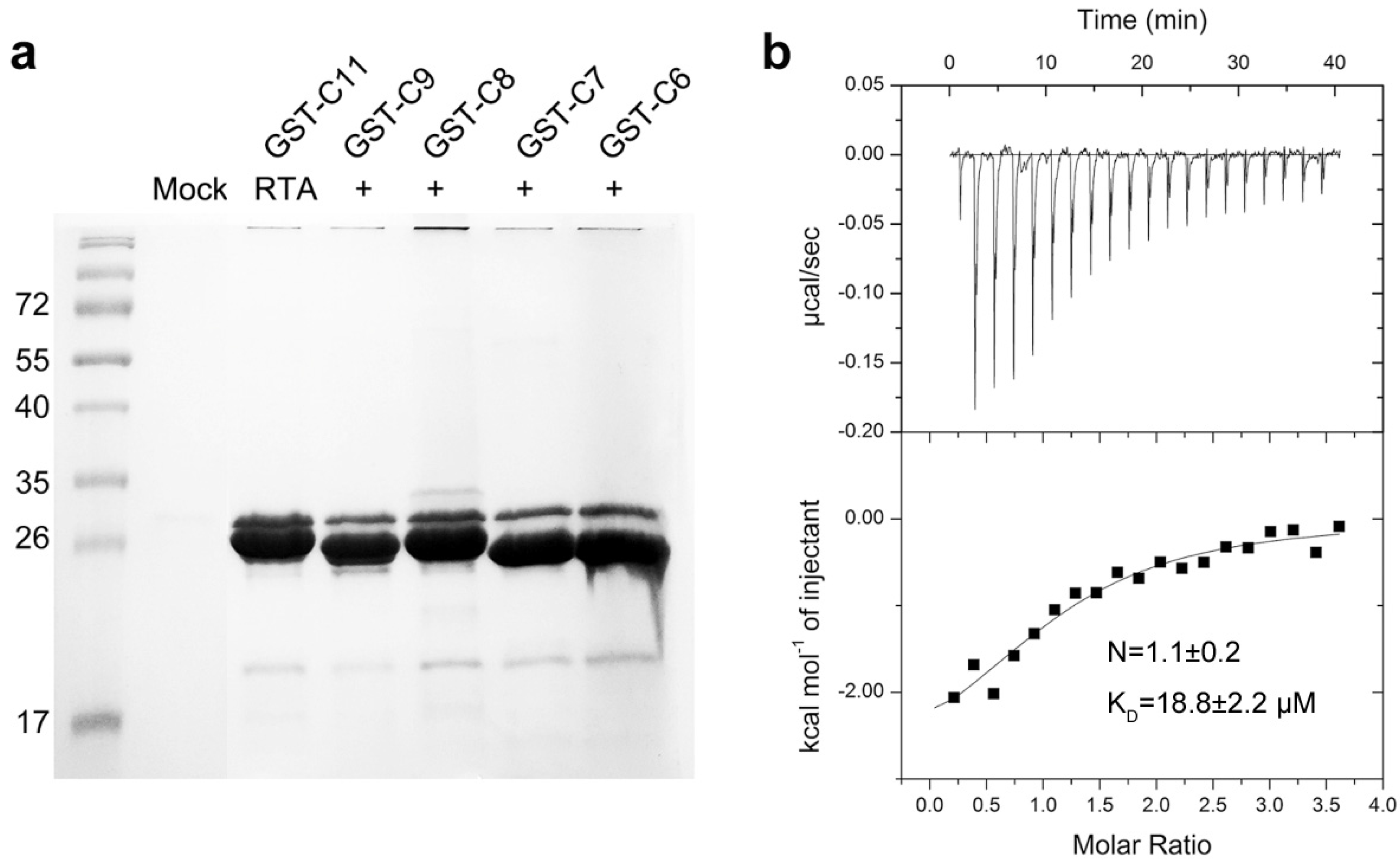
2.1. RTA Interacts with the Conserved C-Terminal Region of Acidic Ribosomal Stalk Protein P
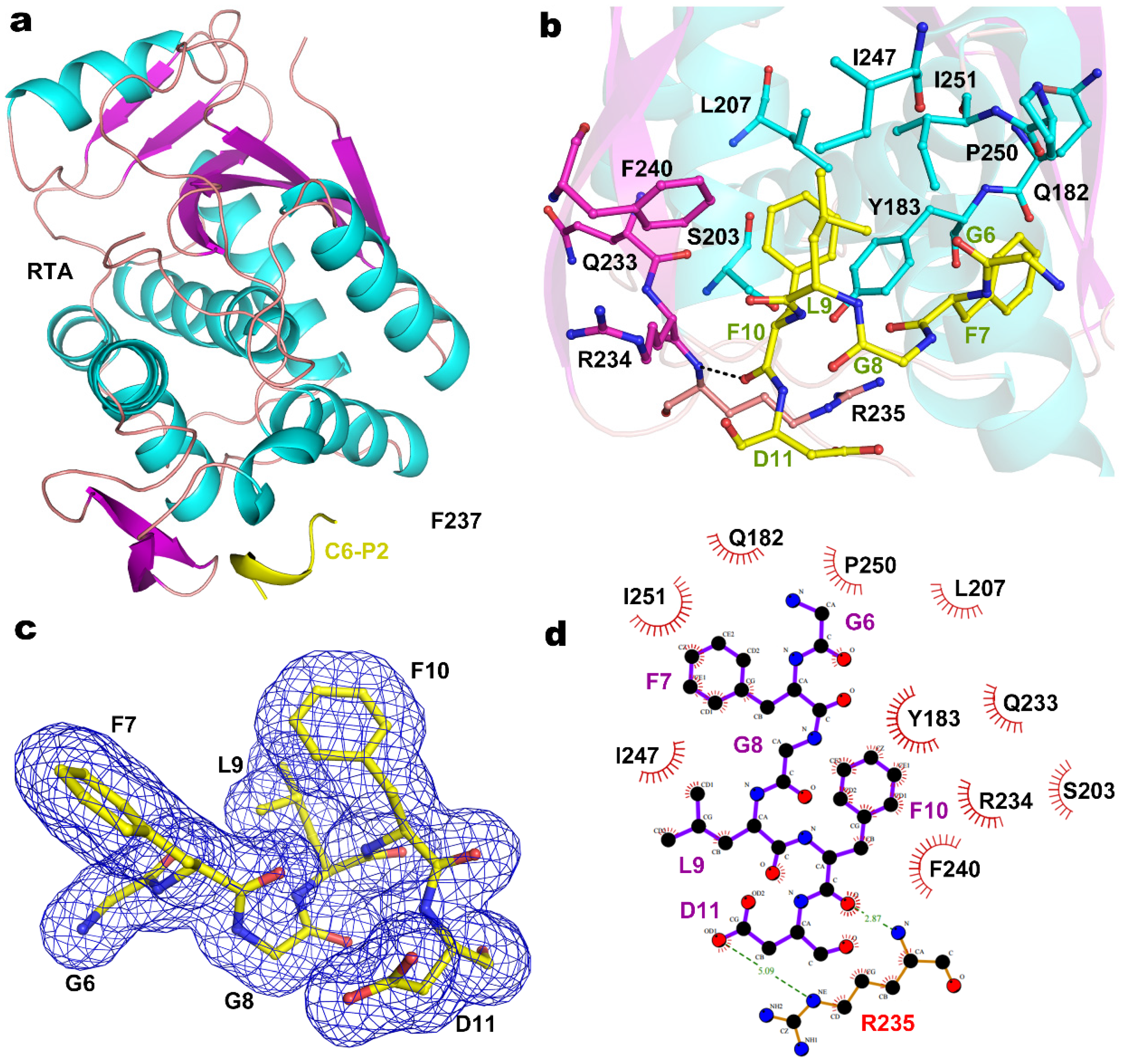
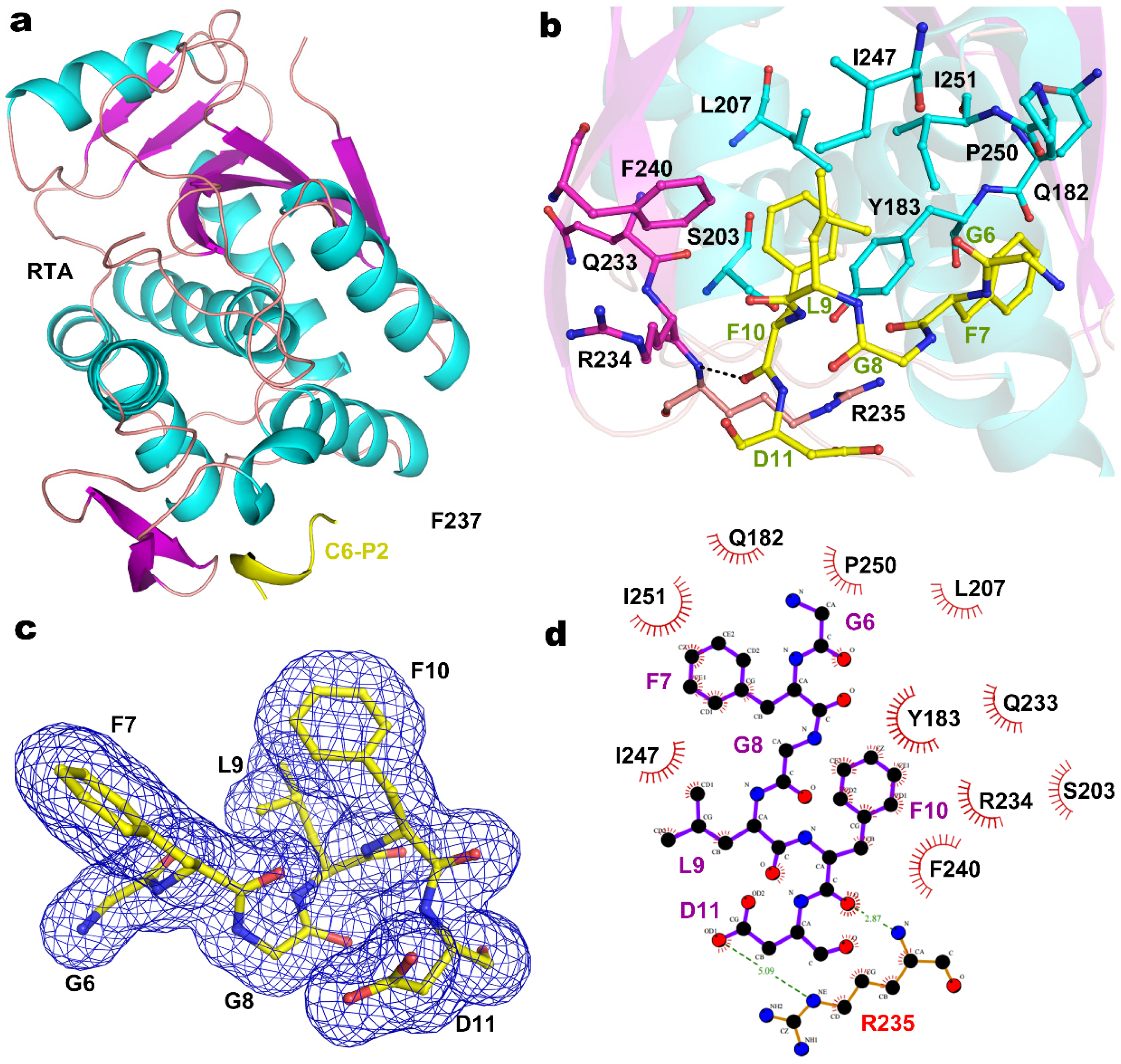
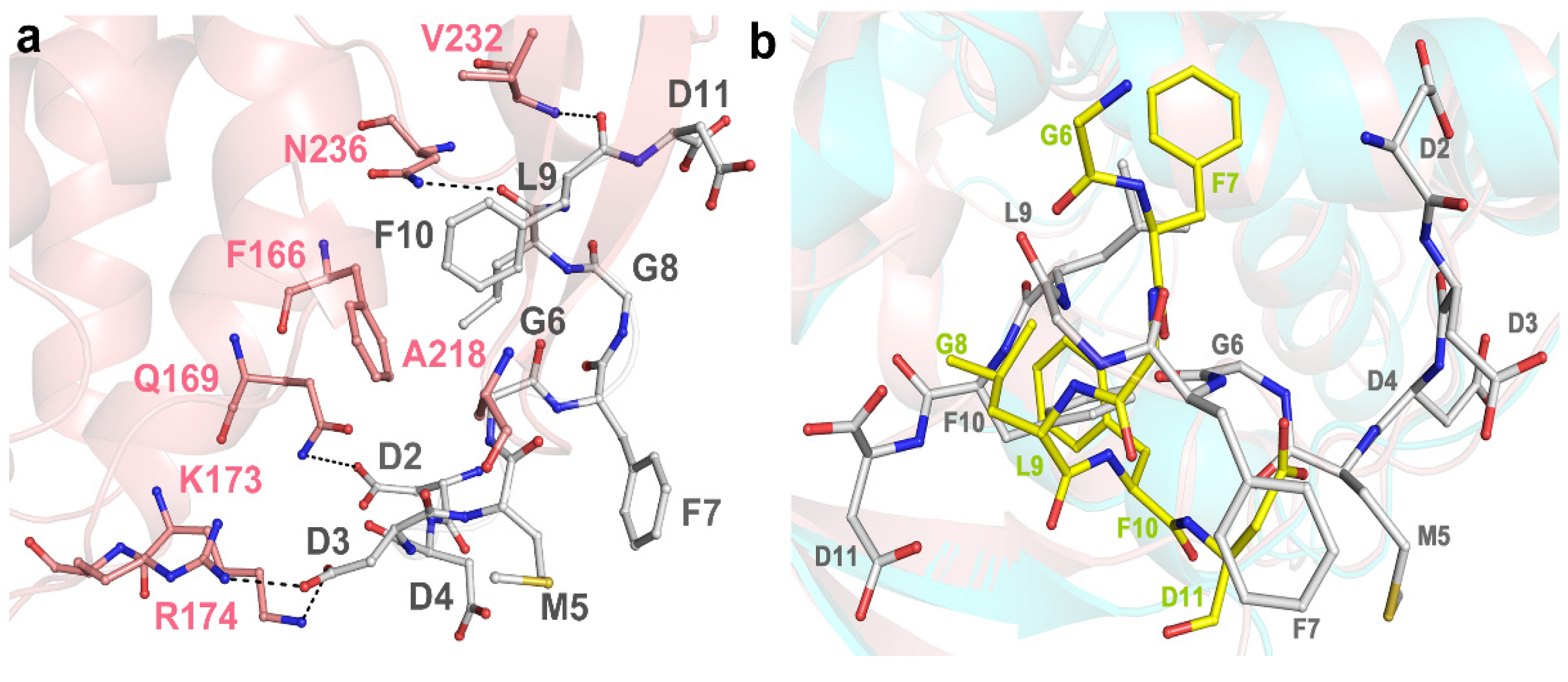
2.2. Structure of RTA-C9-P2 Complex
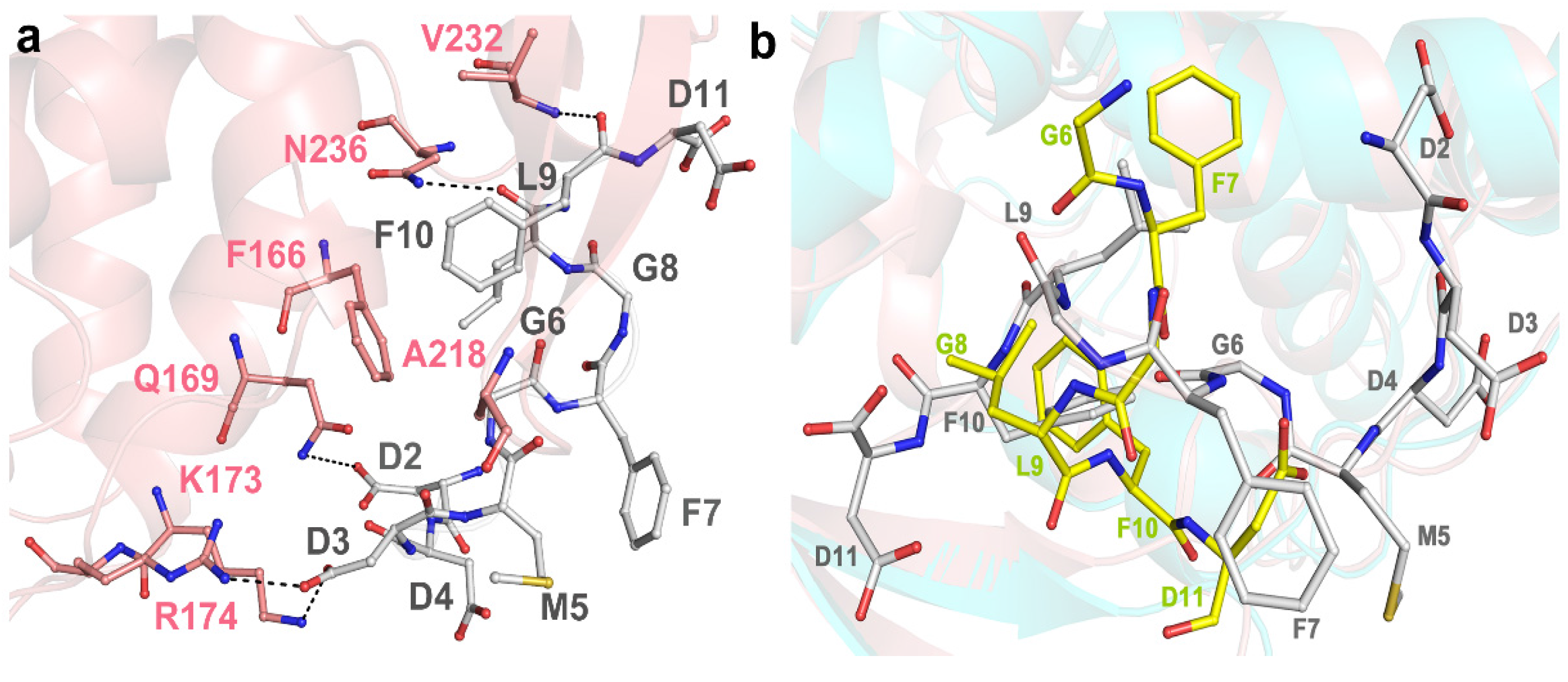
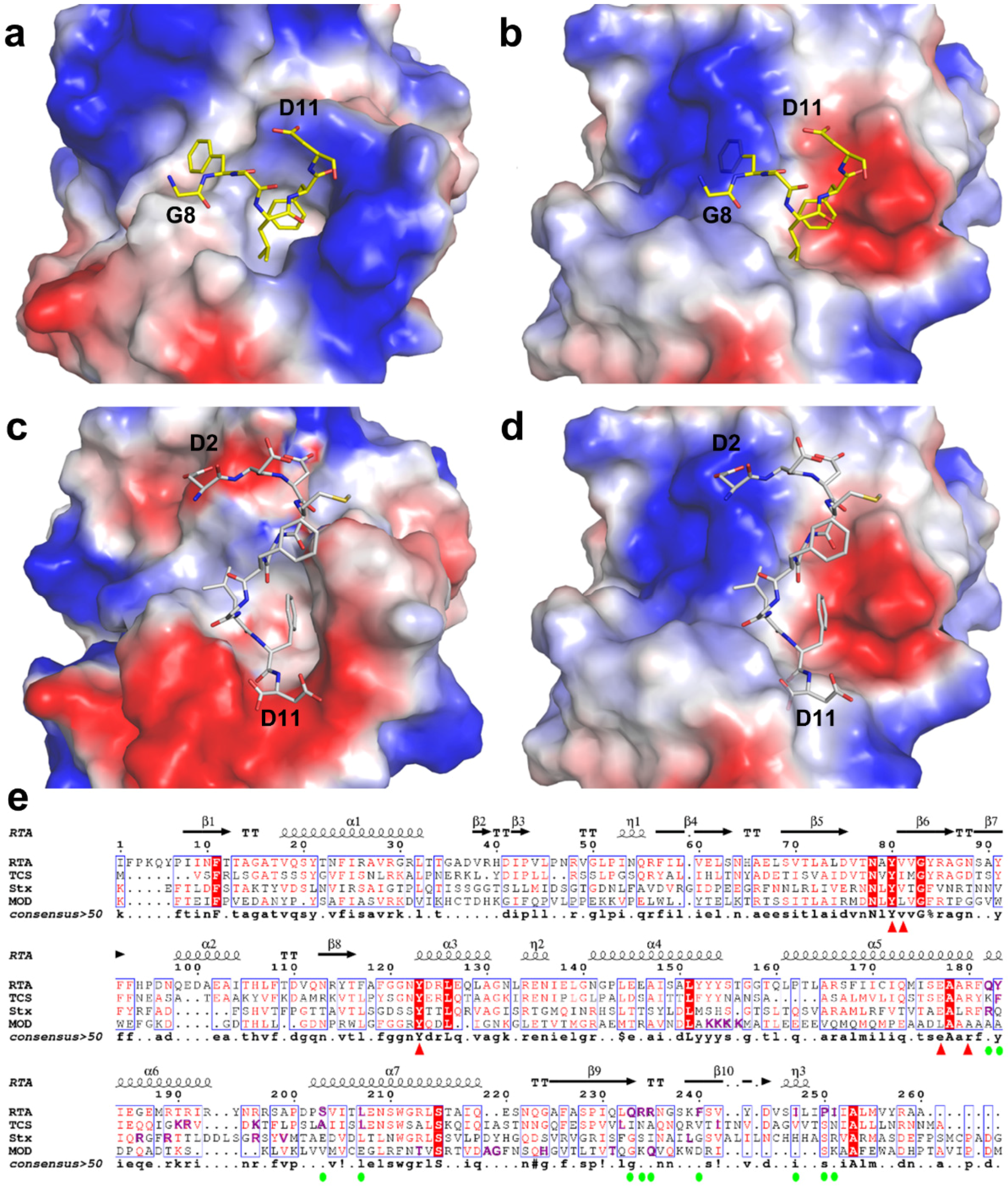
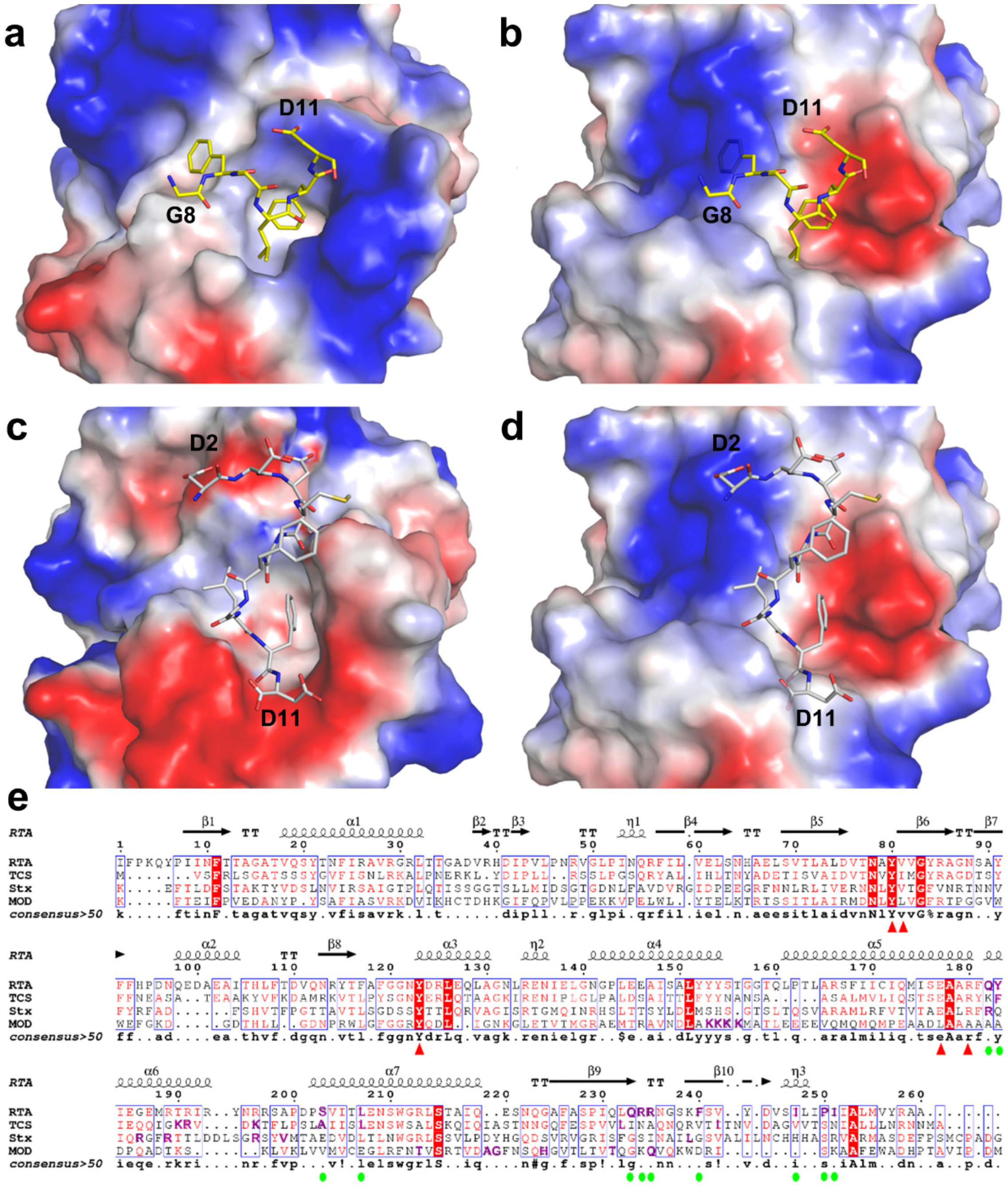
2.3. Structural Analysis and Comparison of P2 Binding Pockets of RTA and TCS
2.4. P2 Protein May Adopt Different Ways of Binding to Different RIPs
3. Discussion
4. Materials and Methods
4.1. Cloning, Expression, and Purification of RTA and P2 Variants
4.2. Crystallization, Data Collection and Processing
4.3. Structure Determination and Refinement
4.4. In Vitro GST-Pull-Down Assays
4.5. Isothermal Titration Calorimetry (ITC)
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Olsnes, S. The history of ricin, abrin and related toxins. Toxicon 2004, 44, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Tsurugi, K. RNA N-glycosidase activity of ricin A-chain. Mechanism of action of the toxic lectin ricin on eukaryotic ribosomes. J. Biol. Chem. 1987, 262, 8128–8130. [Google Scholar] [PubMed]
- Correll, C.C.; Munishkin, A.; Chan, Y.L.; Ren, Z.; Wool, I.G.; Steitz, T.A. Crystal structure of the ribosomal RNA domain essential for binding elongation factors. Proc. Natl. Acad. Sci. USA 1998, 95, 13436–13441. