
Development of an Enzyme-Linked Immunosorbent Assay Method Specific for the Detection of G-Group Aflatoxins


 ,
, 
Abstract
:1. Introduction
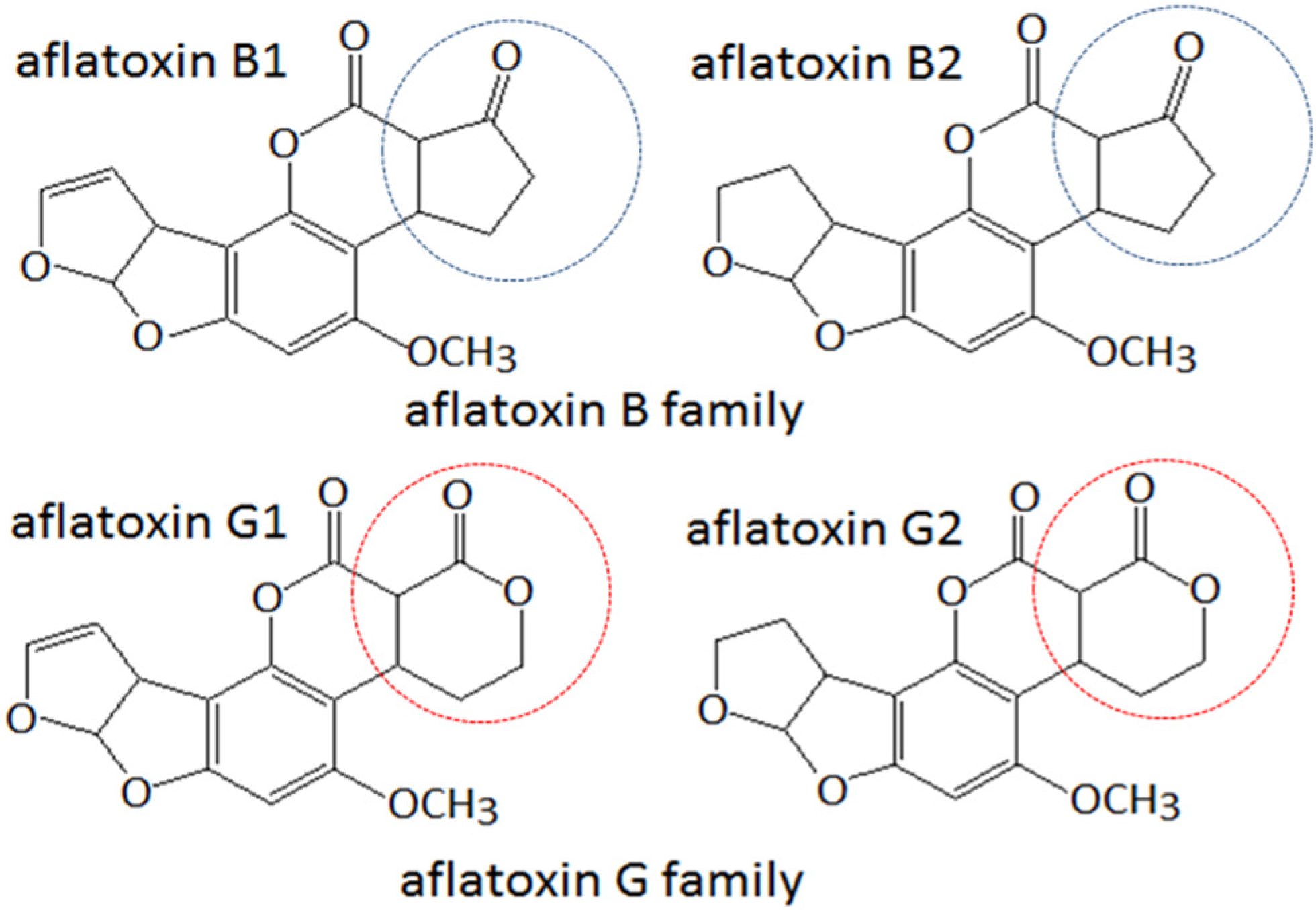
2. Results and Discussion
2.1. Immunization, Cell Fusion, and Screening
2.2. Sensitivity and Specificity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Aflatoxins | 2G6 | 3D4 | 4A4 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| IC50 | CR | Titer | IC50 | CR | Titer | IC50 | CR | Titer | |
| AFB1 | - a | 0 | 2.56 × 105 | - | 0 | 4 × 103 | >500 | <5 | 8 × 103 |
| AFB2 | - | 0 | - | 0 | - | 0 | |||
| AFG1 | 17.18 | 100 | 19.25 | 100 | 20.56 | 100 | |||
| AFG2 | 19.75 | 87 | 29.62 | 65 | 41.96 | 49 | |||
| AFM1 | - | 0 | - | 0 | - | 0 | |||
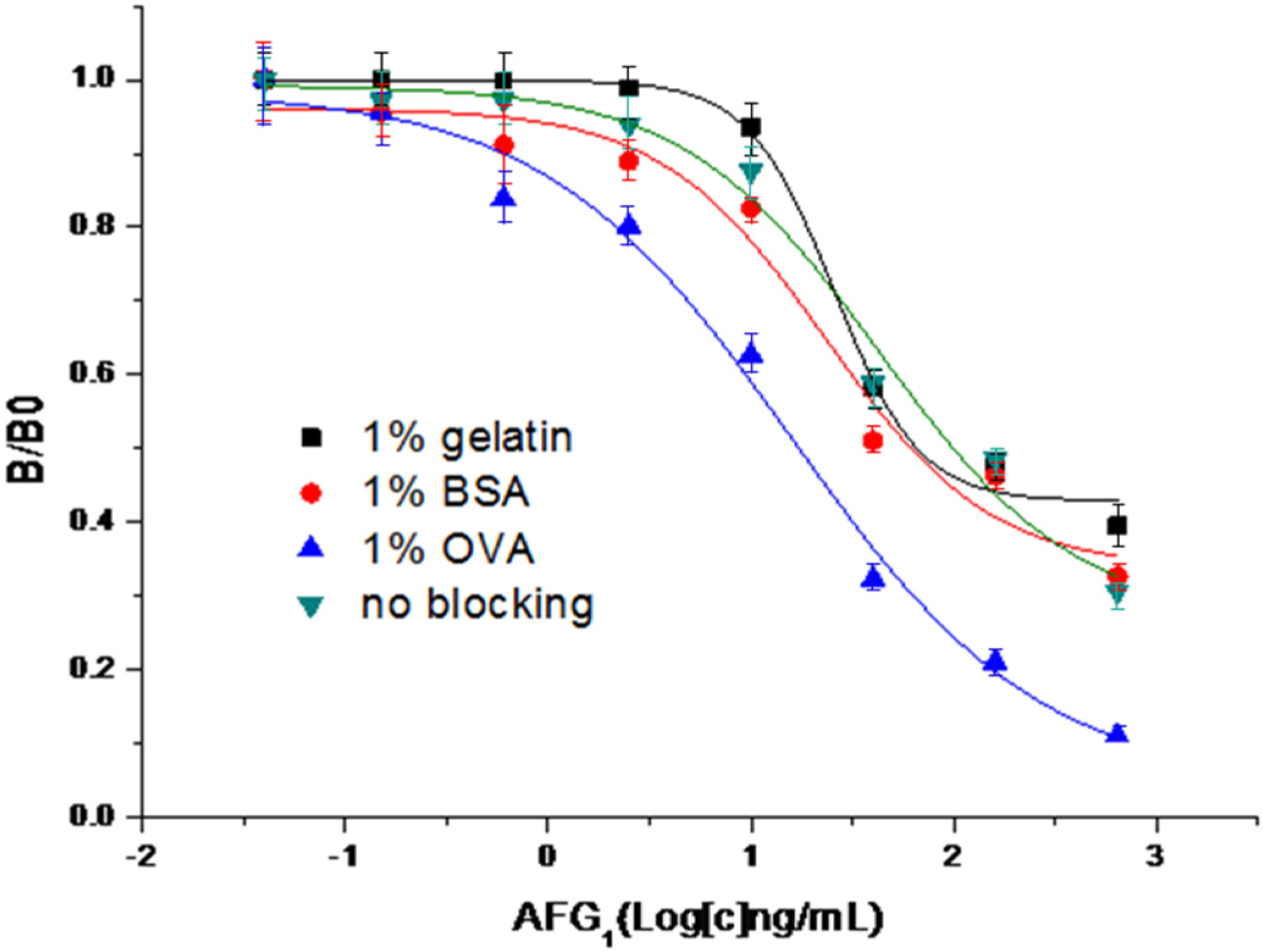
