Direct Activation of Ribosome-Associated Double-Stranded RNA-Dependent Protein Kinase (PKR) by Deoxynivalenol, Anisomycin and Ricin: A New Model for Ribotoxic Stress Response Induction

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
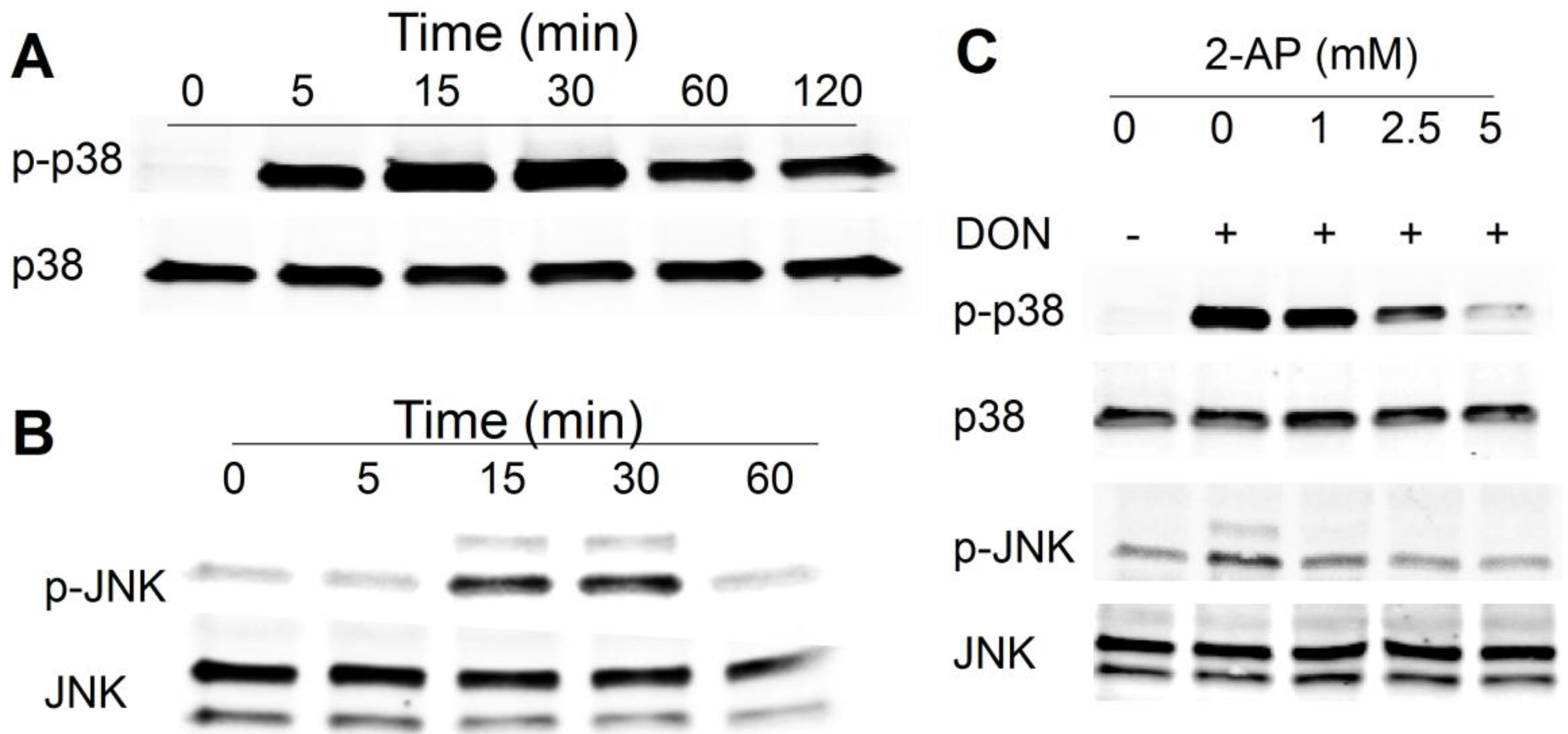
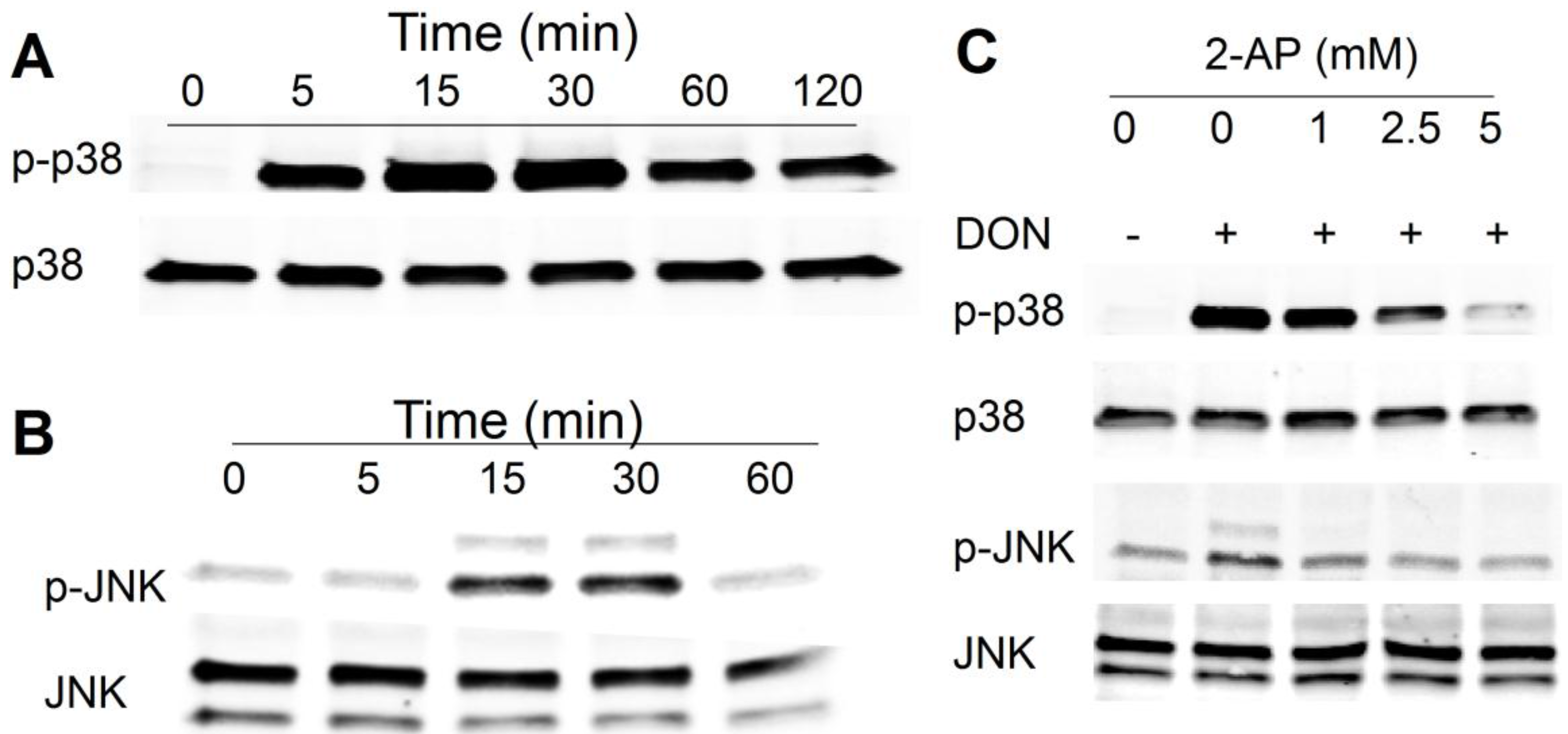
2.1. DON Induces PKR-Dependent MAPK Phosphorylation in HeLa Cells
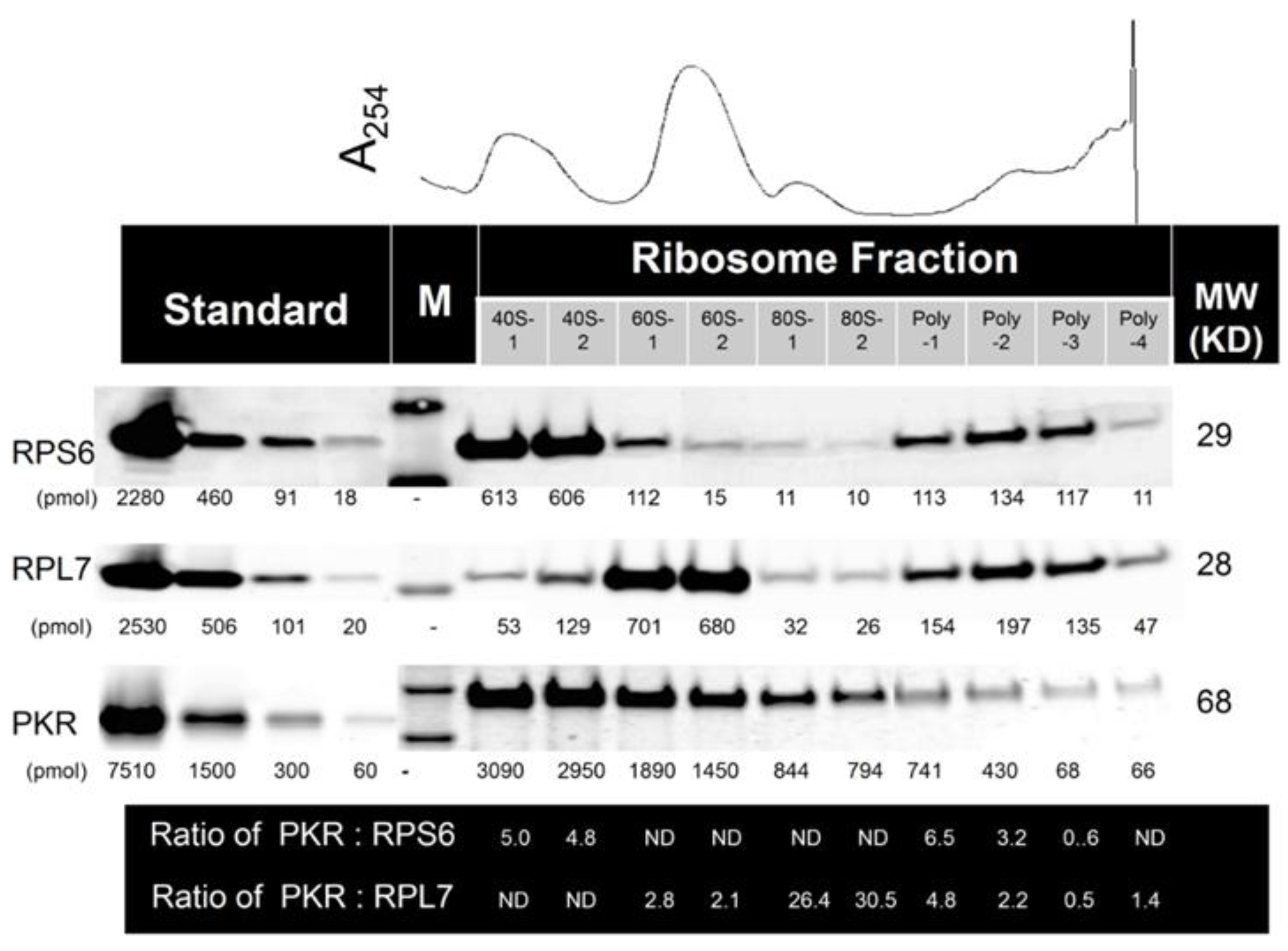
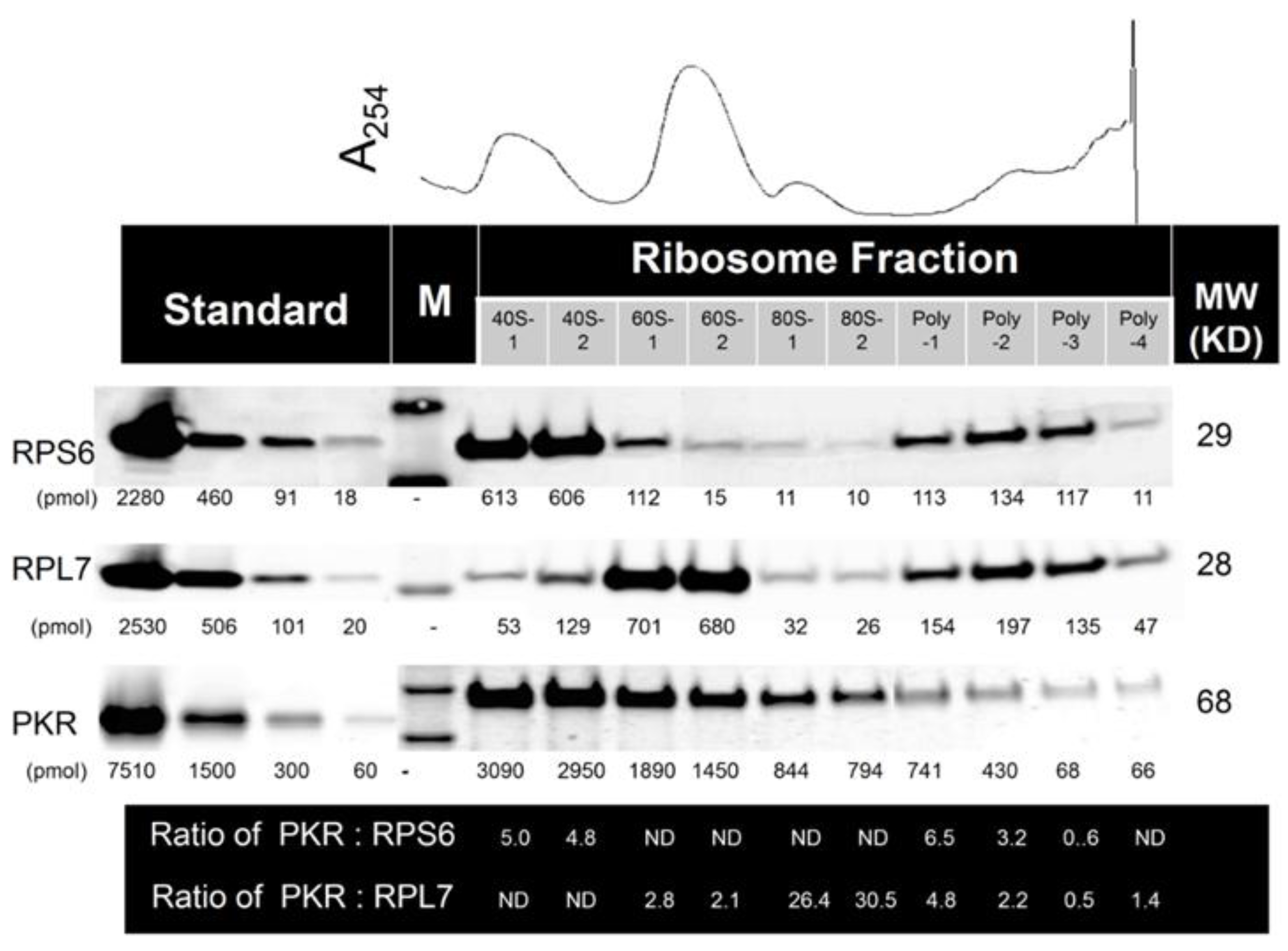
2.2. Multiple PKR Molecules Basally Associate with the 40S, 60S, 80S and Polysomal Fractions of the Ribosome in HeLa Lysate
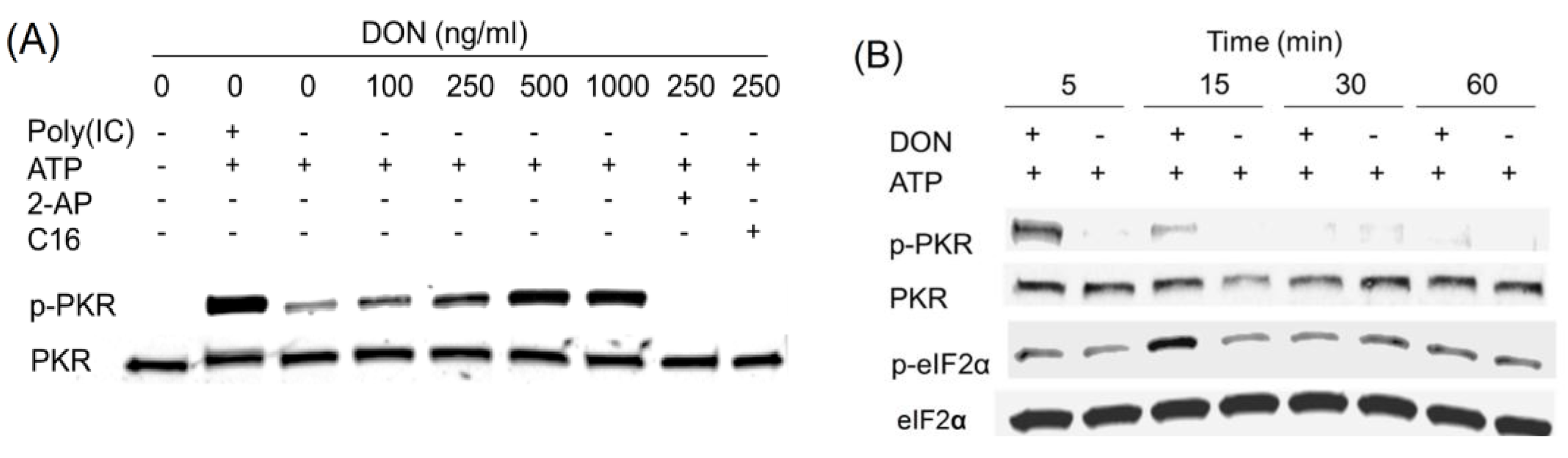
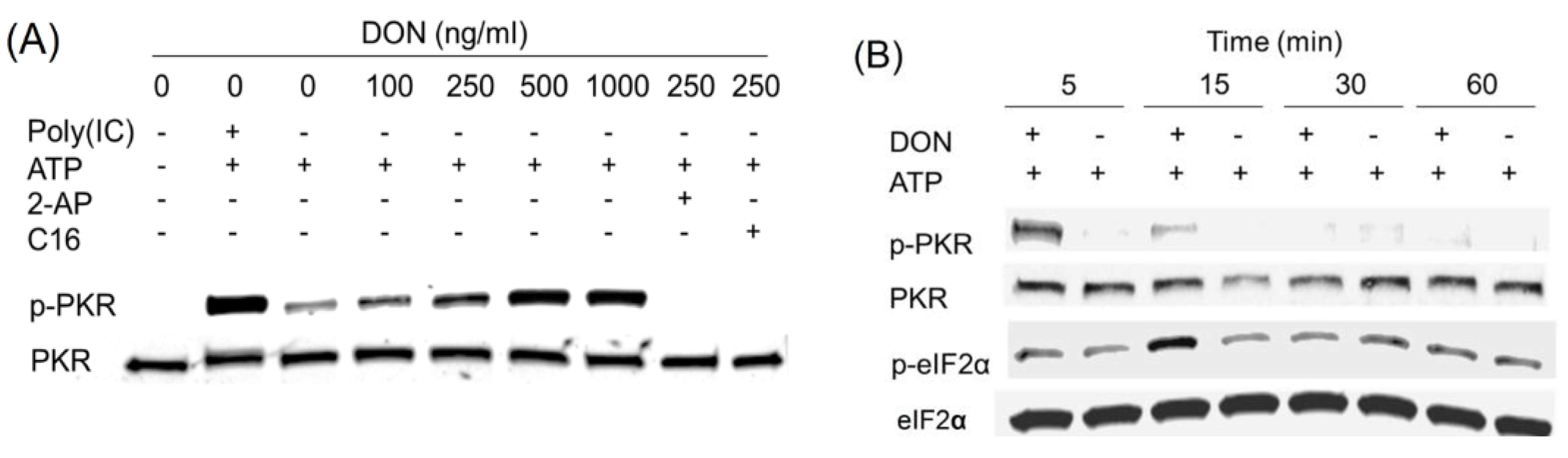
2.3. DON Induces PKR Activation in ATP-Containing HeLa Lysate
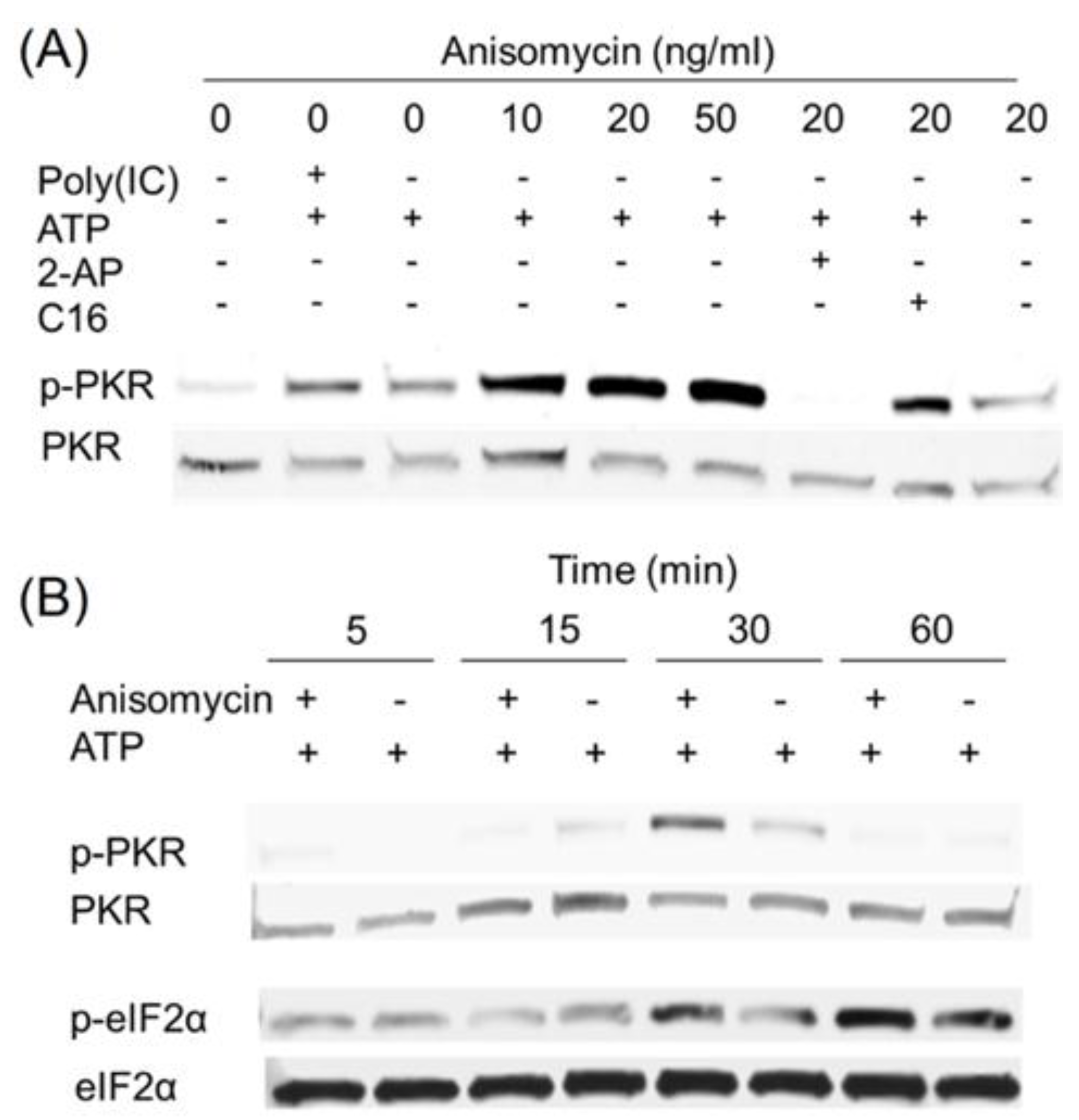
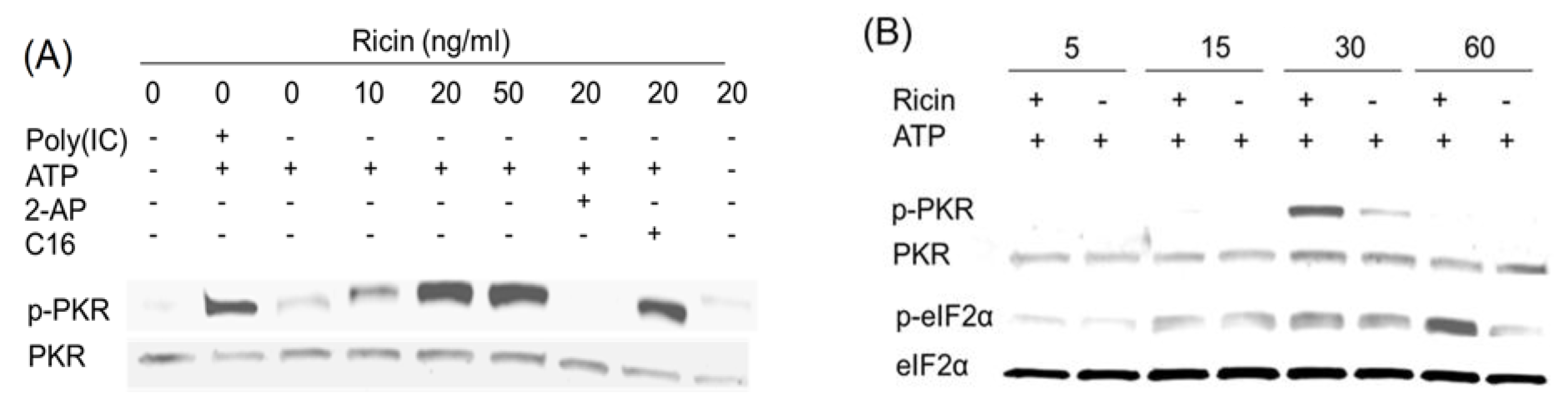
2.4. Anisomycin and Ricin Induce PKR Activation in HeLa Lysate
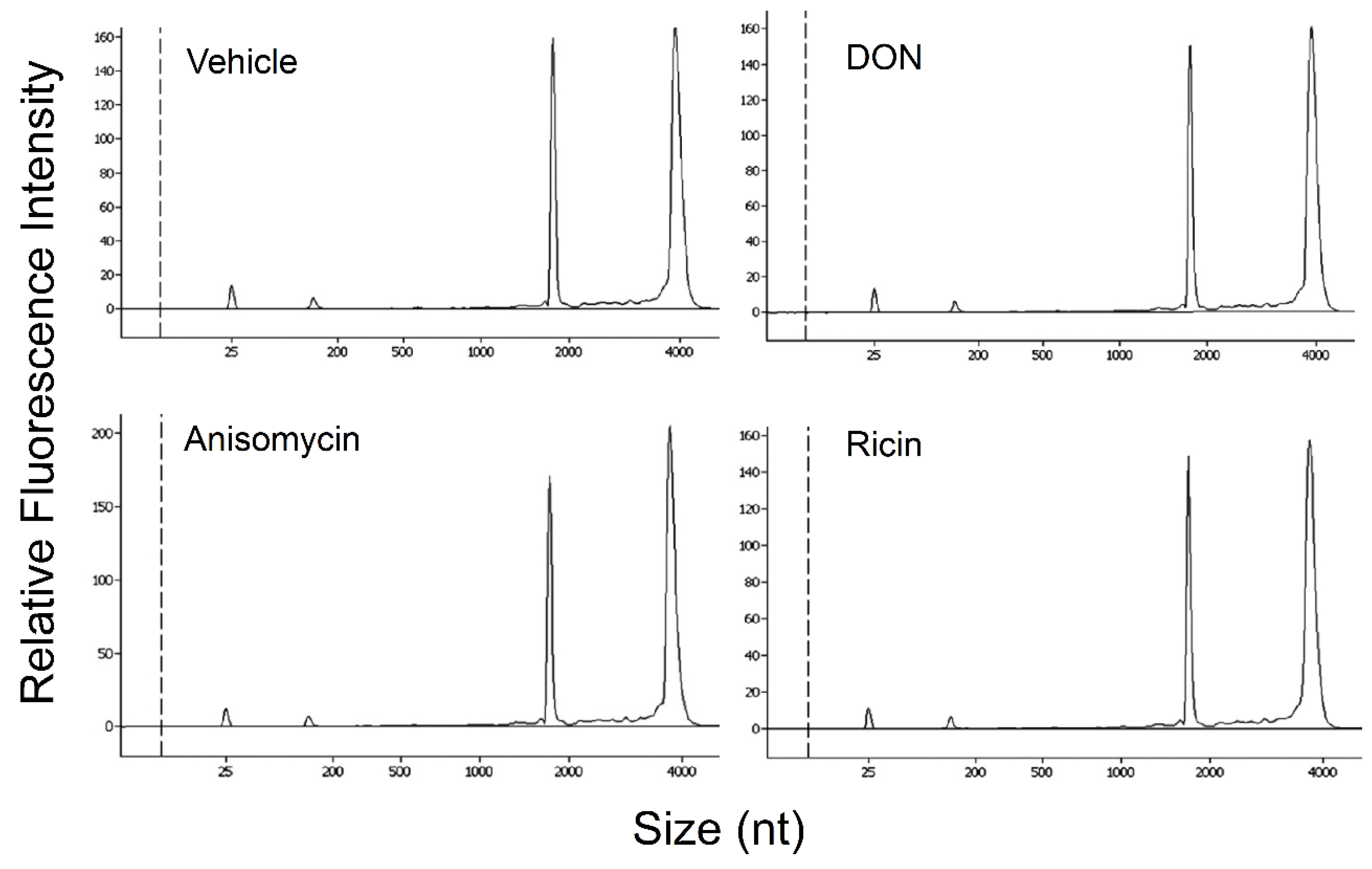
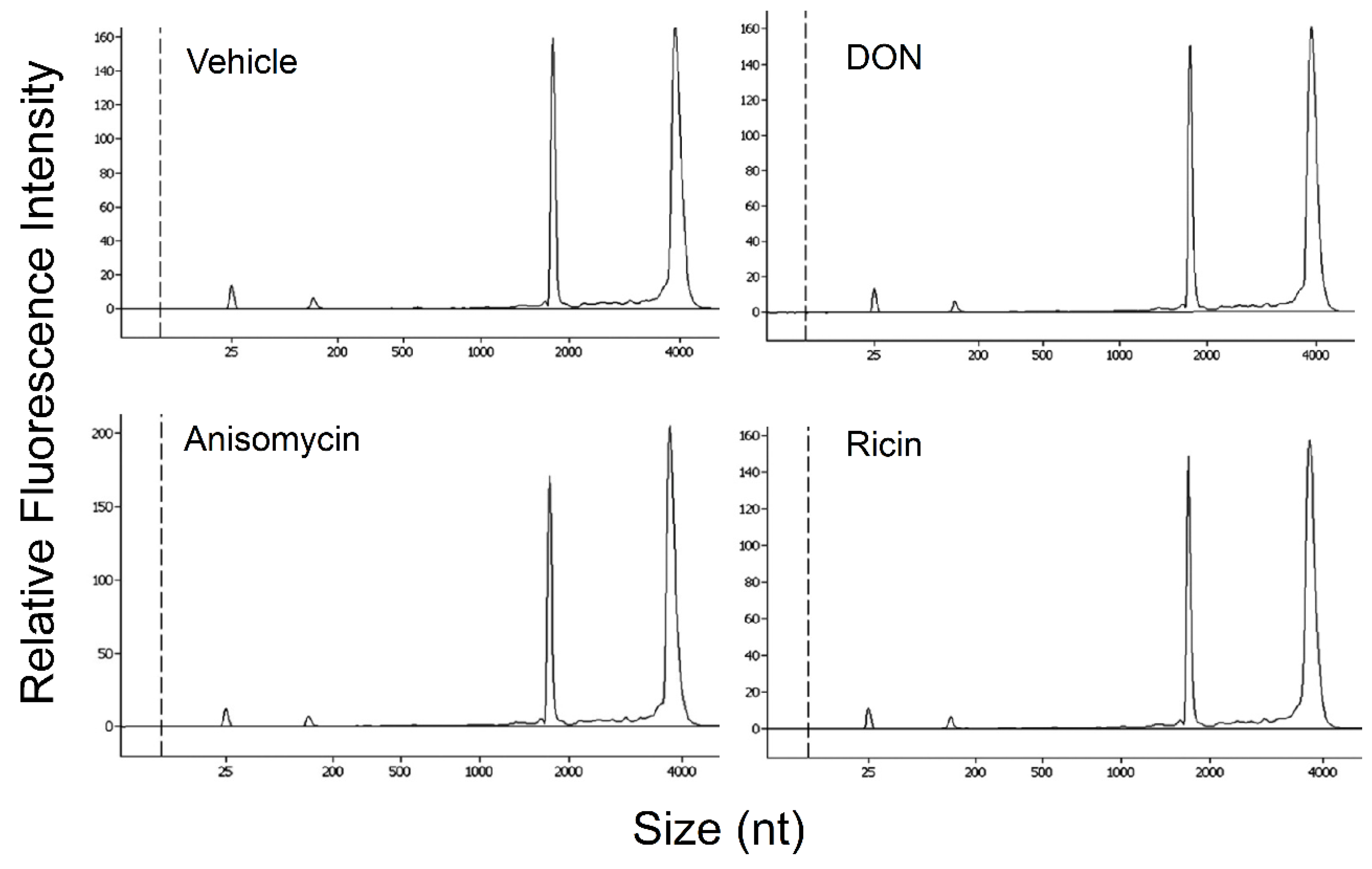
2.5. DON, Anisomycin and Ricin Do Not Affect rRNA Integrity in HeLa Lysate
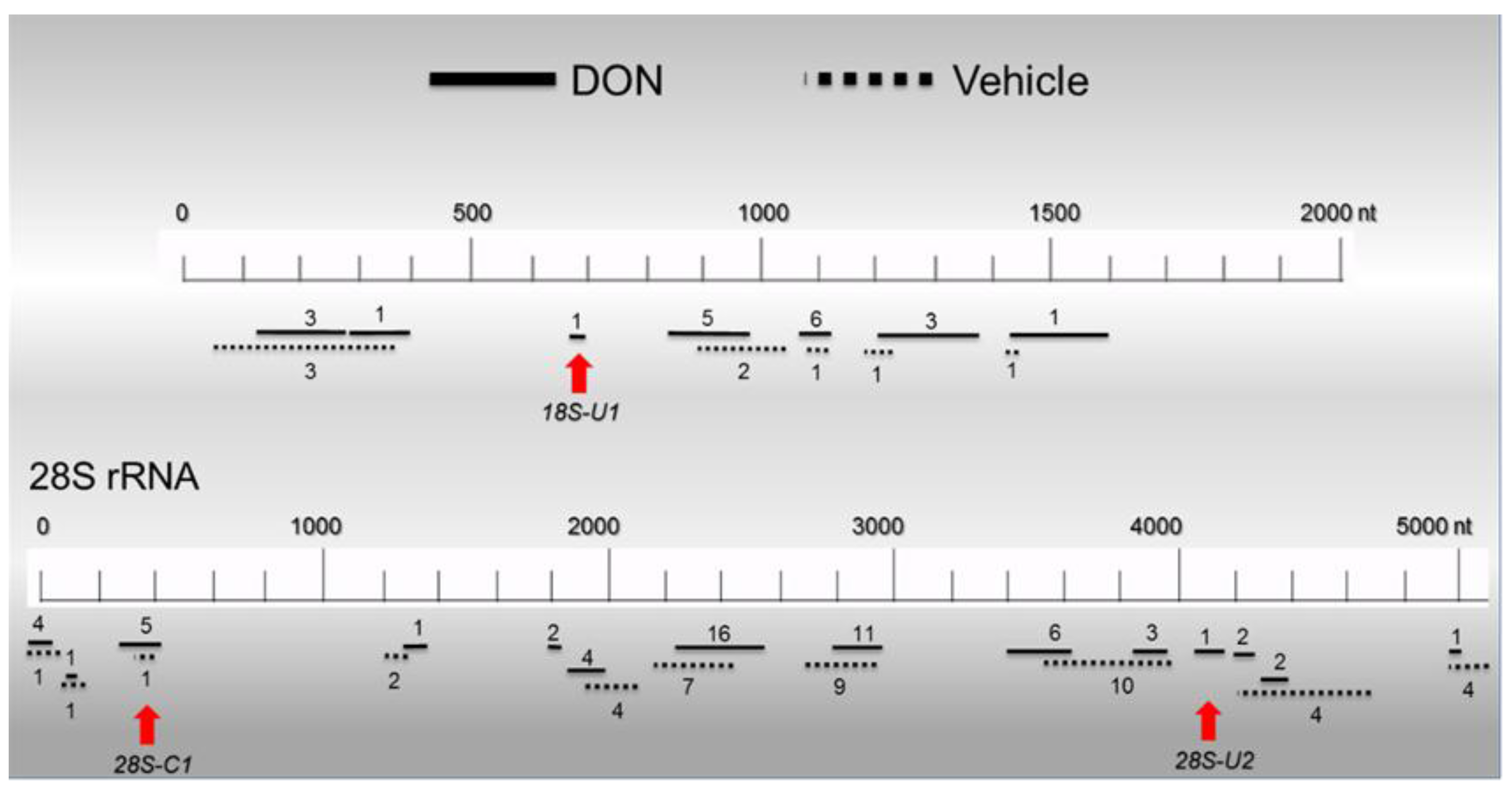
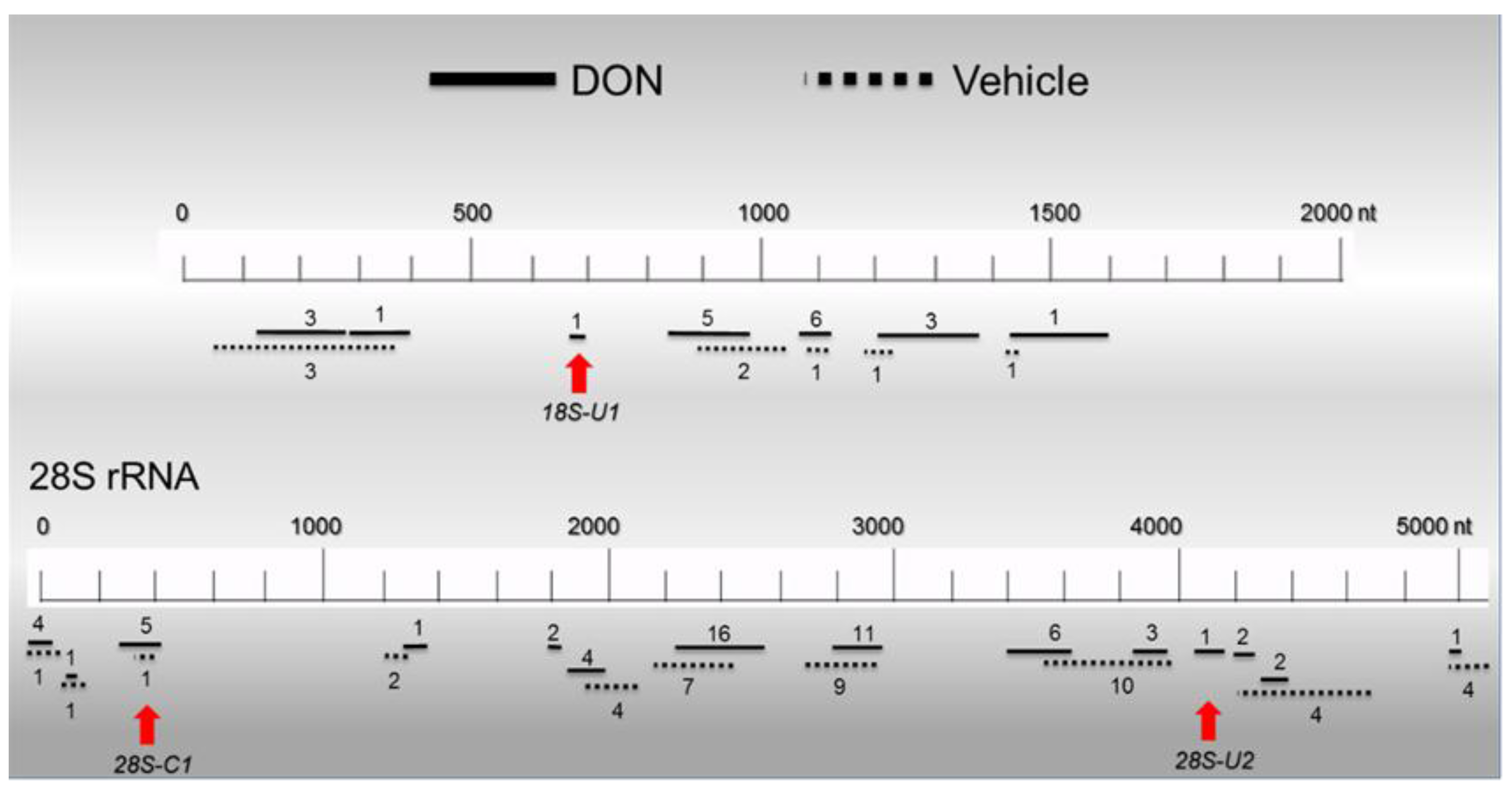
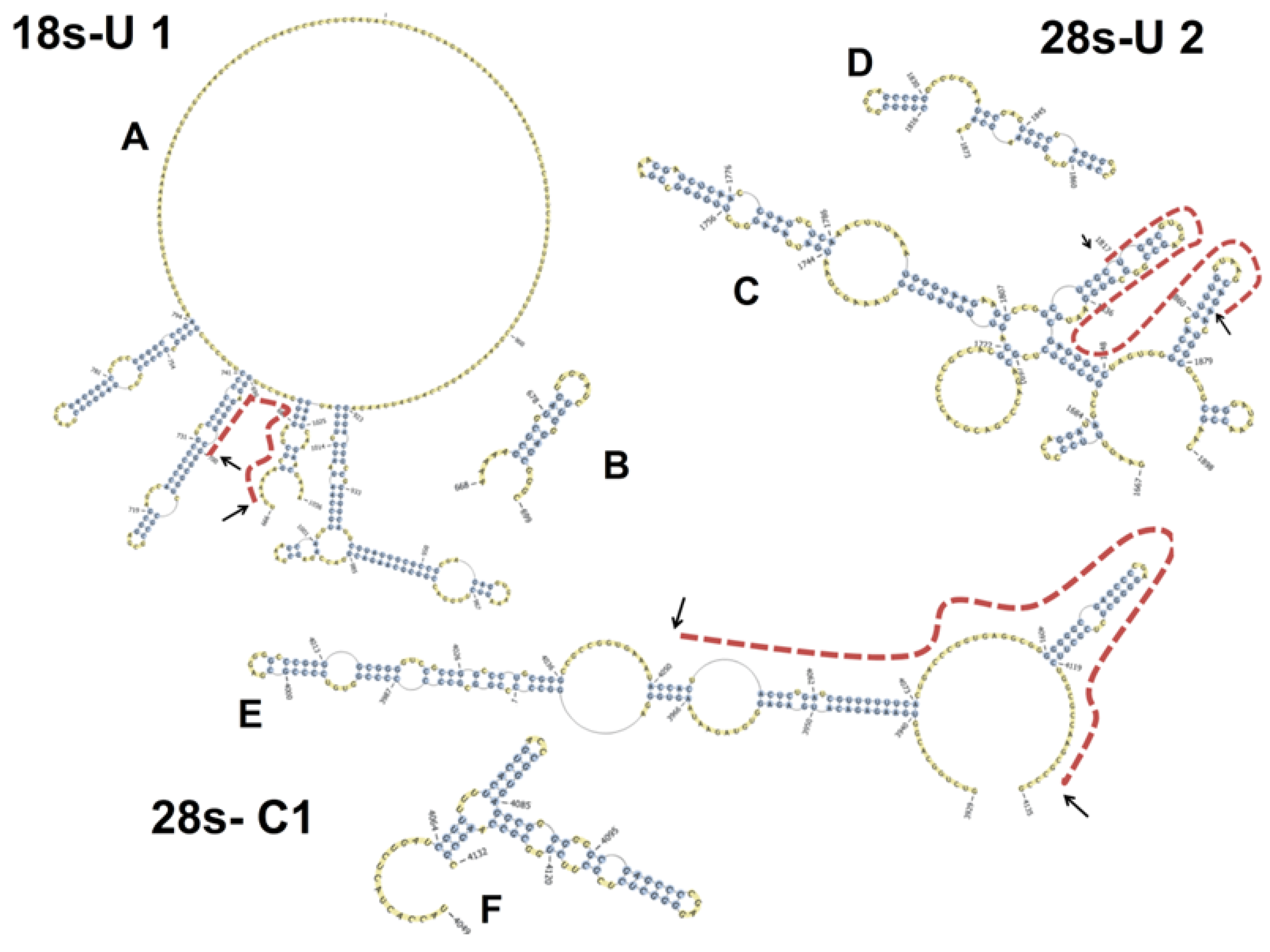
2.6. Basal PKR Association with rRNA in HeLa Lysates Is Unaffected by DON
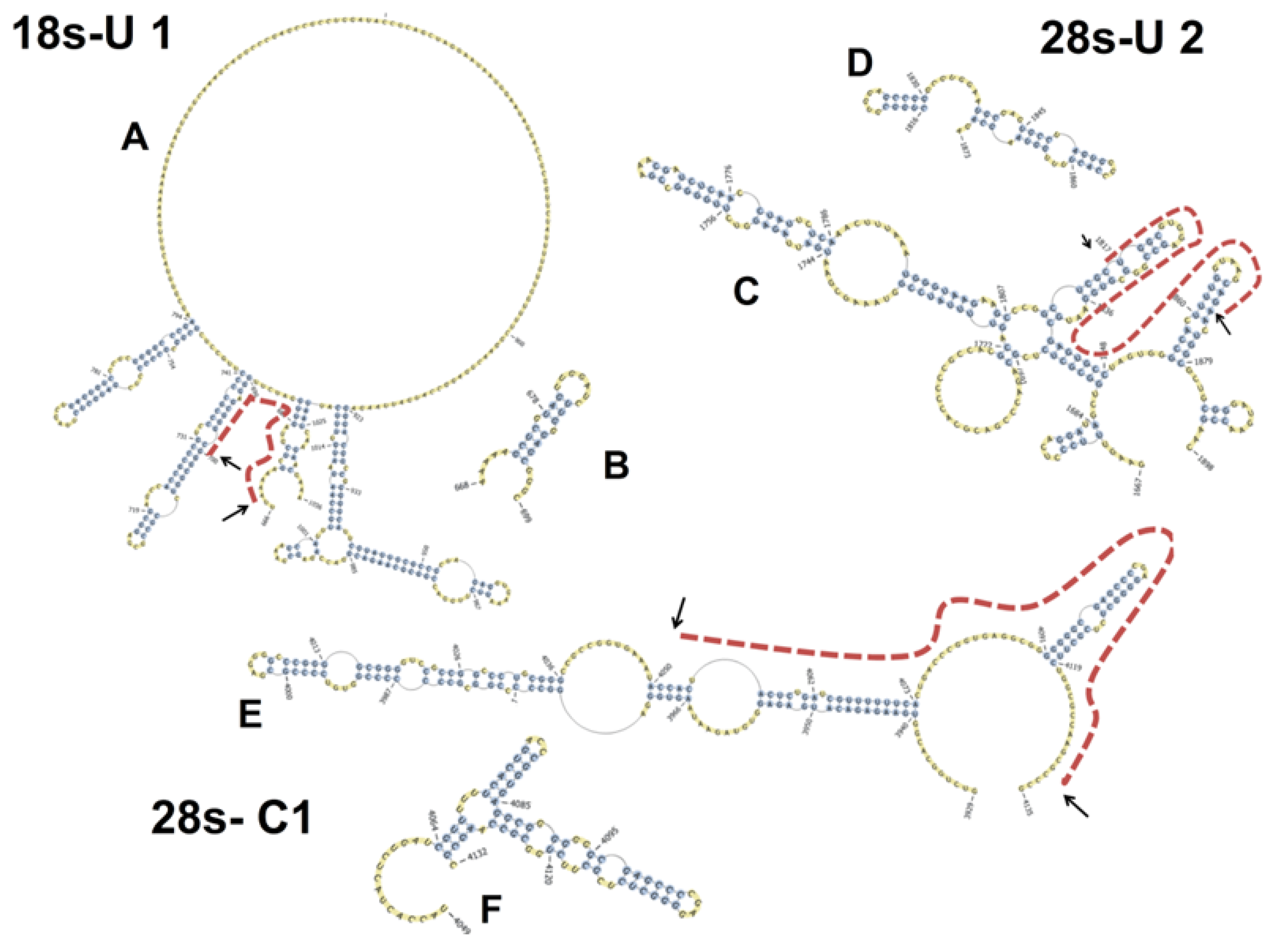

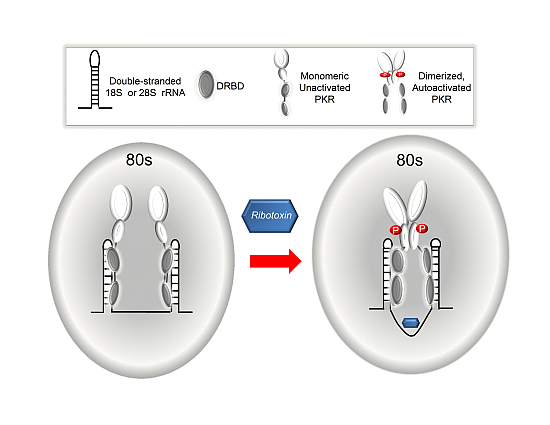
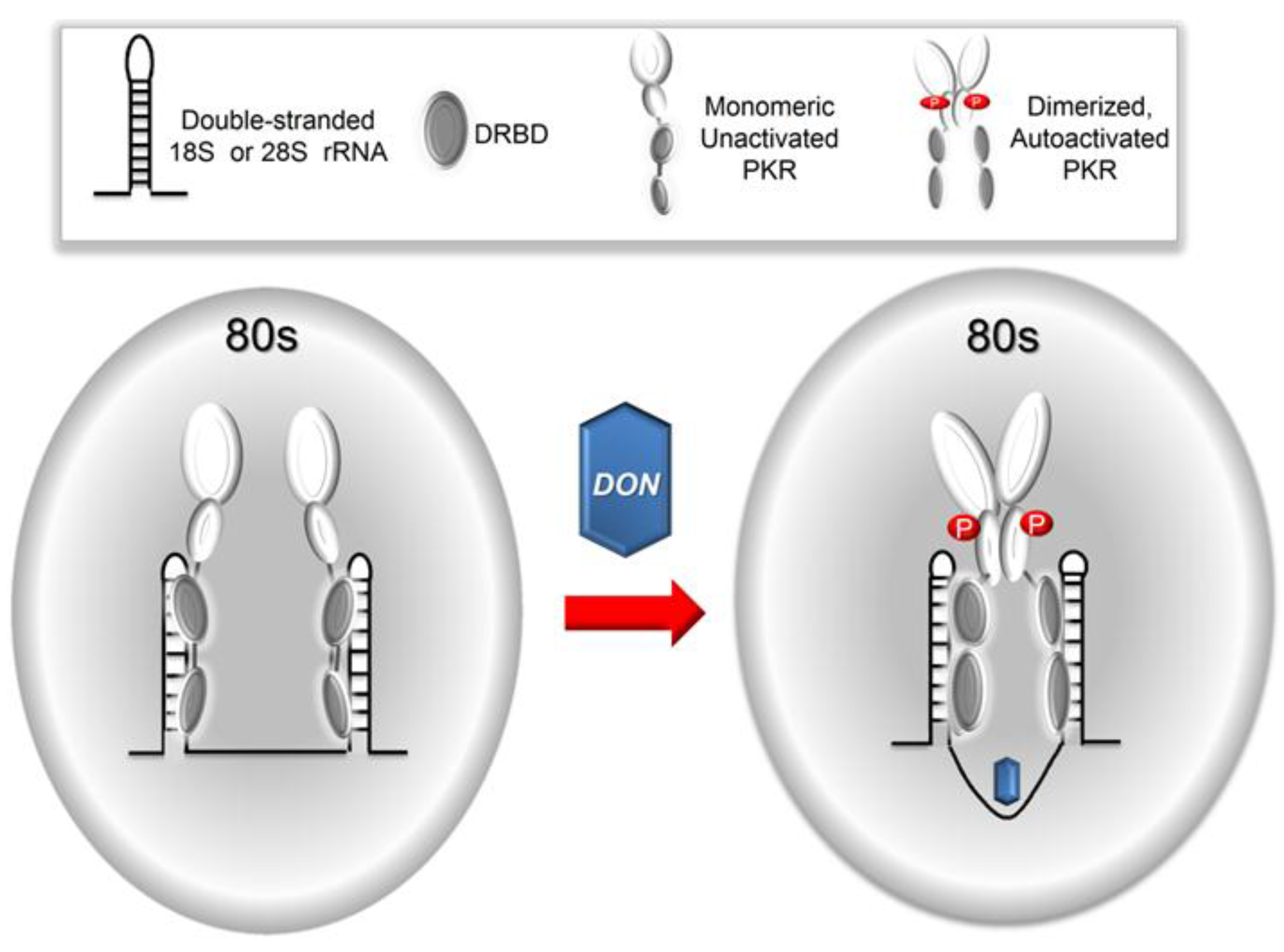
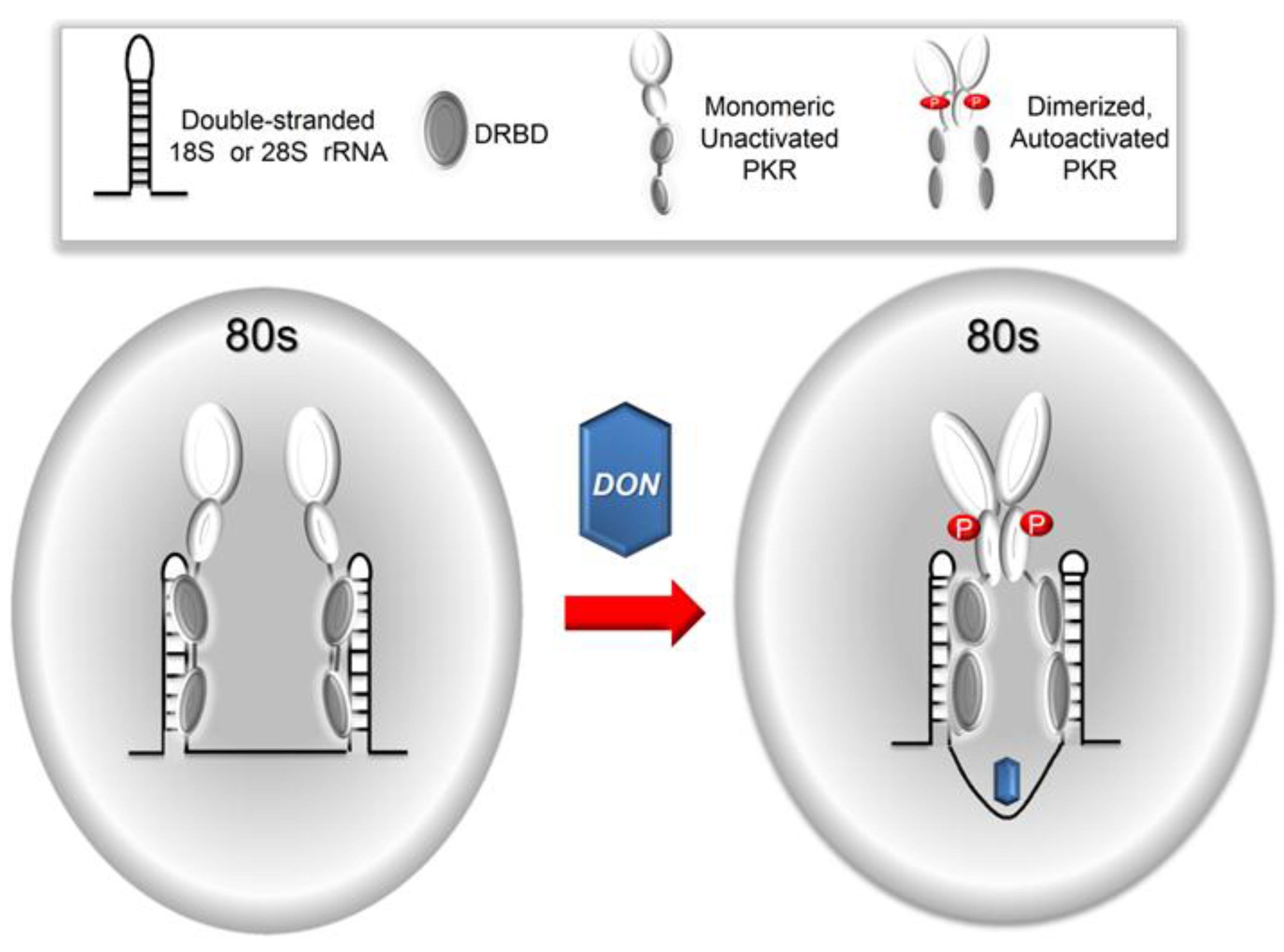
3. Discussion
4. Experimental Section
4.1. Assessment of PKR-Dependent MAPK Activation in HeLa Cells
4.2. Quantitation of PKR: Ribosome Stoichiometry in HeLa Cells
4.3. Ribotoxin-Induced PKR Activation in Translationally-Active Cell-Free HeLa Lysates
4.4. Assessment rRNA Integrity
4.5. RNA Immunoprecipitation (RNA-IP) of rRNA-PKR Complexes
4.6. Cloning and Sequencing of PKR-Associated rRNA Fragments
