Modulation of the Antioxidant System of Caco-2 Cells in the Presence of Aflatoxin B1, Ochratoxin A, and Ferulic Acid

 ,
,  , and
, and 
Abstract
1. Introduction
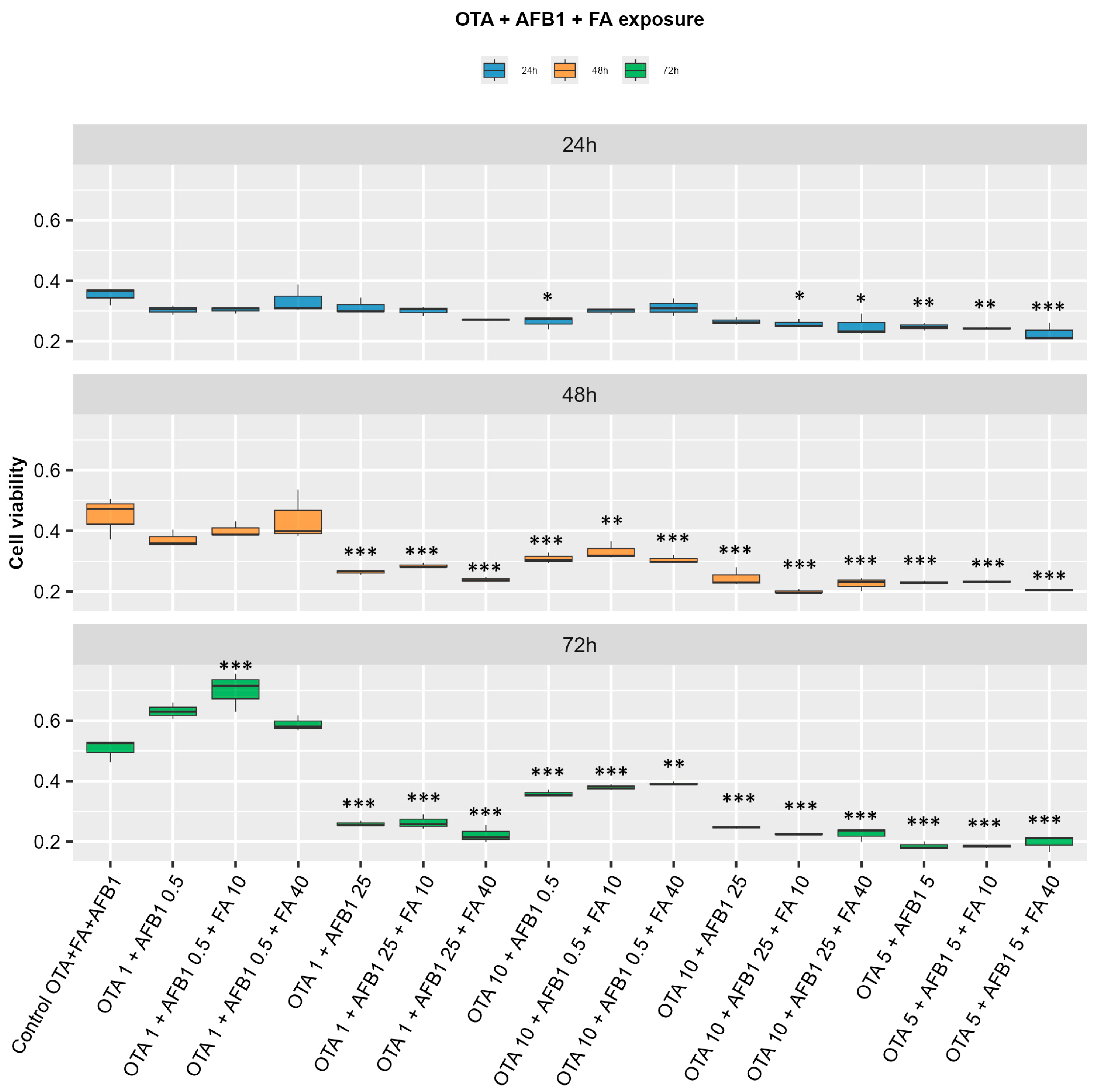
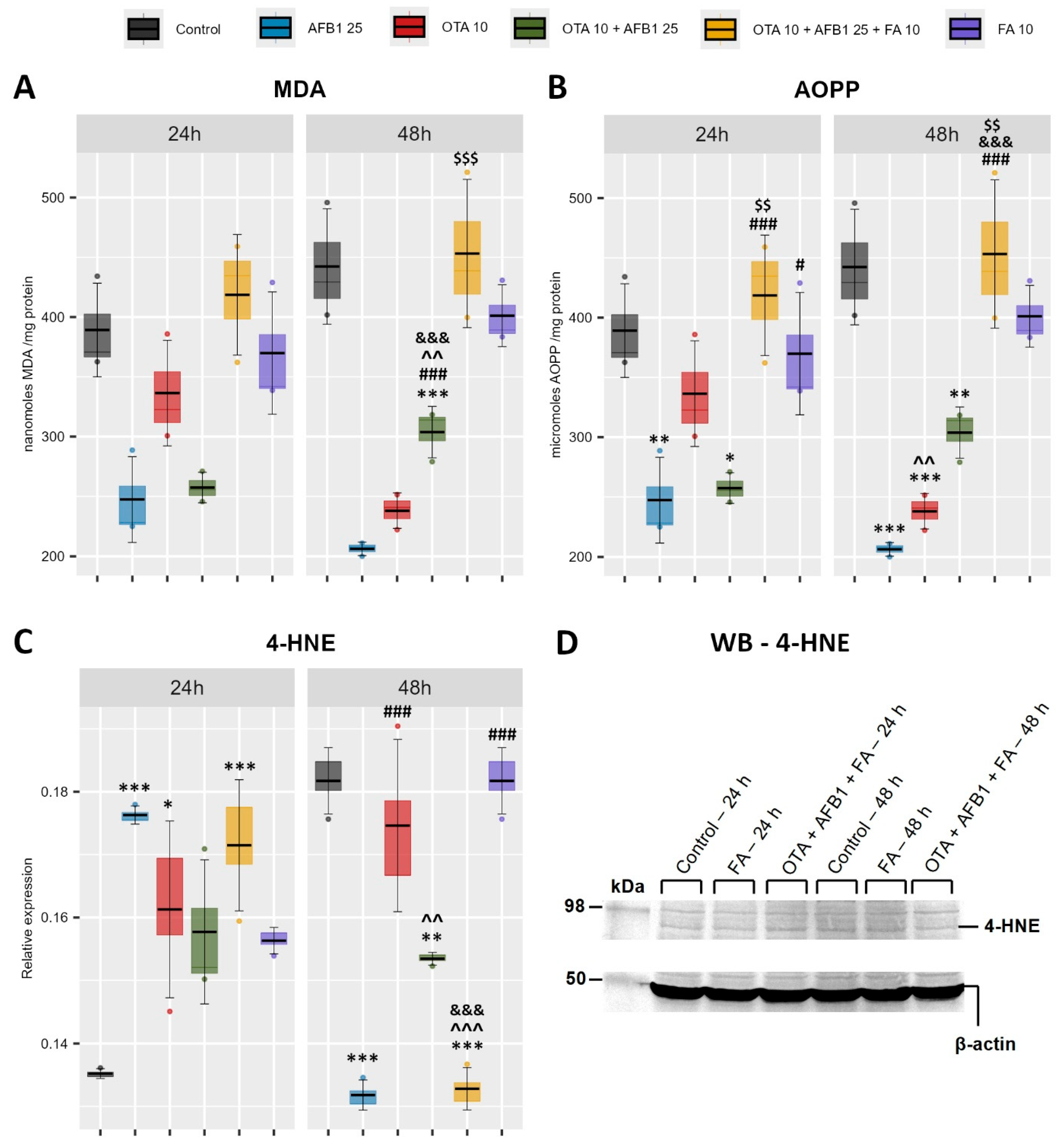
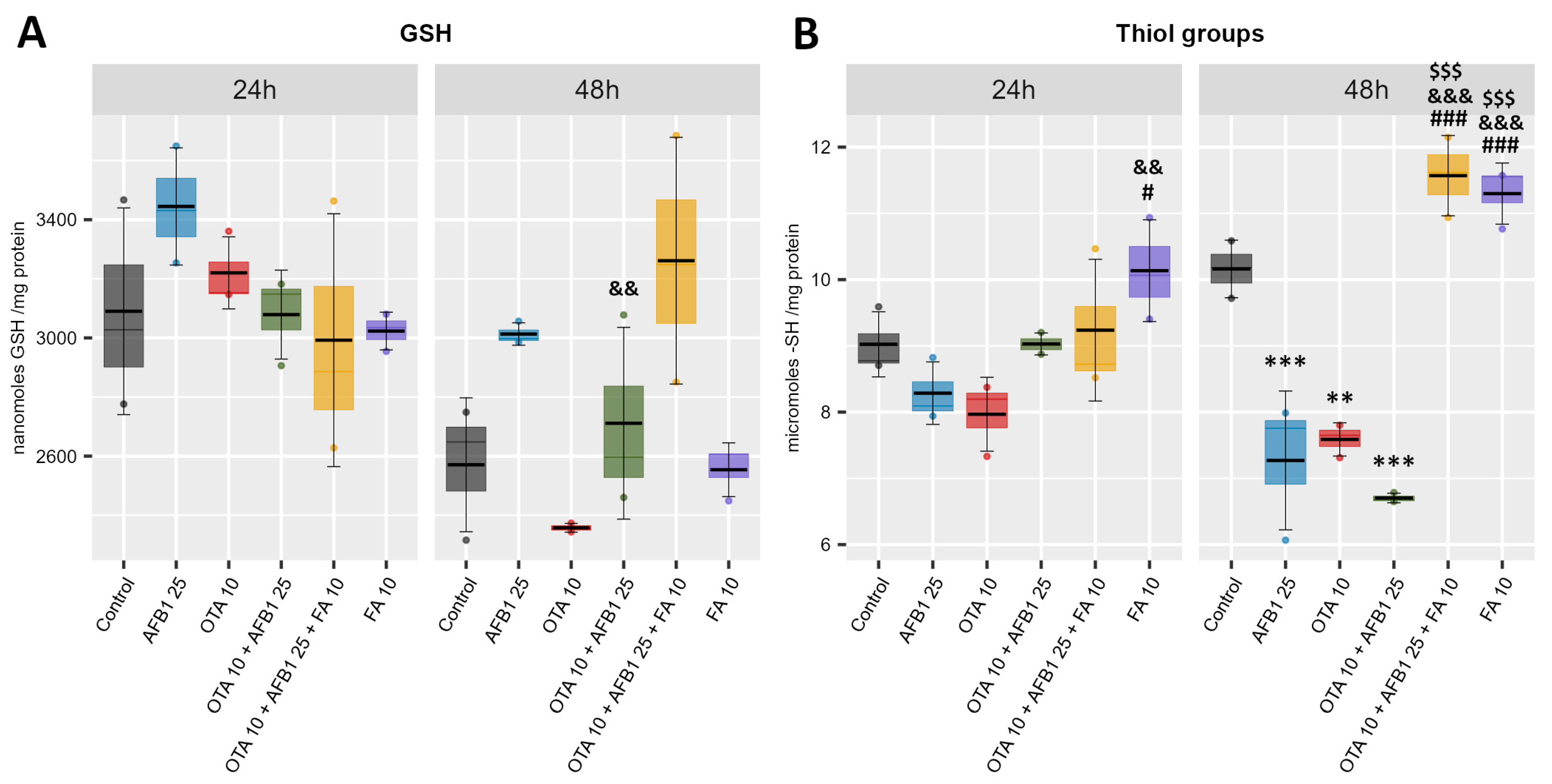
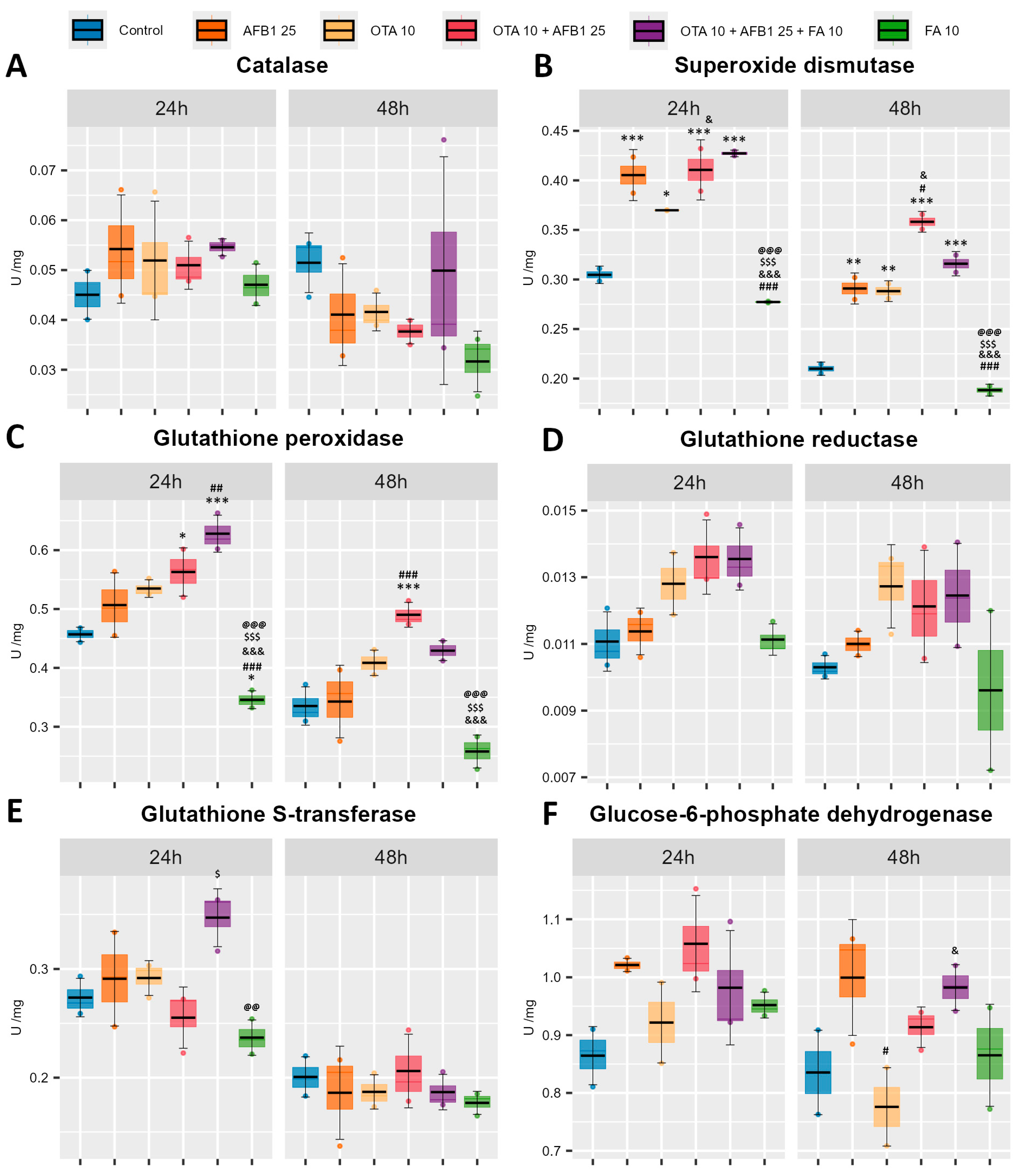
2. Results
2.1. Indicators of Oxidative Stress and Lipid Peroxidation
2.2. Nrf2 Activation as Response to Oxidative Stress
2.3. Non-Enzymatic Antioxidant Defense
2.4. Enzymatic Antioxidant Defense
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Cell Line
5.2. Cells’ Treatment and MTT Assay
5.3. Assessment of Oxidative Stress and Lipid Peroxidation
5.3.1. Detection of Malondialdehyde (MDA) Level
5.3.2. Advanced Oxidation Protein Products (AOPPs)
5.3.3. Western Blot
5.4. Non-Enzymatic Antioxidant Defense
5.4.1. Quantification of Reduced Glutathione (GSH) Concentration
5.4.2. Thiol Groups
5.5. Enzymatic Antioxidant Defense
5.5.1. Catalase
5.5.2. Superoxide Dismutase
5.5.3. Glutathione Peroxidase
5.5.4. Glutathione S-Transferase
5.5.5. Glutathione Reductase
5.5.6. Glucose-6-Phosphate Dehydrogenase
5.6. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Condition Name | Concentration [μM] | ||
|---|---|---|---|
| OTA | AFB1 | FA | |
| Control AFB1 | - | - | - |
| AFB1 0.5 | - | 0.5 | - |
| AFB1 5 | - | 5 | - |
| AFB1 25 | - | 25 | - |
| Control OTA | - | - | - |
| OTA 1 | 1 | - | - |
| OTA 5 | 5 | - | - |
| OTA 10 | 10 | - | - |
| Control FA | - | - | - |
| FA 10 | - | - | 10 |
| FA 40 | - | - | 40 |
| Control AFB1 + OTA | - | - | - |
| OTA 1 + AFB1 0.5 | 1 | 0.5 | - |
| OTA 5 + AFB1 5 | 5 | 5 | - |
| OTA 10 + AFB1 25 | 10 | 25 | - |
| OTA 1 + AFB1 25 | 1 | 25 | - |
| OTA 10 + AFB1 0.5 | 10 | 0.5 | - |
| Control OTA + AFB1 + FA | - | - | - |
| OTA 1 + AFB1 0.5 + FA 10 | 1 | 0.5 | 10 |
| OTA 1 + AFB1 0.5 + FA 40 | 1 | 0.5 | 40 |
| OTA 5 + AFB1 5 + FA 10 | 5 | 5 | 10 |