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, L.; Lambert, N.J.; Maklan, E.J.; Horan, L.H.; Noller, H.F. The sarcin-ricin loop of 23S rRNA is essential for assembly of the functional core of the 50S ribosomal subunit. RNA 2008, 14, 1999–2012. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Tumer, N.E. Pokeweed antiviral protein cleaves double-stranded supercoiled DNA using the same active site required to depurinate rRNA. Nucleic Acids Res. 1999, 27, 1900–1905. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Tsurugi, K. The RNA N-glycosidase activity of ricin A-chain. The characteristics of the enzymatic activity of ricin A-chain with ribosomes and with rRNA. J. Biol. Chem. 1988, 263, 8735–8739. [Google Scholar] [PubMed]
- Vater, C.A.; Bartle, L.M.; Leszyk, J.D.; Lambert, J.M.; Goldmacher, V.S. Ricin A chain can be chemically cross-linked to the mammalian ribosomal proteins L9 and L10e. J. Biol. Chem. 1995, 270, 12933–12940. [Google Scholar] [PubMed]
- Chiou, J.C.; Li, X.P.; Remacha, M.; Ballesta, J.P.; Tumer, N.E. The ribosomal stalk is required for ribosome binding, depurination of the rRNA and cytotoxicity of ricin A chain in Saccharomyces cerevisiae. Mol. Microbiol. 2008, 70, 1441–1452. [Google Scholar] [CrossRef] [PubMed]
- Tchorzewski, M. The acidic ribosomal P proteins. Int. J. Biochem. Cell Biol. 2002, 34, 911–915. [Google Scholar] [CrossRef]
