
Quantitative Analysis of Staphylococcal Enterotoxins A and B in Food Matrices Using Ultra High-Performance Liquid Chromatography Tandem Mass Spectrometry (UPLC-MS/MS)

Abstract
:1. Introduction
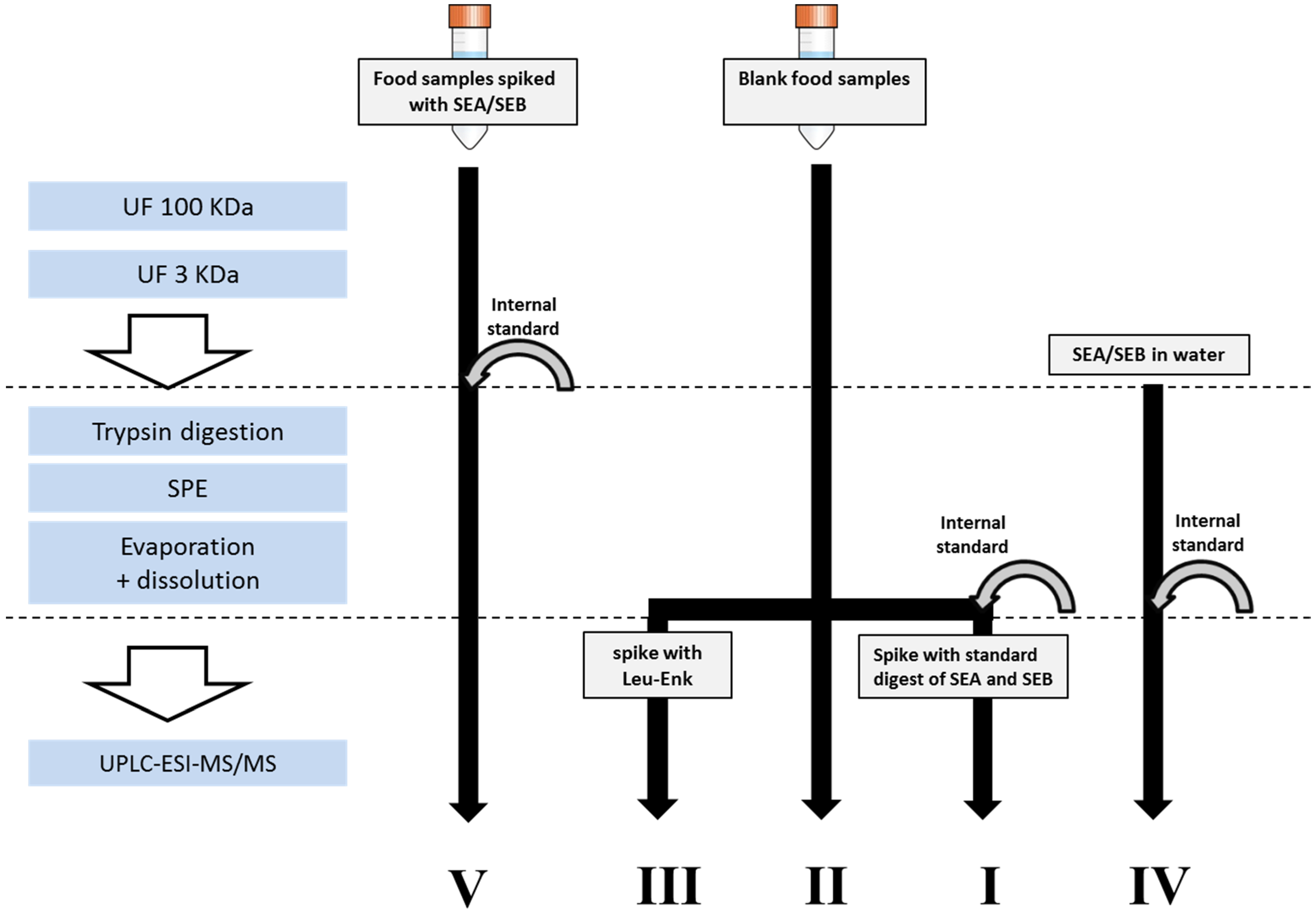
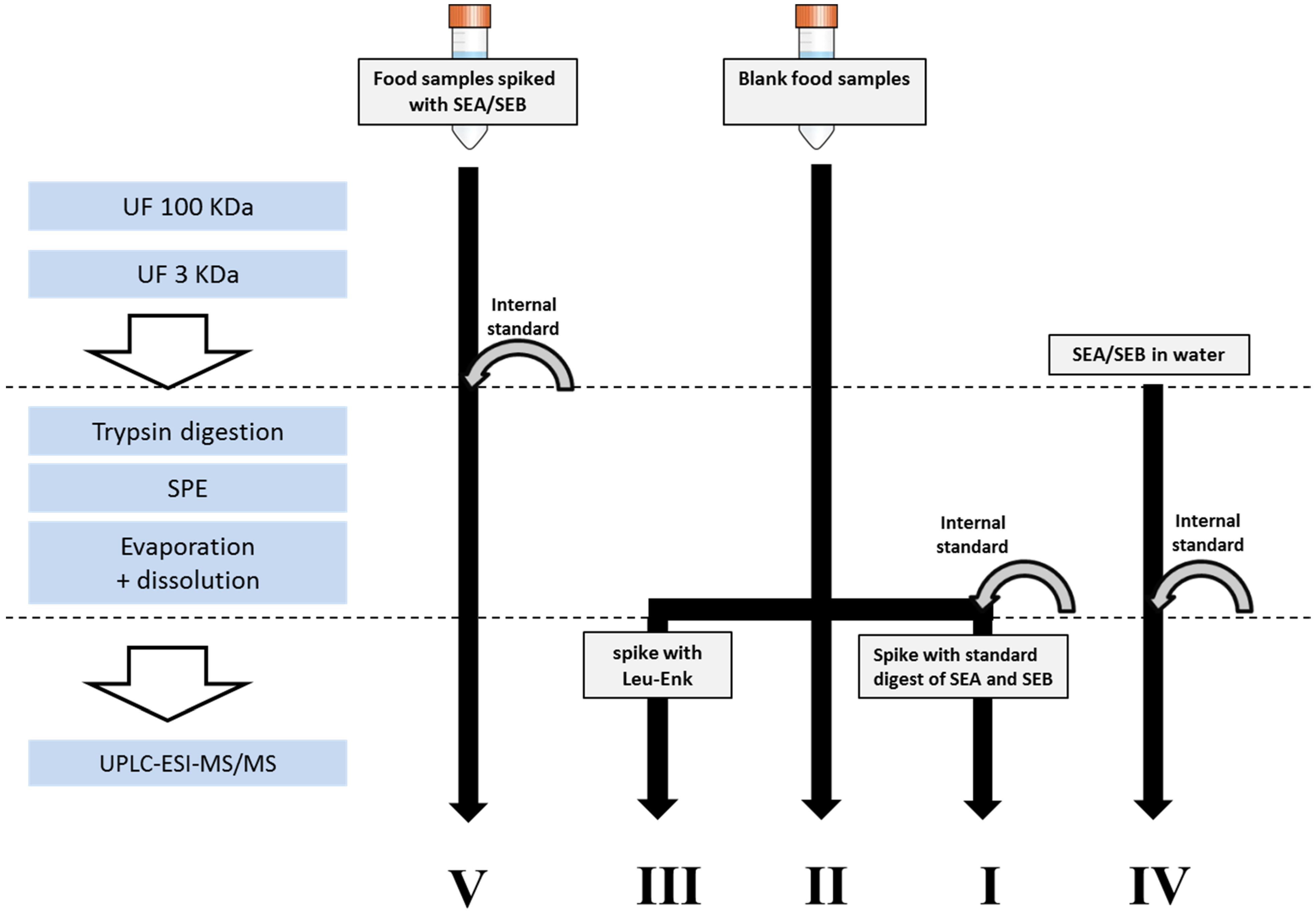
2. Results and Discussion

{kind=link}
{kind=link}
{kind=link}
| Toxin | Peptide Sequence | Peptide Mass | Charge State | Q1 m/z | Q2 m/z | Retention Time (min) |
|---|---|---|---|---|---|---|
| SEA | GLIVFHTSTEPSVNYDLFGA QGQYSNTLLR | 3326.7 | 3+ | 1109.9 | 1454.74 | 9.2 |
| 1307.67 | ||||||
| 1250.65 | ||||||
| 1179.61 | ||||||
| GFFTDHSWYNDLLVDFDSK | 2305.0 | 3+ | 769.4 | 1165.57 | 10.0 | |
| 1051.53 | ||||||
| 936.50 | ||||||
| 823.42 | ||||||
| YNLYNSDVFDGK | 1433.6 | 2+ | 717.83 | 1157.55 | 6.3 | |
| 1044.46 | ||||||
| 881.40 | ||||||
| 767.36 | ||||||
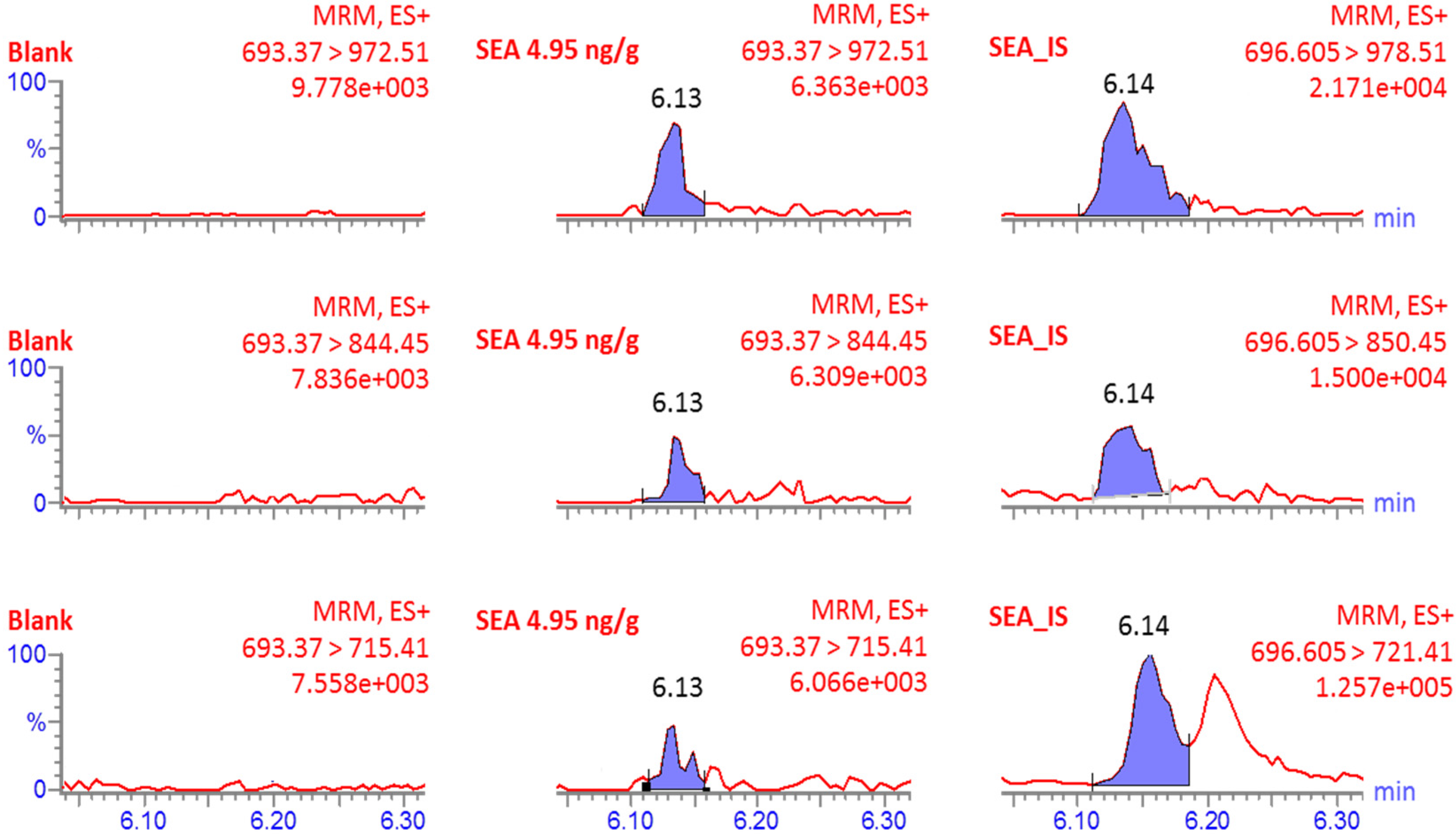
| NVTVQELDLQAR | 1384.7 | 2+ | 693.37 | 1071.58 | 6.0 | |
| Qu. 972.51 | ||||||
| Co. 844.45 | ||||||
| 715.41 | ||||||
| SELQGTALGNLK | 1229.7 | 2+ | 615.84 | 1014.59 | 5.6 | |
| 901.51 | ||||||
| 773.45 | ||||||
| 716.43 | ||||||
| ESHDQFLQHTILFK | 1741.9 | 3+ | 581.63 | 999.60 | 6.8 | |
| 886.51 | ||||||
| 758.46 | ||||||
| 621.40 | ||||||
| VPINLWLDGK | 1153.6 | 2+ | 577.83 | 1055.59 | 7.8 | |
| 958.54 | ||||||
| 845.45 | ||||||
| 731.41 | ||||||
| 618.32 | ||||||
| QNTVPLETVK | 1127.6 | 2+ | 564.82 | 886.52 | 4.8 | |
| 785.48 | ||||||
| 686.41 | ||||||
| 589.36 | ||||||
| Internal standard ISA13C6 | NVTVQELDL[13C6]QAR | 1391.2 | 2+ | 696.60 | 1077.58 | 6.0 |
| Qu. 978.51 | ||||||
| Co. 850.45 | ||||||
| 721.41 |
| Toxin | Peptide Sequence | Peptide Mass | Charge State | Q1 m/z | Q2 m/z | Retention Time (min) |
|---|---|---|---|---|---|---|
| SEB | SIDQFLYFDLIYSIK | 1864.0 | 2+ | 932.99 | 1421.77 | 11.0 |
| 1274.70 | ||||||
| 1161.62 | ||||||
| 998.56 | ||||||
| LYEFNNSPYETGYIK | 1836.9 | 2+ | 919.44 | 1285.61 | 6.6 | |
| 1171.56 | ||||||
| 1057.52 | ||||||
| 970.49 | ||||||
| VLYDDNHVSAINVK | 1585.5 | 2+ | 793.91 | 1211.60 | 5.2 | |
| 1096.57 | ||||||
| 981.55 | ||||||
| 867.50 | ||||||
| VTAQELDYLTR | 1307.7 | 2+ | 654.84 | 1037.53 | 6.2 | |