| OTA 5 + AFB1 5 + FA 40 | 5 | 5 | 40 |
| OTA 10 + AFB1 25 + FA 10 | 10 | 25 | 10 |
| OTA 10 + AFB1 25 + FA 50 | 10 | 25 | 40 |
| OTA 1 + AFB1 25 + FA 10 | 1 | 25 | 10 |
| OTA 1 + AFB1 25 + FA 40 | 1 | 25 | 40 |
| OTA 10 + AFB1 0.5 + FA 10 | 10 | 0.5 | 10 |
| OTA 10 + AFB1 0.5 + FA 40 | 10 | 0.5 | 40 |
References
- Walls, H.; Baker, P.; Chirwa, E.; Hawkins, B. Food security, food safety& healthy nutrition: Are they compatible? Glob. Food Sec. 2019, 21, 69–71. [Google Scholar]
- De Ruyck, K.; Huybrechts, I.; Yang, S.; Arcella, D.; Claeys, L.; Abbeddou, S.; De Keyzer, W.; De Vries, J.; Ocke, M.; Ruprich, J.; et al. Mycotoxin exposure assessments in a multi-center European validation study by 24-h dietary recall and biological fluid sampling. Environ. Int. 2020, 137, 105539. [Google Scholar] [CrossRef]
- Bălan, I.M.; Trasca, T.I.; Tulcan, C.; Belc, N.; Musteţea, G.; Tripon, M.Z.; Lattanzio, V.; Rychlik, M.; Duţă, D. From Safety to Security and to Food Ethics. In Worldwide Megatrends in Food Safety and Food Security; Marc, R.A., Mureşan, C.C., Postolache, A.N., Eds.; IntechOpen: London, UK, 2024; pp. 1–14. [Google Scholar]
- Frugier, C.; Bégin, P. Food toxins. In Encyclopedia of Food Allergy; Sicherer, S.H., Ed.; Elsevier: Amsterdam, The Netherlands, 2024; pp. 814–827. [Google Scholar]
- Goessens, T.; Mouchtaris-Michaelidis, T.; Teofariam, K.; Truong, N.N.; Vertriest, F.; Bader, Y.; De Saeger, S.; Lachat, C.; De Boevre, M. Dietary mycotoxin exposure and human health risks: A protocol for a systematic review. Environ. Int. 2024, 184, 108456. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, A.R.; Mohan, K.; Karthick Rajan, D.; Pillay, A.A.; Palanisami, T.; Sathichkimar, P.; Conterno, I. Distribution, toxicity, interactive effects and detection of ochrtoxin and deoxynivalenol in food. A review. Food Chem. 2022, 378, 131978. [Google Scholar] [CrossRef]