4.7. Prediction of the Secondary Structures of PKR-Bound rRNA Sequences
4.8. RNase Protection Assay
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tscherne, J.S.; Pestka, S. Inhibition of protein-synthesis in intact HeLa cells. Antimicrob. Agents. Chemotherap. 1975, 8, 479–487. [Google Scholar] [CrossRef]
- Iordanov, M.S.; Pribnow, D.; Magun, J.L.; Dinh, T.H.; Pearson, J.A.; Chen, S.L.; Magun, B.E. Ribotoxic stress response: Activation of the stress-activated protein kinase JNK1 by inhibitors of the peptidyl transferase reaction and by sequence-specific RNA damage to the alpha-sarcin/ricin loop in the 28S rRNA. Mol. Cell. Biol. 1997, 17, 3373–3381. [Google Scholar] [PubMed]
- Laskin, J.D.; Heck, D.E.; Laskin, D.L. The ribotoxic stress response as a potential mechanism for MAP kinase activation in xenobiotic toxicity. Toxicol. Sci. 2002, 69, 289–291. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, T. Translation inhibitors and their unique biological properties. Eur. J. Pharmacol. 2012, 676, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Tesh, V.L. Activation of cell stress response pathways by shiga toxins. Cell. Microbiol. 2012, 14, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.H.; Jarvis, B.B.; Chung, Y.J.; Pestka, J.J. Apoptosis induction by the satratoxins and other trichothecene mycotoxins: Relationship to ERK, p38 MAPK, and SAPK/JNK activation. Toxicol. Appl. Pharmacol. 2000, 164, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.R.; Lau, A.S.; Pestka, J.J. Role of double-stranded RNA-activated protein kinase R (PKR) in deoxynivalenol-induced ribotoxic stress response. Toxicol. Sci. 2003, 74, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Islam, Z.; Hegg, C.C.; Bae, H.K.; Pestka, J.J. Satratoxin G-induced apoptosis in PC-12 neuronal cells is mediated by PKR and caspase independent. Toxicol. Sci. 