| Qu. 909.47 | ||||||
| Co. 780.43 | ||||||
| 667.34 | ||||||
| NLLSFDVQTNK | 1277.7 | 2+ | 639.84 | 1051.54 | 6.9 | |
| 938.46 | ||||||
| 851.43 | ||||||
| 704.36 | ||||||
| YLMMYNDNK | 1190.5 | 2+ | 596.26 | 1028.45 | 5.3 | |
| 915.37 | ||||||
| 784.33 | ||||||
| 653.29 | ||||||
| IEVYLTTK | 965.5 | 2+ | 483.78 | 853.47 | 5.5 | |
| 724.42 | ||||||
| 625.36 | ||||||
| LGNYDNVR | 949.5 | 2+ | 475.74 | 837.38 | 3.4 | |
| 780.36 | ||||||
| 666.32 | ||||||
| 503.26 | ||||||
| Internal standard ISB13C6 | VTAQELDYL[13C6]TR | 1314.7 | 2+ | 658.33 | 1043.53 | 6.2 |
| Qu. 915.47 | ||||||
| Co. 786.43 | ||||||
| 673.34 |
2.1. Validation Design
2.1.1. Specificity
2.1.2. Calibration and Linearity
| Enterotoxin | Matrix | Calibration Curve | ||
|---|---|---|---|---|
| Slope | y-intercept | R2 | ||
| SEA | Milk | 0.11 | −0.06 | 0.9582 |
| Milk | 0.10 | 0.01 | 0.9858 | |
| Shrimps | 0.12 | 1 | 0.9563 | |
| Shrimps | 0.11 | 0.62 | 0.9503 | |
| SEB | Milk | 0.11 | −0.08 | 0.9183 |
| Milk | 0.11 | −0.06 | 0.9709 | |
| Shrimps | 0.19 | 0.46 | 0.9645 | |
| Shrimps | 0.13 | 0.06 | 0.9429 | |
2.1.3. Recovery in Sample Preparation and Matrix Effects in ESI-MS
| Milk | SEA Added (ng/g) | Recovery in Sample Preparation (%) | Matrix Effect Suppression in ESI-MS (%) | |
| Calculated for SEs (Va/Ia) | Calculated for SEs (Ia/IVa) | Calculated for IS (Ib/IVb) | ||
| 2.5 | 2.9 | 73 | 73 | |
| 5 | 4.6 | 75 | 75 | |
| 10 | 3.5 | 75 | 75 | |
| 15 | 4.6 | 72 | 74 | |
| SEB added (ng/g) | ||||
| 2.5 | 6.1 | 58 | 64 | |
| 5 | 6.3 | 69 | 65 | |
| 10 | 7.5 | 68 | 75 | |
| 15 | 7.3 | 65 | 65 | |
| Shrimp | SEA added (ng/g) | |||
| 10 | 6.0 | 75 | 77 | |
| 15 | 6.3 | 75 | 76 | |
| SEB added (ng/g) | ||||
| 10 | 6.3 | 72 | 72 | |
| 15 | 6.6 | 74 | 73 | |
2.1.4. Trueness, Reproducibility, LOD and LOQ
| Milk | SEA Added (ng/g) | n | SEA Found Mean (ng/g) | Trueness (%) | In house reproducibility RSD (%) |
| 2.5 | 6 | 1.9 | 74 | 22 | |
| 5 | 6 | 4.6 | 93 | 30 | |
| 10 | 6 | 9.2 | 92 | 21 | |
| 15 | 6 | 15.4 | 103 | 9 | |
| SEB added (ng/g) | |||||
| 2.5 | 6 | 2.2 | 87 | 23 | |
| 5 | 6 | 4.7 | 93 | 8 | |
| 10 | 6 | 10.9 | 109 | 9 | |