- Popescu, R.G.; Rădulescu, A.L.; Georgescu, S.E.; Dinischiotu, A. Aflatoxins in Feed: Types, Metabolism, Health Consequences in Swine and Mitigation Strategies. Toxins 2022, 14, 853. [Google Scholar] [CrossRef] [PubMed]
- Casu, A.; Leggieri, M.C.; Toscano, P.; Battilani, P. Changing climate shifting mycotoxins: A comprehensive review of climate change impact on mycotoxin contamination. Comp. Rev. Food Sci. Food Saf. 2024, 23, e13323. [Google Scholar] [CrossRef]
- Paterson, R.R.M.; Lima, N.; Taniwaki, M.H. Coffee, mycotoxins and climate change. Food Res. Int. 2014, 61, 1–15. [Google Scholar] [CrossRef]
- Nikolić, M.; Savić, I.; Nikolić, A.; Jauković, M.; Kandić, V.; Stevanović, M.; Stanković, S. Toxigenic species Aspergillus parasiticus originating from Maize Kernels grown in Serbia. Toxins 2021, 13, 847. [Google Scholar] [CrossRef]
- Gacem, M.A.; Ould El Hadj-Khelil, A.; Boudjemaa, B.; Gacem, H. Mycotoxins occurrence, toxicity and detection methods. In Sustainable Agriculture Reviews; Springer: Cham, Switzerland, 2020; Volume 40. [Google Scholar]
- Omer, R.E.; Kuijsten, A.; Kadaru, A.M.; Kok, F.J.; Idris, M.O.; El Khidir, I.M. Population-attributable risk of dietary aflatoxins and hepatitis B virus infection with respect to hepatocellular carcinoma. Nutr. Cancer 2004, 48, 15–21. [Google Scholar] [CrossRef]
- Turner, P.C.; Collinson, A.C.; Cheung, Y.B.; Gong, Y.Y.; Hall, A.J.; Prentice, A.M.; Wild, C.P. Aflatoxin exposure in utero causes growth falling in Gambian infants. Int. J. Epidemiol. 2007, 36, 1119–1125. [Google Scholar] [CrossRef]
- Ding, L.; Han, M.; Wang, X.; Guo, Y. Ochratoxin A: Overview of Prevention, Removal and Detoxification Methods. Toxins 2023, 15, 565. [Google Scholar] [CrossRef] [PubMed]
- Pfohl-Lezkowicz, A.; Manderville, R.A.; Ochratoxin, A. An overview on toxicity and carcinogenicity in animals and humans. Mol. Nutr. Food Res. 2007, 51, 61–99. [Google Scholar] [CrossRef]