2.3. Optimization of CI-ELISA

2.4. Analysis of Spiked Samples and Naturally Contaminated Samples
| Theoretical (ng·mL−1) a | CI-ELISA | HPLC | ||||
|---|---|---|---|---|---|---|
| Measured (ng·mL−1) | Recovery (%) | Mean ± SD (%) | Measured (ng·mL−1) | Recovery (%) | Mean ± SD (%) | |
| 1 | 1.05 | 105.0 | 101 ± 4.6 | 0.96 | 96.2 | 92.2 ± 3.0 |
| 0.96 | 96.0 | 0.89 | 89.1 | |||
| 1.02 | 102.0 | 0.91 | 91.3 | |||
| 10 | 9.59 | 95.9 | 94.2 ± 3.3 | 9.64 | 96.4 | 96.3 ± 1.6 |
| 8.96 | 89.6 | 9.43 | 94.3 | |||
| 9.70 | 97.0 | 9.81 | 98.1 | |||
| 40 | 41.32 | 103.3 | 102.8 ± 2.6 | 41.79 | 104.5 | 101.7 ± 3.1 |
| 42.28 | 105.7 | 39.01 | 97.5 | |||
| 39.79 | 99.5 | 40.98 | 102.5 | |||
| Samples | CI-ELISA (μg·kg−1) | HPLC (μg·kg−1) | Two Methods RSD (%) | ||
|---|---|---|---|---|---|