| 15 | 6 | 18.0 | 120 | 11 | |
| Shrimp | SEA added (ng/g) | ||||
| 2.5 | 2 | 7 | 281 | 9 | |
| 10 | 4 | 10.6 | 106 | 15 | |
| 15 | 4 | 17.4 | 116 | 5 | |
| SEB added (ng/g) | |||||
| 2.5 | 2 | 3.6 | 143 | 41 | |
| 10 | 4 | 8.5 | 85 | 25 | |
| 15 | 4 | 14.8 | 99 | 9 |
| Matrix | Nominal Concentration (ng/g) | UPLC-ESI-MS/MS (NFA) (ng/g) | ELISA (ANSES) (ng/g) |
|---|---|---|---|
| Milk | 2.47 | 2.98 | 1.80 |
| Milk | 4.95 | 3.55 | 3.77 |
| Cream dessert | 9.89 | 2.30 | 9.03 |
| Ready-to-eat-food | 0.22 | 2.68 | 0.14 |

| Author of the Method | Matrix | Toxin | Extraction | Detection | Standards | Analyte | LOD | LOQ |
|---|---|---|---|---|---|---|---|---|
| Kientz et al., (1997) [35] | Water with sodium phosphate | SEB | Dialysis, digestion | QqQ | N/A | Proteotypic peptides | 100 ppb | N/A |
| Nedelkov et al., (2003) [36] | Mushroom | SEB | Centrifugation, spiking of supernatant, Immunocapture (on sensor chip) | MALDI-TOF | N/A | Whole protein | 1 ppb (in extract) | N/A |
| Callahan et al., (2006) [21] | Apple juice | SEB | UF (MWCO 5 and 10 kDa), digestion | QTOF | Surrogate internal standard. | Proteotypic peptides | 60 ppb | 100 ppb |
| QqQ | ||||||||
| Dupuis et al., (2008) [22] | Cheese, Coco- pearls | 13 SEs | Precipitation, Dialysis, immunocapture, SDS-PAGE, in-gel digestion | QTOF | PSAQ (full-length isotope labeled SEs) | Proteotypic peptides | 1.5 ppb | 1.5 ppb |
| Sospedra et al., (2011) [23] | Milk | SEA | SDS-PAGE, Digestion | MALDI-TOF | Peptide calibration standards | Proteotypic peptides | N/A | N/A |
| Bao et al., (2011) [24] | Raw chicken meat | SEB | Protein precipitation, digestion, UF (MWCO 10 kDa) | QIT | Acetic anhydrid label surrogate standards | Proteotypic peptides | 6 ppb | 6 ppb |
| Sospedra et al., (2012) [25] | Milk, Apple juice, Orange juice | SEA, SEB | Precipitation | QqQ | Standard curve (external calibration) | Whole protein | 25 ppb | 50 ppb |
| Present method | Milk, Shrimps | SEA, SEB | Precipitation, UF (MWCO 100 and 3 kDa), digestion | QqQ | Synthetic 13C-labeled proteotypic peptides as internal standards | Proteotypic peptides | 2.5 ppb | Milk: 2.5 ppb |