- Pavlović, N.M. Balkan endemic nephropathy-Current status and future perspectives. Clin. Kidney J. 2013, 6, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Furlong, E.B.; Buffon, J.G.; Cerqueira, M.B.; Kupski, L. Mitigation of Mycotoxins in Food-Is it Possible? Foods 2024, 13, 1112. [Google Scholar] [CrossRef]
- Galvano, F.; Piva, A.; Ritieni, J.; Galvano, G. Dietary Strategies to Counteract the Effects of Mycotoxins: A Review. J. Food Prot. 2001, 64, 120–131. [Google Scholar] [CrossRef]
- da Silva, E.O.; Bracarense, A.P.F.I.; Oswald, L.P. Mycotoxins and oxidative stress: Where are we? World Mycotoxins J. 2018, 11, 113–133. [Google Scholar] [CrossRef]
- Cory, H.; Passarelli, S.; Szeto, J.; Tamez, M.; Mattei, J. The Role of Polyphenols in Human Health and Foos Systems: A Mini-Review. Front. Nutr. 2018, 5, 87. [Google Scholar] [CrossRef] [PubMed]
- Singla, R.; Dubey, A.K.; Garg, A.; Sharma, R.K.; Fiorino, M.; Ameen, S.M.; Haddad, M.A.; Al-Hiary, M. Natural Polyphenols: Chemical Classification, Definition of Classes, Subcategories and Structure. JAOAC Int. 2019, 102, 1397–1400. [Google Scholar]
- Srinivasan, M.; Sudheer, A.R.; Menon, V.P. Ferulic Acid: Therapeutic Potential Through Its Antioxidant Property. J. Clin. Biochem. Nutr. 2007, 40, 92–100. [Google Scholar] [CrossRef]
- Song, Y.; Wu, T.; Yang, Q.; Chen, X.; Wang, M.; Wang, Y. Ferulic acid alleviates the symptoms of diabetes in obese rats. J. Funct. Food 2014, 9, 141–147. [Google Scholar] [CrossRef]
- Yu, L.J.; Wu, F.; Jiang, M.; Yang, C.G.; Liu, W.; Tian, J. Ferulic acid: A natural compound as an efficient feed additive for GIFT (Oreochromis niloticus). Aquac. Nutr. 2018, 24, 27–35. [Google Scholar] [CrossRef]
- Ramar, M.; Manikandan, B.; Raman, T.; Priyadarsini, A.; Palanisamy, S.; Velayudam, M. Protective effect of ferulic acid and resveratrol against alloxan-induced diabetes in mice. Eur. J. Pharmacol. 2012, 690, 226–235. [Google Scholar] [CrossRef]