2008, 105, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Bae, H.; Gray, J.S.; Li, M.; Vines, L.; Kim, J.; Pestka, J.J. Hematopoietic cell kinase associates with the 40S ribosomal subunit and mediates the ribotoxic stress response to deoxynivalenol in mononuclear phagocytes. Toxicol. Sci. 2010, 115, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Gray, J.S.; Bae, H.K.; Li, J.C.; Lau, A.S.; Pestka, J.J. Double-stranded RNA-activated protein kinase mediates induction of interleukin-8 expression by deoxynivalenol, shiga toxin 1, and ricin in monocytes. Toxicol. Sci. 2008, 105, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.A.; Gil, J.; Ventoso, I.; Guerra, S.; Domingo, E.; Rivas, C.; Esteban, M. Impact of protein kinase PKR in cell biology: From antiviral to antiproliferative action. Microbiol. Mol. Biol. Rev. 2006, 70, 1032–1060. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Tang, D. PKR-dependent inflammatory signals. Sci. signal. 2012, 5. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.R. Signal integration via PKR. Sci. STKE 2001, 89. [Google Scholar] [CrossRef]
- Sudhakar, A.; Ramachandran, A.; Ghosh, S.; Hasnain, S.E.; Kaufman, R.J.; Ramaiah, K.V. Phosphorylation of serine 51 in initiation factor 2 alpha (eIF2 alpha) promotes complex formation between eIF2alpha and eIF2B and causes inhibition in the guanine nucleotide exchange activity of eIF2B. Biochemistry 2000, 39, 12929–12938. [Google Scholar] [CrossRef] [PubMed]
- Verma, I.M.; Stevenson, J.K.; Schwarz, E.M.; Van Antwerp, D.; Miyamoto, S. Rel/NF-kappa B/I kappa B family: Intimate tales of association and dissociation. Genes Dev. 1995, 9, 2723–2735. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.R. PKR; a sentinel kinase for cellular stress. Oncogene 1999, 18, 6112–6120. [Google Scholar] [CrossRef] [PubMed]