| Shrimp: 5 ppb |
3. Experimental Section
3.1. Reagents
3.2. Materials
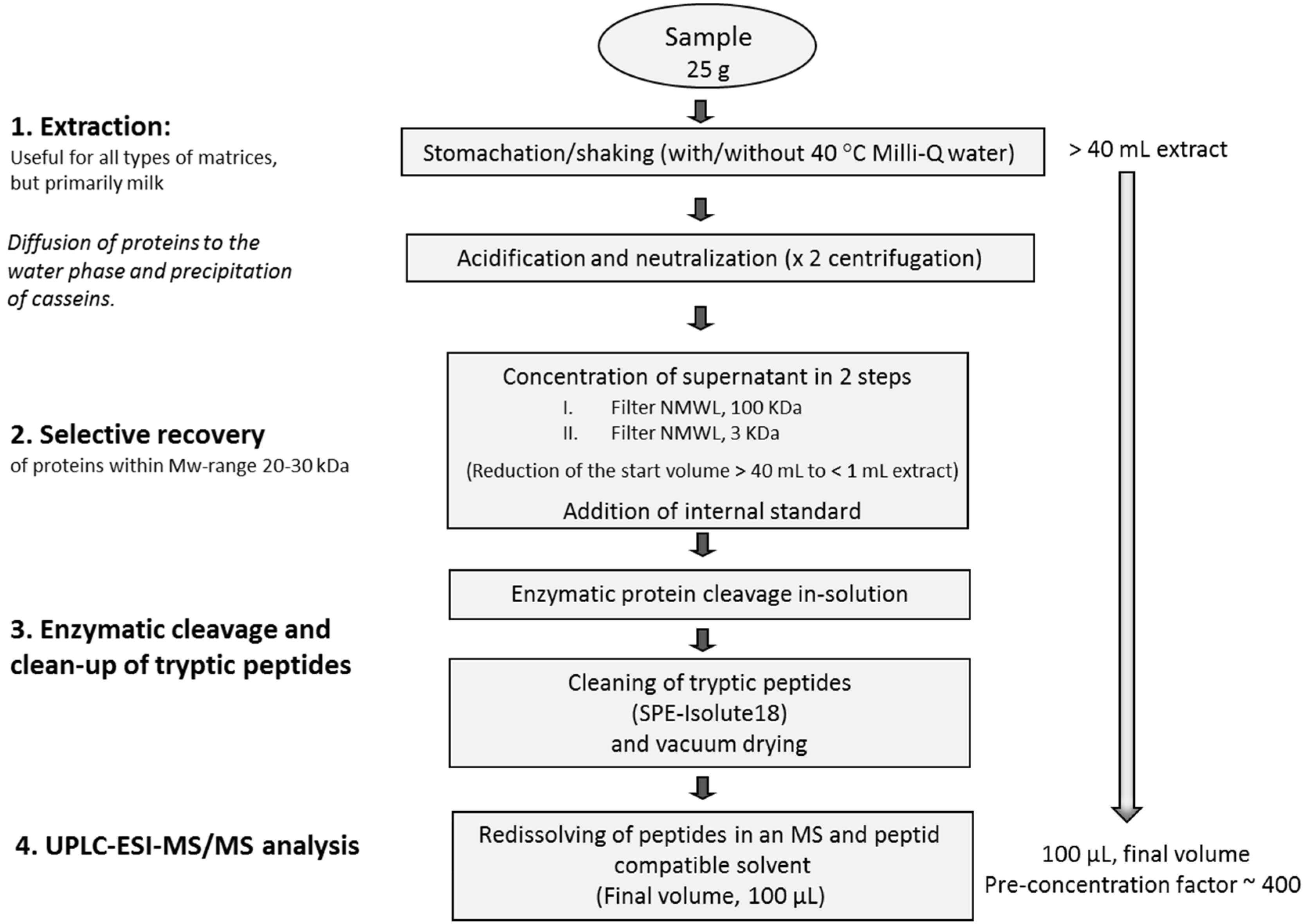
3.3. Sample Preparation
3.3.1. Extraction and Concentration of Enterotoxins
3.3.2. Enzymatic Digestion and Cleaning
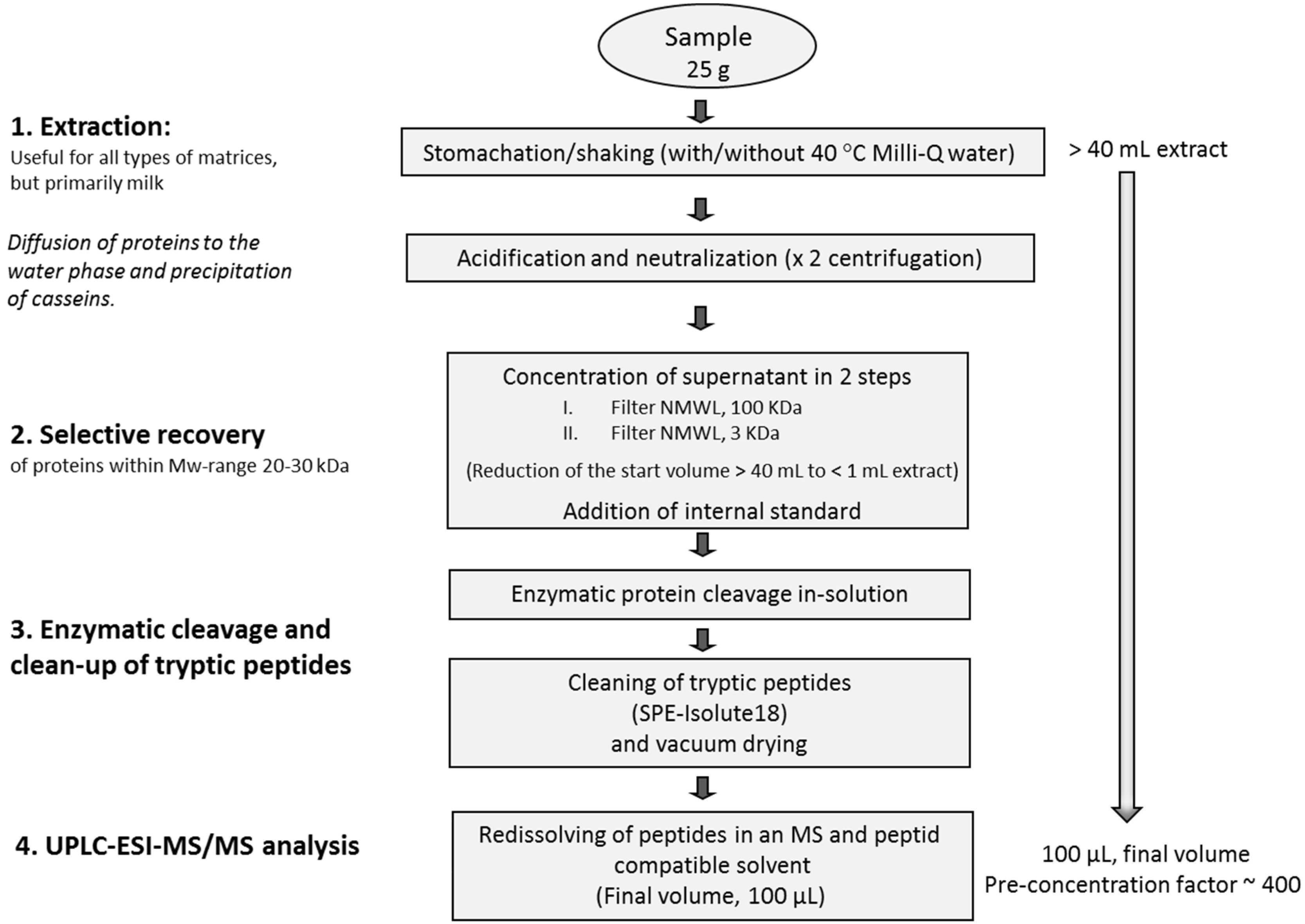
3.4. UPLC-ESI-MS/MS
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mead, P.S.; Slutsker, L.; Dietz, V.; McCaig, L.F.; Bresee, J.S.; Shapiro, C.; Griffin, P.M.; Tauxe, R.V. Food-related illness and death in the United States. Emerg. Infect. Dis. 1999, 5, 607–625. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, T.; Tamate, N.; Yamaguchi, K.; Makino, S. Mass outbreak of food poisoning disease caused by small amounts of staphylococcal enterotoxins A and H. Appl. Environ. Microbiol. 2005, 71, 2793–2795. [Google Scholar] [CrossRef] [PubMed]
- Asao, T.; Kumeda, Y.; Kawai, T.; Shibata, T.; Oda, H.; Haruki, K.; Nakazawa, H.; Kozaki, S. An extensive outbreak of staphylococcal food poisoning due to low-fat milk in Japan: Estimation of enterotoxin A in the incriminated milk and powdered skim milk. Epidemiol. Infect. 2003, 130, 33–40. [Google Scholar] [CrossRef] [PubMed]
- European Food Safety Authority (EFSA). The Community Summary Report on Trends and Sources of Zoonoses, Zoonotic Agents and food-borne outbreaks in the European Union in 2008. EFSA J. 2010, 8, 1496. [Google Scholar] [CrossRef]
- Ler, S.G.; Lee, F.K.; Gopalakrishnakone, P. Trends in detection of warfare agents. Detection methods for ricin, staphylococcal enterotoxin B and T-2 toxin. J. Chromatogr. A 2006, 1133, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Greenfield, R.A.; Brown, B.R.; Hutchins, J.B.; Iandolo, J.J.; Jackson, R.; Slater, L.N.; Bronze, M.S. Microbiological, biological, and chemical weapons of warfare and terrorism. Am. J. Med. Sci. 2002, 323, 326–340. [Google Scholar] [CrossRef] [PubMed]
- Hennekinne, J.A.; Ostyn, A.; Guillier, F.; Herbin, S.; Prufer, A.L.; Dragacci, S. How should staphylococcal food poisoning outbreaks be characterized? Toxins 2010, 2, 2106–2116. [Google Scholar] [CrossRef] [PubMed]