- Roy, S.; Metya, S.K.; Rahaman, N.; Sannigrahi, S.; Ahmed, F. Ferulic acid in the treatment of post-diabetes testicular damage: Relevance to the down regulation of apoptosis correlates with antioxidant status via modulation of TGF-β1, IL-1β and Akt signalling. Cell Biochem. Funct. 2014, 32, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Salazar-López, N.J.; Astiazarán-García, H.; González-Aguilar, G.A.; Loarca-Piña, G.; Ezquerra-Brauer, J.M.; Domínguez Avila, J.A.; Robles-Sánchez, M. Ferulic acid on glucose dysregulation, dyslipidemia, and inflammation in diet-induced obese rats: An integrated study. Nutrients 2017, 9, 675. [Google Scholar] [CrossRef]
- Wang, O.; Liu, J.; Cheng, Q.; Guo, X.; Wang, Y.; Zhao, L. Effects of ferulic acid and γ-Oryzanol on high-fat and high-fructose diet-induced metabolic syndrome in rats. PLoS ONE 2015, 10, e0118135. [Google Scholar] [CrossRef]
- Li, Y.; Li, L.; Li, J.; Zhang, L.; Gao, F.; Zhou, G. Effects of dietary supplementation with ferulic acid or vitamin E individually or in combination on meat quality and antioxidant capacity of finishing pigs. Asian-Australas J. Anim. Sci. 2015, 28, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Valadez-García, K.M.; Avendaño-Reyes, L.; Meza-Herrera, C.A.; Mellado, M.; Díaz-Molina, R.; González-Ríos, H.; Macías-Cruz, U. Ferulic acid in animal feeding: Mechanisms of action, productive benefits, and future perspectives in meat production. Food Biosci. 2021, 43, 101247. [Google Scholar] [CrossRef]
- Hussein, A.M.; Abbas, K.M.; Abulseoud, O.A.; El-Hussainy, E.H.M.A. Effects of ferulic acid on oxidative stress, heat shock protein 70, connexin 43, and monoamines in the hippocampus of pentylenetetrazole-kindled rats. Can. J. Physiol. Pharmacol. 2017, 95, 732–742. [Google Scholar] [CrossRef]
- Ma, Z.; Hong, Q.; Wang, Y.; Liang, Q.; Tan, H.; Xiao, C. Ferulic acid induces heme oxygenase-1 via activation of ERK and Nrf2. Drug Discov. Ther. 2011, 5, 299–305. [Google Scholar] [CrossRef]