- May, K.L.; Li, X.P.; Martinez-Azorin, F.; Ballesta, J.P.; Grela, P.; Tchorzewski, M.; Tumer, N.E. The P1/P2 proteins of the human ribosomal stalk are required for ribosome binding and depurination by ricin in human cells. FEBS J. 2012, 279, 3925–3936. [Google Scholar] [CrossRef] [PubMed]
- Nomura, N.; Honda, T.; Baba, K.; Naganuma, T.; Tanzawa, T.; Arisaka, F.; Noda, M.; Uchiyama, S.; Tanaka, I.; Yao, M.; et al. Archaeal ribosomal stalk protein interacts with translation factors in a nucleotide-independent manner via its conserved C terminus. Proc. Natl. Acad. Sci. USA 2012, 109, 3748–3753. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.S.; Chu, L.O.; Lee, K.M.; Too, P.H.; Ma, K.W.; Sze, K.H.; Zhu, G.; Shaw, P.C.; Wong, K.B. Interaction between trichosanthin, a ribosome-inactivating protein, and the ribosomal stalk protein P2 by chemical shift perturbation and mutagenesis analyses. Nucleic Acids Res. 2007, 35, 1660–1672. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.H.; Hung, F.S.; Chan, D.S.; Shaw, P.C. Trichosanthin interacts with acidic ribosomal proteins P0 and P1 and mitotic checkpoint protein MAD2B. Eur. J. Biochem. 2001, 268, 2107–2112. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Mak, A.N.; Shaw, P.C.; Sze, K.H. Solution structure of an active mutant of maize ribosome-inactivating protein (MOD) and its interaction with the ribosomal stalk protein P2. J. Mol. Biol. 2010, 395, 897–907. [Google Scholar] [CrossRef] [PubMed]