| AFG Group Mean ± SD | AFG1 Mean ± SD | AFG2 Mean ± SD | AFG1 + AFG2 | ||
| 1 | 12.6 ± 1.3 | 9.8 ± 0.7 | No | 9.8 | 17.7 |
| 2 | 17.3 ± 1.5 | 13.5 ± 1.0 | 1.2 ± 0.1 | 14.7 | 11.5 |
| 3 | 16.0 ± 1.3 | 13.3 ± 0.6 | 0.9 ± 0.1 | 14.2 | 8.4 |
| 4 | n.d. a | n.d. | n.d. | n.d. | - |
| 5 | n.d. | n.d. | n.d. | n.d. | - |
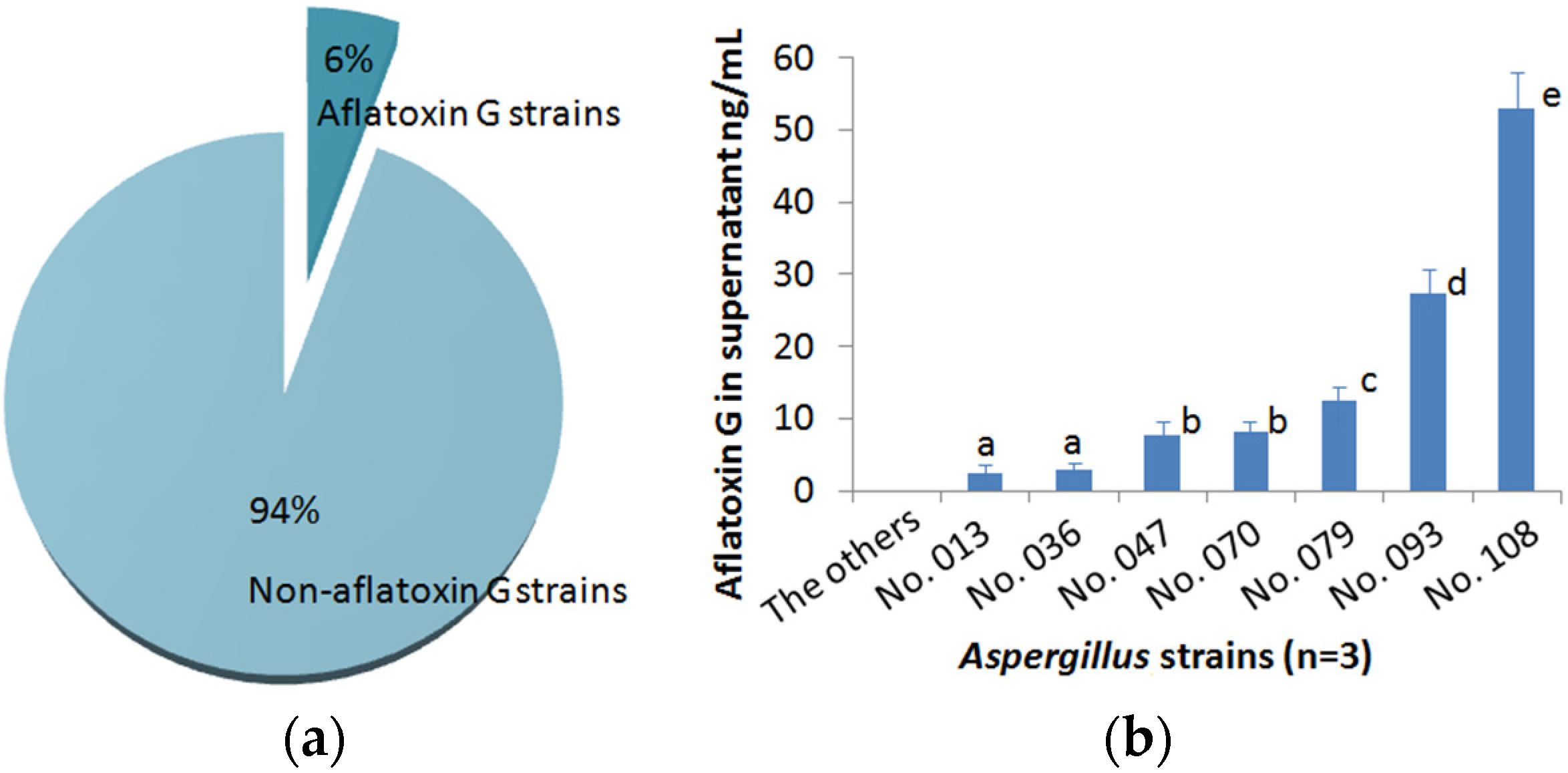
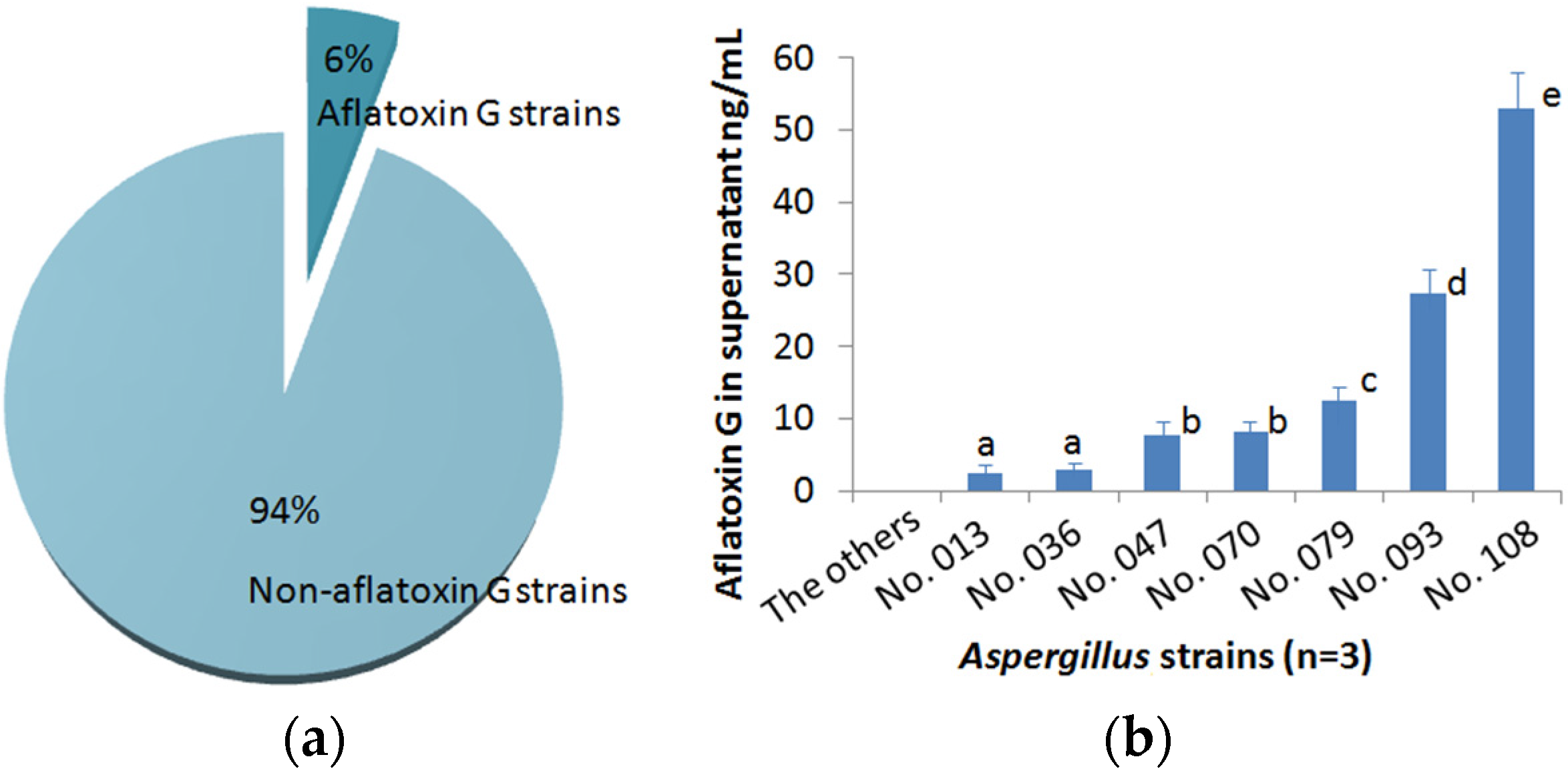
2.5. Identification of Aspergillus Strains that Produce G-Group Aflatoxins
3. Experimental Section
3.1. Chemicals and Instruments
3.2. Solutions, Buffers, and Growth Media
3.3. Preparation of Artificial Antigen
3.4. Immunization
3.5. Cell Fusion and Cloning
3.6. ELISA Screening
3.7. Determination of Isotype, Sensitivity, and Specificity
3.8. Optimization of CI-ELISA
3.9. Analysis of Peanut Samples Spiked with and Naturally Contaminated by Aflatoxin G1