- Chu, W.M.; Ostertag, D.; Li, Z.W.; Chang, L.; Chen, Y.; Hu, Y.; Williams, B.; Perrault, J.; Karin, M. JNK2 and IKKβ are required for activating the innate response to viral infection. Immunity 1999, 11, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Koromilas, A.E.; Roy, S.; Barber, G.N.; Katze, M.G.; Sonenberg, N. Malignant transformation by a mutant of the IFN-inducible dsRNA dependent protein kinase. Science 1992, 257, 1685–1689. [Google Scholar] [CrossRef] [PubMed]
- Lengyel, P. Tumor-suppressor genes: News about the interferon connection. Proc. Natl. Acad. Sci. USA 1993, 90, 5893–5895. [Google Scholar] [CrossRef] [PubMed]
- Pestka, J.J.; Zhou, H.R.; Moon, Y.; Chung, Y.J. Cellular and molecular mechanisms for immune modulation by deoxynivalenol and other trichothecenes: Unraveling a paradox. Toxicol. Lett. 2004, 153, 61–73. [Google Scholar] [CrossRef]
- Shi, Y.; Pestka, J.J. Mechanisms for suppression of interleukin-6 expression in peritoneal macrophages from docosahexaenoic acid-fed mice. J. Nutr. Biochem. 2009, 20, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Ben-Shem, A.; Garreau de Loubresse, N.; Melnikov, S.; Jenner, L.; Yusupova, G.; Yusupov, M. The structure of the eukaryotic ribosome at 3.0 a resolution. Science 2011, 334, 1524–1529. [Google Scholar]
- Zhu, S.; Romano, P.R.; Wek, R.C. Ribosome targeting of PKR is mediated by two double-stranded RNA-binding domains and facilitates in vivo phosphorylation of eukaryotic initiation factor-2. J. Biol. Chem. 1997, 272, 14434–14441. [Google Scholar] [CrossRef] [PubMed]
- Bae, H.K.; Shinozuka, J.; Islam, Z.; Pestka, J.J. Satratoxin G interaction with 40s and 60s ribosomal subunits precedes apoptosis in the macrophage. Toxicol. Appl. Pharmacol. 2009, 237, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, N.; Gorman, A.M.; Gupta, S.; Samali, A. The eIF2α kinases: Their structures and functions. Cell. Mol. Life Sci. 2013, 70, 3493–3511. [Google Scholar] [CrossRef] [PubMed]