- McCluskey, A.J.; Poon, G.M.; Bolewska-Pedyczak, E.; Srikumar, T.; Jeram, S.M.; Raught, B.; Gariepy, J. The catalytic subunit of Shiga-like toxin 1 interacts with ribosomal stalk proteins and is inhibited by their conserved C-terminal domain. J. Mol. Biol. 2008, 378, 375–386. [Google Scholar] [CrossRef] [PubMed]
- McCluskey, A.J.; Bolewska-Pedyczak, E.; Jarvik, N.; Chen, G.; Sidhu, S.S.; Gariepy, J. Charged and hydrophobic surfaces on the a chain of Shiga-like toxin 1 recognize the C-terminal domain of ribosomal stalk proteins. PLoS ONE 2012, 7, e31191. [Google Scholar] [CrossRef] [PubMed]
- Too, P.H.; Ma, M.K.; Mak, A.N.; Wong, Y.T.; Tung, C.K.; Zhu, G.; Au, S.W.; Wong, K.B.; Shaw, P.C. The C-terminal fragment of the ribosomal P protein complexed to trichosanthin reveals the interaction between the ribosome-inactivating protein and the ribosome. Nucleic Acids Res. 2009, 37, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.M.; Yu, C.W.; Chan, D.S.; Chiu, T.Y.; Zhu, G.; Sze, K.H.; Shaw, P.C.; Wong, K.B. Solution structure of the dimerization domain of ribosomal protein P2 provides insights for the structural organization of eukaryotic stalk. Nucleic Acids Res. 2010, 38, 5206–5216. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.M.; Yu, C.W.; Chiu, T.Y.; Sze, K.H.; Shaw, P.C.; Wong, K.B. Solution structure of the dimerization domain of the eukaryotic stalk P1/P2 complex reveals the structural organization of eukaryotic stalk complex. Nucleic Acids Res. 2012, 40, 3172–3182. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.M.; Yusa, K.; Chu, L.O.; Yu, C.W.; Oono, M.; Miyoshi, T.; Ito, K.; Shaw, P.C.; Wong, K.B.; Uchiumi, T. Solution structure of human P1*P2 heterodimer provides insights into the role of eukaryotic stalk in recruiting the ribosome-inactivating protein trichosanthin to the ribosome. Nucleic Acids Res. 2013, 41, 8776–8787. [Google Scholar] [CrossRef] [PubMed]
- Bargis-Surgey, P.; Lavergne, J.P.; Gonzalo, P.; Vard, C.; Filhol-Cochet, O.; Reboud, J.P. Interaction of elongation factor eEF-2 with ribosomal p proteins. Eur. J. Biochem. 1999, 262, 606–611. [Google Scholar] [CrossRef] [PubMed]
- Helgstrand, M.; Mandava, C.S.; Mulder, F.A.; Liljas, A.; Sanyal, S.; Akke, M. The ribosomal stalk binds to translation factors IF2, EF-Tu, EF-G and RF3 via a conserved region of the L12 C-terminal domain. J. Mol. Biol. 2007, 365, 468–479. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, M.; Kitamyo, M.; Miyoshi, T.; Ito, K.; Uchiumi, T. Analysis of chimeric ribosomal stalk complexes from eukaryotic and bacterial sources: Structural features responsible for specificity of translation factors. Genes Cells 2012, 17, 273–284. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.K.; Wong, E.C.; Lee, K.M.; Wong, K.B. Structures of eukaryotic ribosomal stalk proteins and its complex with trichosanthin, and their implications in recruiting ribosome-inactivating