- Ma, Z.C.; Hong, Q.; Wang, Y.G.; Tan, H.L.; Xiao, C.R.; De Liang, Q. Effects of ferulic acid on hematopoietic cell recovery in whole-body gamma irradiated mice. Int. J. Radiat. Biol. 2011, 87, 499–505. [Google Scholar] [CrossRef]
- Hasanein, P.; Fazeli, F.; Parviz, M.; Roghani, M. Ferulic acid prevents lead-induced testicular oxidative stress and suppressed spermatogenesis in rats. Andrologia 2018, 50, e12798. [Google Scholar] [CrossRef] [PubMed]
- Salma, M.; Abdel, F.; Karam, H.; El Hameed, M. The potential protective effect of ferulic acid against gamma irradiation induced ovarian failure in rats. Egypt. J. Radiat. Sci. Appl. 2019, 32, 1–12. [Google Scholar]
- Tanihara, F.; Hirata, M.; Nhien, N.T.; Hirano, T.; Kunihara, T.; Otoi, T. Effect of ferulic acid supplementation on the developmental competence of porcine embryos during in vitro maturation. J. Vet. Med. Sci. 2018, 80, 1007–1011. [Google Scholar] [CrossRef]
- Wang, X.; Yang, F.; Na, L.; Jia, M.; Ishfaq, M.; Zhang, Y.; Wu, C. Ferulic acid alleviates AFB1-induced duodenal barrier damage in rats via up-regulating tight junction proteins, down-regulating ROCK, competing CYP450 enzyme and activating GST. Ecotoxicol. Environ. Saf. 2022, 241, 113805. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Yu, W.; Duan, N.; Wang, Z.; Shen, Y.; Wu, S. Protective Effects of Ferulic Acid on deoxynivalenol-Induced Toxicity in IPEC-J2 Cells. Toxins 2022, 14, 275. [Google Scholar] [CrossRef]
- Frangiamone, M.; Lázaro, A.; Cimbala, A.; Font, G.; Manyes, L. In vitro and in vivo assessment of AFB1 and OTA toxic effects and the beneficial role of bioactive compounds. A system review. Food Chem. 2024, 447, 138909. [Google Scholar] [CrossRef] [PubMed]
- Fato, M. In vitro models replicating the human intestinal epithelium for absorption and metabolism studies: A systematic review. J. Contr. Rel. 2021, 335, 247–268. [Google Scholar]