- Husain, B.; Mukerji, I.; Cole, J.L. Analysis of high-affinity binding of protein kinase R to double-stranded RNA. Biochemistry 2012, 51, 8764–8770. [Google Scholar] [CrossRef] [PubMed]
- Sadler, A.J. Orchestration of the activation of protein kinase R by the RNA-binding motif. J. Interferon Cyt. Res 2010, 30, 195–204. [Google Scholar] [CrossRef]
- Sadler, A.J.; Williams, B.R. Structure and function of the protein kinase R. Curr. Top. Microbiol. Immunol. 2007, 316, 253–292. [Google Scholar] [PubMed]
- Bevilacqua, P.C.; Blose, J.M. Structures, kinetics, thermodynamics, and biological functions of RNA hairpins. Annu. Rev. Phys. Chem. 2008, 59, 79–103. [Google Scholar] [CrossRef] [PubMed]
- Manche, L.; Green, S.R.; Schmedt, C.; Mathews, M.B. Interactions between double-stranded RNA regulators and the protein kinase DAI. Mol. Cell. Biol. 1992, 12, 5238–5248. [Google Scholar] [PubMed]
- Wu, S.; Kumar, K.U.; Kaufman, R.J. Identification and requirement of three ribosome binding domains in dsRNA dependent protein kinase (PKR). Biochemistry 1998, 37, 13816–13826. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Nishimura, S.; Matsunaga, S.; Fusetani, N.; Horinouchi, S.; Yoshida, M. Inhibition of protein synthesis and activation of stress-activated protein kinases by onnamide A and theopederin B, antitumor marine natural products. Cancer Sci. 2005, 96, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Pace, C.N.; Vajdos, F.; Fee, L.; Grimsley, G.; Gray, T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995, 4, 2411–2423. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Whitten, D.A.; Wilkerson, C.G.; Pestka, J.J. Dynamic changes in ribosome-associated proteome and phosphoproteome during deoxynivalenol-induced translation inhibition and ribotoxic stress. Toxicol. Sci. 2013, 138, 217–233. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.C.; von Hippel, P.H. Calculation of protein extinction coefficients from amino acid sequence data. Anal. Biochem. 1989, 182, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Calvet, J.P.; Pederson, T. Secondary structure of heterogeneous nuclear RNA: Two classes of double-stranded RNA in native ribonucleoprotein. Proc. Natl. Acad. Sci. USA 1977, 74, 3705–3709. [Google Scholar] [CrossRef] [PubMed]
- Fedoroff, N.; Wellauer, P.K.; Wall, R. Intermolecular duplexes in heterogeneous nuclear RNA from HeLa cells. Cell 1977, 10, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Ben-Asouli, Y.; Banai, Y.; Pel-Or, Y.; Shir, A.; Kaempfer, R. Human interferon-gamma mRNA autoregulates its translation through a pseudoknot that activates the interferon-inducible protein kinase PKR. Cell 2002, 108, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, W.; Mangir, M.; Lochmann, E.R. The effect of pentachlorophenol and its metabolite tetrachlorohydroquinone on RNA, protein, and ribosome synthesis in Saccharomyces cells. Ecotoxicol. Environ. Saf. 1987, 13, 7–12. [Google Scholar] [PubMed]
- Li, J.; Petryshyn, R.A. Activation of the double-stranded RNA-dependent eIF-2 alpha kinase by cellular RNA from 3T3-F442A cells. Eur. J. Biochem. 1991, 195, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Toroney, R.; Bevilacqua, P.C. PKR and the ribosome compete for mRNA. Nat. Chem. Biol. 2009, 5, 873–874. [Google Scholar] [CrossRef] [PubMed]
- Rabl, J.; Leibundgut, M.; Ataide, S.F.; Haag, A.; Ban, N. Crystal structure of the eukaryotic 40S ribosomal subunit in complex with initiation factor 1. Science 2011, 331, 730–736. [Google Scholar] [CrossRef] [PubMed]