proteins to the ribosomes. Toxins 2015, 7, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Li, X.P.; Kahn, P.C.; Kahn, J.N.; Grela, P.; Tumer, N.E. Arginine residues on the opposite side of the active site stimulate the catalysis of ribosome depurination by ricin A chain by interacting with the P-protein stalk. J. Biol. Chem. 2013, 288, 30270–30284. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.; Ballesta, J.P. The highly conserved protein P0 carboxyl end is essential for ribosome activity only in the absence of proteins P1 and P2. J. Biol. Chem. 1995, 270, 20608–20614. [Google Scholar] [CrossRef] [PubMed]
- Mlsna, D.; Monzingo, A.F.; Katzin, B.J.; Ernst, S.; Robertus, J.D. Structure of recombinant ricin A chain at 2.3 A. Protein Sci. 1993, 2, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Robertus, J.D.; Monzingo, A.F. The structure of ribosome inactivating proteins. Mini Rev. Med. Chem. 2004, 4, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.T.; Ng, Y.M.; Mak, A.N.; Sze, K.H.; Wong, K.B.; Shaw, P.C. Maize ribosome-inactivating protein uses Lys158-Lys161 to interact with ribosomal protein P2 and the strength of interaction is correlated to the biological activities. PLoS ONE 2012, 7, e49608. [Google Scholar] [CrossRef] [PubMed]
- Korennykh, A.V.; Correll, C.C.; Piccirilli, J.A. Evidence for the importance of electrostatics in the function of two distinct families of ribosome inactivating toxins. RNA 2007, 13, 1391–1396. [Google Scholar] [CrossRef] [PubMed]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Macromol. Crystallogr. 1997, 276, 307–326. [Google Scholar]