3.10. Identification of G-Group Aflatoxin-Producing Fungal Strains
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hoffmeister, D.; Keller, N.P. Natural products of filamentous fungi: Enzyme, genes, and their regulation. Nat. Prod. Rep. 2007, 24, 393–416. [Google Scholar] [CrossRef] [PubMed]
- Velazhahan, R.; Vijayanandraj, S.; Vijayasamundeeswari, A.; Paranidharan, V.; Samiyappan, R.; Iwamoto, T.; Friebe, B.; Muthukrishnan, S. Detoxification of aflatoxins by seed extracts of the medicinal plant, Trachyspermum ammi (L.) Sprague ex Turrill-Structural analysis and biological toxicity of degradation product of aflatoxin G1. Food Control 2010, 21, 719–725. [Google Scholar] [CrossRef]
- Sherif, S.O.; Salama, E.E.; Abdel-Wahhab, M.A. Mycotoxins and child health: The need for health risk assessment. Int. J. Hyg. Environ. Health 2010, 212, 347–368. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, K.C.; Kobbeman, K.; Montalbano, B.G.; Cotty, P.J. Aflatoxin-producing Aspergillus species from Thailand. Int. J. Food Microbiol. 2007, 114, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Khayoon, W.S.; Saad, B.; Yan, C.B.; Hashim, N.H.; Ali, A.S.M.; Salleh, M.I.; Salleh, B. Determination of aflatoxins in animal feeds by HPLC with multifunctional column clean-up. Food Chem. 2010, 118, 882–886. [Google Scholar] [CrossRef]
- Codex Alimentarius Commission. Joint FAO/WHO food standards programme, codex committee on food additives and contaminants. In Proceedings of the Thirtythird Session CODEX, Hague, The Netherlands, 12–16 March 2001.