- Jones, R.M.; Mercante, J.W.; Neish, A.S. Reactive Oxygen Production Induced by the Gut Microbiota: Pharmatherapeutic Implications. Curr. Med. Chem. 2012, 19, 1519–1529. [Google Scholar] [CrossRef]
- Petterson, M.D.; Mooseker, M.S. Characterization of the enterocyte-like brush border cytoskeleton of C2BBE clones of the human intestinal cell line, Caco-2. J. Cell Sci. 1992, 102, 581–600. [Google Scholar] [CrossRef]
- Andrade, M.E.; Araujo, R.S.; de Barros, P.A.; Soares, A.D.; Abrantes, F.A.; Generoso, S.V.; Fernandes, S.O.; Cardoso, V.N. The role of immunomodulators on intestinal barrier homeostasis in experimental models. Clin. Nutr. 2015, 34, 1080–1087. [Google Scholar] [CrossRef]
- Hagelberg, S.; Hult, K.; Fuchs, R. Toxicokinetics of Ochratoxin A in several species and its plasma-binding properties. J. Appl. Toxicol. 1989, 9, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Roth, A.; Chakor, K.; Creppy, E.E.; Kane, A.; Roschenthaler, R.; Dirheimer, G. Evidence for an enterohepatic circulation of Ochratoxin A in mice. Toxicol 1988, 48, 293–308. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Liang, J.; Wu, D.; Guo, X.; Cao, H.; Zhang, C.; Liu, P.; Hu, R.; Hu, G.; Zhuang, Y. Blunting ROS/TRPML1 pathway protects AFB1-induced porcine intestinal epithelial cells apoptosis by restoring impaired autophagic flux. Ecotox. Environ. Saf. 2023, 257, 114942. [Google Scholar] [CrossRef]
- Wang, H.; Zhai, N.; Chen, Y.; Fu, C.; Huang, K. OTA induces intestinal epithelial barrier dysfunction and tight junction disruption in IPEC-J2 cells through ROS/Ca2+- mediated MLCK activation. Environ. Pollut. 2018, 242, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Toskulkao, C.; Glinsukon, T. Hepatic mitochondrial function and lysosomal enzyme activity in ethanol-potentiated aflatoxin B1 hepatotoxicity. Toxicol. Lett. 1990, 52, 179–190. [Google Scholar] [CrossRef]