- Collaborative Computational Project, Number 4. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 1994, 50, 760–763. [Google Scholar] [CrossRef] [PubMed]
- Murshudov, G.N.; Skubak, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. Refmac5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- Davis, I.; Leaver-Fay, A.; Chen, V.; Block, J.; Kapral, G.; Wang, X.; Murray, L.; Arendall, W. Molprobity: All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007, 35, W375–W383. [Google Scholar] [CrossRef] [PubMed]
- Laskowski, R.; Macarthur, M.; Moss, D.; Thornton, J. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- DeLano, W. The PyMOL Molecular Graphics System; De Lano Scientific: San Carlos, CA, USA, 2002. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RTA-C6-P2 | |
|---|---|
| Data collection | |
| Space group | P 1 21 1 |
| Unit cell a, b, c (Å) α, β, γ (°) | 67.45, 59.88, 67.50 90.00, 99.89, 90.00 |
| Resolution range (Å) | 25.00–1.55 (1.61–1.55) a |
| Unique reflections | 76937 |
| Completeness (%) | 97.8 (96.4) |
| <I/σ(I)> | 24.0 (3.5) |
| Rmerge b (%) | 33.9 (4.6) |
| Average redundancy | 3.6 (3.6) |
| Structure refinement | |
| Resolution range (Å) | 24.65–1.55 |
| R-factor c/R-free d (%) | 25.6/21.7 |
| Number of protein atoms | 4366 |