- Commission Regulation (EU). No 165/2010 of 26 February 2010 Amending Regulation (EC) No 1881/2006 Setting Maximum Levels for Certain Contaminants in Foodstuffs as Regards Aflatoxins. Available online: https://www.fsai.ie/uploadedFiles/Reg165_2010.pdf (accessed on 15 December 2015).
- Kamkar, A. A study on the occurrence of aflatoxin M1 in Iranian Feta cheese. Food Control 2006, 17, 768–775. [Google Scholar] [CrossRef]
- Manetta, A.C.; di Giuseppe, L.; Giammarco, M.; Fusaro, I.; Simonella, A.; Gramenzi, A.; Formigoni, A. High-performance liquid chromatography with post-column derivatisation and fluorescence detection for sensitive determination of aflatoxin M1 in milk and cheese. J. Chromatogr. A 2005, 1083, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Edinboro, L.E.; Karnes, H.T. Determination of aflatoxin B1 in sidestream cigarette smoke by immunoaffinity column extraction coupled with liquid chromatography/mass spectrometry. J. Chromatogr. A 2005, 1083, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, P. Rapid screening of mycotoxins in liquid milk and milk powder by automated size-exclusion SPE-UPLC-MS/MS and quantification of matrix effects over the whole chromatographic run. Food Chem. 2015, 173, 897–904. [Google Scholar] [CrossRef] [PubMed]
- Dzantiev, B.B.; Byzova, N.A.; Urusov, A.E.; Zherdev, A.V. Immunochromatographic methods in food analysis. Trends Anal. Chem. 2014, 55, 81–93. [Google Scholar] [CrossRef]
- Eslami, M.; Mashak, Z.; Heshmati, A.; Shokrzadeh, M.; Sasan, A.; Nejad, M. Determination of aflatoxin B1 levels in Iranian rice by ELISA method. Toxin Rev. 2015, 34, 125–128. [Google Scholar] [CrossRef]
- Christoforidou, S.; Malissiova, E.; Gortzi, O.; Hadjichristodoulou, C. Comparative evaluation of ELISA kits′ reliability for the aflatoxin M1 determination in goat milk. Eur. Food Res. Technol. 2015, 240, 701–706. [Google Scholar] [CrossRef]
- Zhang, D.; Li, P.; Zhang, Q.; Zhang, W.; Huang, Y.; Ding, X.; Jiang, J. Production of ultrasensitive generic monoclonal antibodies against major aflatoxins using a modified two-step screening procedure. Anal. Chim. Acta 2009, 636, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Holtzapple, C.K.; Carlin, R.J.; Rose, B.G.; Kubena, L.F.; Stanker, L.H. Characterization of monoclonal antibodies to aflatoxin M1 and molecular modeling studies of related aflatoxins. Mol. Immunol. 1996, 33, 939–946. [Google Scholar] [CrossRef]
- Zhang, J.; Li, P.; Zhang, W.; Zhang, Q.; Ding, X.; Chen, X.; Wu, W.; Zhang, X. Production and characterization of monoclonal antibodies against aflatoxin G1. Hybridoma 2009, 28, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Santacroce, M.P.; Conversano, M.C.; Casalino, E.; Lai, O.; Zizzadoro, C.; Centoducati, G.; Crescenzo, G. Aflatoxins in aquatic species: Metabolism, toxicity and perspectives. Rev. Fish Biol. Fish. 2008, 18, 99–130. [Google Scholar] [CrossRef]
- Lee, N.A.; Wang, S.; Allan, R.D.; Kennedy, I.R. A rapid aflatoxin B1 ELISA: Development and validation with reduced matrix effects for peanuts, corn, pistachio and soybean. J. Agric. Food Chem. 2004, 52, 2746–2755. [Google Scholar] [CrossRef] [PubMed]
- Chu, F.S.; Steinert, B.W.; Gaur, P.K. Production and characterization of antibody against aflatoxin G1. J. Food Saf. 1985, 7, 161–170. [Google Scholar] [CrossRef]