- Yoshihama, M.; Uechi, T.; Asakawa, S.; Kawasaki, K.; Kato, S.; Higa, S.; Maeda, N.; Minoshima, S.; Tanaka, T.; Shimizu, N.; et al. The human ribosomal protein genes: Sequencing and comparative analysis of 73 genes. Genome Res. 2002, 12, 379–390. [Google Scholar]
- Langland, J.O.; Jacobs, B.L. Cytosolic double-stranded RNA-dependent protein kinase is likely a dimer of partially phosphorylated Mr = 66,000 subunits. J. Biol. Chem. 1992, 267, 10729–10736. [Google Scholar] [PubMed]
- He, K.; Zhou, H.R.; Pestka, J.J. Targets and intracellular signaling mechanisms for deoxynivalenol-induced ribosomal RNA cleavage. Toxicol. Sci. 2012, 127, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Pestka, J.J. Comparative induction of 28S ribosomal RNA cleavage by ricin and the trichothecenes deoxynivalenol and T-2 toxin in the macrophage. Toxicol. Sci. 2008, 105, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Schutz-Geschwender, A.; Zhang, Y.; Holt, T.; McDermitt, D.; Olive, D.M. Quantitative, two-color western blot detection with infrared fluorescence. Li-Cor Biosci. 2004. Available online: http://cdn.licor.com/bio/PDF/IRquant.pdf (accessed on 9 December 2014).
- Jammi, N.V.; Whitby, L.R.; Beal, P.A. Small molecule inhibitors of the RNA-dependent protein kinase. Biochem. Biophys. Res. Commun. 2003, 308, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Demeshkina, N.; Repkova, M.; Ven’yaminova, A.; Graifer, D.; Karpova, G. Nucleotides of 18S rRNA surrounding mRNA codons at the human ribosomal A, P, and E sites: A crosslinking study with mRNA analogs carrying an aryl azide group at either the uracil or the guanine residue. RNA 2000, 6, 1727–1736. [Google Scholar] [CrossRef] [PubMed]