| Number of water atoms | 234 |
| RMSD e bond lengths (Å) | 0.0261 |
| RMSD bond angles (°) | 1.500 |
| Ramachandran plot f (residues, %) | - |
| Most favored (%) | 95.8 |
| Additional allowed (%) | 4.2 |
| Outliers (%) | 0 |
| PDB entry | 5GU4 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shi, W.-W.; Tang, Y.-S.; Sze, S.-Y.; Zhu, Z.-N.; Wong, K.-B.; Shaw, P.-C. Crystal Structure of Ribosome-Inactivating Protein Ricin A Chain in Complex with the C-Terminal Peptide of the Ribosomal Stalk Protein P2. Toxins 2016, 8, 296. https://doi.org/10.3390/toxins8100296
Shi W-W, Tang Y-S, Sze S-Y, Zhu Z-N, Wong K-B, Shaw P-C. Crystal Structure of Ribosome-Inactivating Protein Ricin A Chain in Complex with the C-Terminal Peptide of the Ribosomal Stalk Protein P2. Toxins. 2016; 8(10):296. https://doi.org/10.3390/toxins8100296
Chicago/Turabian StyleShi, Wei-Wei, Yun-Sang Tang, See-Yuen Sze, Zhen-Ning Zhu, Kam-Bo Wong, and Pang-Chui Shaw. 2016. "Crystal Structure of Ribosome-Inactivating Protein Ricin A Chain in Complex with the C-Terminal Peptide of the Ribosomal Stalk Protein P2" Toxins 8, no. 10: 296. https://doi.org/10.3390/toxins8100296
APA StyleShi, W.-W., Tang, Y.-S., Sze, S.-Y., Zhu, Z.-N., Wong, K.-B., & Shaw, P.-C. (2016). Crystal Structure of Ribosome-Inactivating Protein Ricin A Chain in Complex with the C-Terminal Peptide of the Ribosomal Stalk Protein P2. Toxins, 8(10), 296. https://doi.org/10.3390/toxins8100296