- Galfre, G.; Milstein, C. Preparation of monoclonal antibodies: Strategies and procedures. Methods Enzymol. 1981, 73, 3–46. [Google Scholar] [PubMed]
- Yu, K.O.A.; Im, J.S.; Illarianov, P.A.; Ndonye, R.M.; Howell, A.R.; Besra, G.S.; Porcelli, S.A. Production and characterization of monoclonal antibodies against complexes of the NKT cell ligand α-galactosylceramide bound to mouse CD1d. J. Immunol. Methods 2007, 323, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Cervino, C.; Weber, E.; Knopp, D.; Niessner, R. Comparison of hybridoma screening methods for the efficient detection of high-affinity hapten-specific monoclonal antibodies. J. Immunol. Methods 2008, 329, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Gao, A.; Chen, Q.; Cheng, Y.; Lei, J.; Zeng, L. Preparation of monoclonal antibodies against a derivative of semicarbazide as a metabolic target of nitrofurazone. Anal. Chim. Acta 2007, 592, 58–63. [Google Scholar] [CrossRef]
- Groopman, J.D.; Trudelt, L.J.; Donahuet, P.R.; Marshak-rothstein, A.; Wogant, G.N. High-affinity monoclonal antibodies for aflatoxins and their application to solid-phase immunoassays. Proc. Natl. Acad. Sci. USA 1984, 81, 7728–7731. [Google Scholar] [CrossRef] [PubMed]
- Magliulo, M.; Mirasoli, M.; Simoni, P.; Lelli, R.; Portanti, O.; Roda, A. Development and validation of an ultrasensitive chemiluminescent enzyme immunoassay for aflatoxin M1 in milk. J. Agric. Food Chem. 2005, 53, 3300–3305. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Sun, Q.; Hu, B.; Shen, Q.; Yang, G.; Liang, X.; Sun, X.; Liu, F. Development of a sensitive ELISA for the analysis of the organophosphorous insecticide fenthion in fruit samples. Food Chem. 2008, 106, 1278–1284. [Google Scholar] [CrossRef]
- Sapsford, K.E.; Taitt, C.R.; Fertig, S.; Moore, M.H.; Lassman, M.E.; Maragos, C.M.; Shriver-Lake, L.C. Indirect competitive immunoassay for detection of aflatoxin B1 in corn and nut products using the array biosensor. Biosens. Bioelectron. 2006, 21, 2298–2305. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, P.; Zhang, Y.; Shao, M. Determination Aflatoxins Content in Liquid Chromatography and Fluorometer. GB/T 18979-2003. Available online: http://d.g.wanfangdata.com.cn/ExternalResource-xdyq201003026%5E2.aspx (accessed on 17 December 2015). (In Chinese)
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, P.; Zhou, Q.; Wang, T.; Zhou, H.; Zhang, W.; Ding, X.; Zhang, Z.; Chang, P.-K.; Zhang, Q. Development of an Enzyme-Linked Immunosorbent Assay Method Specific for the Detection of G-Group Aflatoxins. Toxins 2016, 8, 5. https://doi.org/10.3390/toxins8010005
Li P, Zhou Q, Wang T, Zhou H, Zhang W, Ding X, Zhang Z, Chang P-K, Zhang Q. Development of an Enzyme-Linked Immunosorbent Assay Method Specific for the Detection of G-Group Aflatoxins. Toxins. 2016; 8(1):5. https://doi.org/10.3390/toxins8010005
Chicago/Turabian StyleLi, Peiwu, Qian Zhou, Ting Wang, Haiyan Zhou, Wen Zhang, Xiaoxia Ding, Zhaowei Zhang, Perng-Kuang Chang, and Qi Zhang. 2016. "Development of an Enzyme-Linked Immunosorbent Assay Method Specific for the Detection of G-Group Aflatoxins" Toxins 8, no. 1: 5. https://doi.org/10.3390/toxins8010005
APA StyleLi, P., Zhou, Q., Wang, T., Zhou, H., Zhang, W., Ding, X., Zhang, Z., Chang, P.-K., & Zhang, Q. (2016). Development of an Enzyme-Linked Immunosorbent Assay Method Specific for the Detection of G-Group Aflatoxins. Toxins, 8(1), 5. https://doi.org/10.3390/toxins8010005