- Evenson, M.L.; Hinds, M.W.; Bernstein, R.S.; Bergdoll, M.S. Estimation of human dose of staphylococcal enterotoxin A from a large outbreak of staphylococcal food poisoning involving chocolate milk. Int. J. Food Microbiol. 1988, 7, 311–316. [Google Scholar] [CrossRef]
- Lee, Y.D.; Moon, B.Y.; Park, J.H.; Chang, H.I.; Kim, W.J. Expression of enterotoxin genes in Staphylococcus aureus isolates based on mRNA analysis. J. Microbiol. Biotechnol. 2007, 17, 461–467. [Google Scholar] [PubMed]
- Akineden, O.; Hassan, A.A.; Schneider, E.; Usleber, E. Enterotoxigenic properties of Staphylococcus aureus isolated from goats’ milk cheese. Int. J. Food Microbiol. 2008, 124, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Duquenne, M.; Fleurot, I.; Aigle, M.; Darrigo, C.; Borezee-Durant, E.; Derzelle, S.; Bouix, M.; Deperrois-Lafarge, V.; Delacroix-Buchet, A. Tool for quantification of staphylococcal enterotoxin gene expression in cheese. Appl. Environ. Microbiol. 2010, 76, 1367–1374. [Google Scholar] [CrossRef] [PubMed]
- Bennett, R.W. Staphylococcal enterotoxin and its rapid identification in foods by enzyme-linked immunosorbent assay-based methodology. J. Food Prot. 2005, 68, 1264–1270. [Google Scholar] [PubMed]
- Vernozy-Rozand, C.; Mazuy-Cruchaudet, C.; Bavai, C.; Richard, Y. Comparison of three immunological methods for detecting staphylococcal enterotoxins from food. Lett. Appl. Microbiol. 2004, 39, 490–494. [Google Scholar] [CrossRef] [PubMed]
- Atanassova, V.; Meindl, A.; Ring, C. Prevalence of Staphylococcus aureus and staphylococcal enterotoxins in raw pork and uncooked smoked ham—A comparison of classical culturing detection and RFLP-PCR. Int. J. Food Microbiol. 2001, 68, 105–113. [Google Scholar] [CrossRef]
- Rose, S.A.; Bankes, P.; Stringer, M.F. Detection of staphylococcal enterotoxins in dairy products by the reversed passive latex agglutination (SET-RPLA) kit. Int. J. Food Microbiol. 1989, 8, 65–72. [Google Scholar] [CrossRef]
- Ostyn, A.; de Buyser, M.L.; Guillier, F.; Groult, J.; Felix, B.; Salah, S.; Delmas, G.; Hennekinne, J.A. First evidence of a food poisoning outbreak due to staphylococcal enterotoxin type E, France, 2009. Euro Surveill. 2010, 15, 1–4. [Google Scholar]