- Gorski, J.L.; Gonzalez, I.L.; Schmickel, R.D. The secondary structure of human 28S rRNA: The structure and evolution of a mosaic rRNA gene. J. Mol. Evol. 1987, 24, 236–251. [Google Scholar] [CrossRef] [PubMed]
- Iordanov, M.S.; Pribnow, D.; Magun, J.L.; Dinh, T.H.; Pearson, J.A.; Magun, B.E. Ultraviolet radiation triggers the ribotoxic stress response in mammalian cells. J. Biol. Chem. 1998, 273, 15794–15803. [Google Scholar] [CrossRef] [PubMed]
- Wurtmann, E.J.; Wolin, S.L. RNA under attack: Cellular handling of RNA damage. Crit. Rev. Biochem. Mol. Biol. 2009, 44, 34–49. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, H.-R.; He, K.; Landgraf, J.; Pan, X.; Pestka, J.J. Direct Activation of Ribosome-Associated Double-Stranded RNA-Dependent Protein Kinase (PKR) by Deoxynivalenol, Anisomycin and Ricin: A New Model for Ribotoxic Stress Response Induction. Toxins 2014, 6, 3406-3425. https://doi.org/10.3390/toxins6123406
Zhou H-R, He K, Landgraf J, Pan X, Pestka JJ. Direct Activation of Ribosome-Associated Double-Stranded RNA-Dependent Protein Kinase (PKR) by Deoxynivalenol, Anisomycin and Ricin: A New Model for Ribotoxic Stress Response Induction. Toxins. 2014; 6(12):3406-3425. https://doi.org/10.3390/toxins6123406
Chicago/Turabian StyleZhou, Hui-Ren, Kaiyu He, Jeff Landgraf, Xiao Pan, and James J. Pestka. 2014. "Direct Activation of Ribosome-Associated Double-Stranded RNA-Dependent Protein Kinase (PKR) by Deoxynivalenol, Anisomycin and Ricin: A New Model for Ribotoxic Stress Response Induction" Toxins 6, no. 12: 3406-3425. https://doi.org/10.3390/toxins6123406
APA StyleZhou, H.-R., He, K., Landgraf, J., Pan, X., & Pestka, J. J. (2014). Direct Activation of Ribosome-Associated Double-Stranded RNA-Dependent Protein Kinase (PKR) by Deoxynivalenol, Anisomycin and Ricin: A New Model for Ribotoxic Stress Response Induction. Toxins, 6(12), 3406-3425. https://doi.org/10.3390/toxins6123406