- Hennekinne, J.A.; Guillier, F.; Perelle, S.; de Buyser, M.L.; Dragacci, S.; Krys, S.; Lombard, B. Intralaboratory validation according to the EN ISO 16 140 Standard of the Vidas SET2 detection kit for use in official controls of staphylococcal enterotoxins in milk products. J. Appl. Microbiol. 2007, 102, 1261–1272. [Google Scholar] [CrossRef] [PubMed]
- Schlievert, P.M.; Case, L.C. Molecular analysis of staphylococcal superantigens. Methods Mol. Biol. 2007, 391, 113–126. [Google Scholar] [PubMed]
- Park, C.E.; Akhtar, M.; Rayman, M.K. Nonspecific reactions of a commercial enzyme-linked immunosorbent assay kit (TECRA) for detection of staphylococcal enterotoxins in foods. Appl. Environ. Microbiol. 1992, 58, 2509–2512. [Google Scholar] [PubMed]
- Wieneke, A.A. Comparison of four kits for the detection of staphylococcal enterotoxin in foods from outbreaks of food poisoning. Int. J. Food Microbiol. 1991, 14, 305–312. [Google Scholar] [CrossRef]
- Callahan, J.H.; Shefcheck, K.J.; Williams, T.L.; Musser, S.M. Detection, confirmation, and quantification of staphylococcal enterotoxin B in food matrixes using liquid chromatography—Mass spectrometry. Anal. Chem. 2006, 78, 1789–1800. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, A.; Hennekinne, J.A.; Garin, J.; Brun, V. Protein Standard Absolute Quantification (PSAQ) for improved investigation of staphylococcal food poisoning outbreaks. Proteomics 2008, 8, 4633–4636. [Google Scholar] [CrossRef] [PubMed]
- Sospedra, I.; Soler, C.; Manes, J.; Soriano, J.M. Analysis of staphylococcal enterotoxin A in milk by matrix-assisted laser desorption/ionization-time of flight mass spectrometry. Anal. Bioanal. Chem. 2011, 400, 1525–1531. [Google Scholar] [CrossRef] [PubMed]
- Bao, K.D.; Letellier, A.; Beaudry, F. Analysis of Staphylococcus enterotoxin B using differential isotopic tags and liquid chromatography quadrupole ion trap mass spectrometry. Biomed. Chromatogr. 2012, 26, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Sospedra, I.; Soler, C.; Manes, J.; Soriano, J.M. Rapid whole protein quantitation of staphylococcal enterotoxins A and B by liquid chromatography/mass spectrometry. J. Chromatogr. A 2012, 1238, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Stahl-Zeng, J.; Lange, V.; Ossola, R.; Eckhardt, K.; Krek, W.; Aebersold, R.; Domon, B. High sensitivity detection of plasma proteins by multiple reaction monitoring of N-glycosites. Mol. Cell. Proteom. 2007, 6, 1809–1817. [Google Scholar] [CrossRef] [PubMed]
- Dittrich, J.; Becker, S.; Hecht, M.; Ceglarek, U. Sample preparation strategies for targeted proteomics via proteotypic peptides in human blood using liquid chromatography tandem mass spectrometry. Proteom. Clin. Appl. 2015, 9, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Zuberovic, A.; Hanrieder, J.; Hellman, U.; Bergquist, J.; Wetterhall, M. Proteome profiling of human cerebrospinal fluid: Exploring the potential of capillary electrophoresis with surface modified capillaries for analysis of complex biological samples. Eur. J. Mass Spectrom. 2008, 14, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Hustoft, H.K.; Malerod, H.; Wilson, S.R.; Reubsaet, L.; Lundanes, E.; Greibrokk, T. A critical review of trypsin digestion for LC-MS based proteomics. In Integrative Proteomics; Leung, H.-C.E., Ed.; InTech: Rijeka, Croatia, 2012; pp. 73–92. [Google Scholar]
- Pinchuk, I.V.; Beswick, E.J.; Reyes, V.E. Staphylococcal enterotoxins. Toxins 2010, 2, 2177–2197. [Google Scholar] [CrossRef] [PubMed]
- European Community Commission Regulation. No. 1441/2007 of 5 December 2007. Off. J. Eur. Union 2007, L322, 12–29.
- Paulson, L.; Persson, R.; Karlsson, G.; Silberring, J.; Bierczynska-Krzysik, A.; Ekman, R.; Westman-Brinkmalm, A. Proteomics and peptidomics in neuroscience. Experience of capabilities and limitations in a neurochemical laboratory. J. Mass Spectrom. 2005, 40, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Fillmore, T.L.; Gao, Y.; Zhao, R.; He, J.; Schepmoes, A.A.; Nicora, C.D.; Wu, C.; Chambers, J.L.; Moore, R.J. Long-gradient separations coupled with selected reaction monitoring for highly sensitive, large scale targeted protein quantification in a single analysis. Anal. Chem. 2013, 85, 9196–9203. [Google Scholar] [CrossRef] [PubMed]
- Kientz, C.E.; Hulst, A.G.; Wils, E.R. Determination of staphylococcal enterotoxin B by on-line (micro) liquid chromatography-electrospray mass spectrometry. J. Chromatogr. A 1997, 757, 51–64. [Google Scholar] [CrossRef]
- Nedelkov, D.; Nelson, R.W. Detection of Staphylococcal enterotoxin B via biomolecular interaction analysis mass spectrometry. Appl. Environ. Microbiol. 2003, 69, 5212–5215. [Google Scholar] [CrossRef] [PubMed]
- UniProt Knowledgebase (UniProt KB). Available online: http://www.uniprot.org (accessed from May 2012–September 2014).
- WADA (World Anti-Doping Agency) Laboratory Committee. WADA Technical Document—TD2010IDCR. Version 1.0. 2010, pp. 5–9. Available online: https://www.wada-ama.org (accessed on 9 September 2015).
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muratovic, A.Z.; Hagström, T.; Rosén, J.; Granelli, K.; Hellenäs, K.-E. Quantitative Analysis of Staphylococcal Enterotoxins A and B in Food Matrices Using Ultra High-Performance Liquid Chromatography Tandem Mass Spectrometry (UPLC-MS/MS). Toxins 2015, 7, 3637-3656. https://doi.org/10.3390/toxins7093637
Muratovic AZ, Hagström T, Rosén J, Granelli K, Hellenäs K-E. Quantitative Analysis of Staphylococcal Enterotoxins A and B in Food Matrices Using Ultra High-Performance Liquid Chromatography Tandem Mass Spectrometry (UPLC-MS/MS). Toxins. 2015; 7(9):3637-3656. https://doi.org/10.3390/toxins7093637
Chicago/Turabian StyleMuratovic, Aida Zuberovic, Thomas Hagström, Johan Rosén, Kristina Granelli, and Karl-Erik Hellenäs. 2015. "Quantitative Analysis of Staphylococcal Enterotoxins A and B in Food Matrices Using Ultra High-Performance Liquid Chromatography Tandem Mass Spectrometry (UPLC-MS/MS)" Toxins 7, no. 9: 3637-3656. https://doi.org/10.3390/toxins7093637
APA StyleMuratovic, A. Z., Hagström, T., Rosén, J., Granelli, K., & Hellenäs, K.-E. (2015). Quantitative Analysis of Staphylococcal Enterotoxins A and B in Food Matrices Using Ultra High-Performance Liquid Chromatography Tandem Mass Spectrometry (UPLC-MS/MS). Toxins, 7(9), 3637-3656. https://doi.org/10.3390/toxins7093637