- Ren, X.-L.; Han, P.; Meng, Y. Aflatoxin B1-induced COX-2 expression promotes mitophagy and contributes to lipid accumulation in hepatocytes in vitro and in vivo. Int. J. Toxicol. 2020, 39, 594–604. [Google Scholar] [CrossRef]
- Li, Q.; Dong, Z.; Lian, W.; Cui, J.; Wang, J.; Shen, H.; Liu, W.; Yang, J.; Zhang, X.; Cui, H. Ochratoxin A causes mitochondrial dysfunction apoptotic and autophagic cell death and also induces mitochondrial biogenesis in human gastric epithelium cells. Arch. Toxicol. 2019, 93, 1141–1155. [Google Scholar] [CrossRef]
- Gayathri, L.; Dhivya, R.; Dhanasekaran, D.; Periasamy, V.S.; Alshatwi, A.A.; Akbarsha, M.A. Hepatotoxic effect of ochratoxin A and citrinin, alone and in combination, and protective effect of vitamin E: In vitro study in HepG2 cell. Food Chem. Toxicol. 2015, 83, 151–163. [Google Scholar] [CrossRef]
- Parlakgül, G.; Pang, S.; Artico, L.L.; Min, N.; Cagampan, E.; Villa, R.; Goncalves, R.L.S.; Lee, G.Y.; Xu, C.S.; Hotamisligil, G.S.; et al. Spatial mapping of hepatic ER and mitochondrion architecture reveals zonated remodeling in fasting and obesity. Nat. Commun. 2024, 15, 3982. [Google Scholar] [CrossRef]
- Dinkova-Kostova, A.T.; Massiah, M.A.; Bozak, R.E.; Hicks, R.J.; Talalay, P. Potency of Michael reaction acceptors as inducers of enzymes that protect against carcinogenesis depends on their reactivity with sulfhydryl groups. Proc. Natl. Acad. Sci. USA 2001, 98, 3404–3409. [Google Scholar] [CrossRef]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef] [PubMed]
- Mu, K.; Kitts, D.D. Intestinal polyphenol antioxidant activity involves redox signaling mechanisms facilitated by aquaporin activity. Redox. Biol. 2023, 68, 102948. [Google Scholar] [CrossRef]
- Itagaki, S.; Kurokawa, T.; Nakata, C.; Saita, Y.; Oikawa, S.; Kobayashi, M.; Hirana, T.; Iseki, K. In vitro and in vivo antioxidant properties of ferulic acid: A comparative study with other natural oxidation inhibitors. Food Chem. 2009, 114, 466–470. [Google Scholar] [CrossRef]
- Baker, A.; Lin, C.-C.; Lett, C.; Karpinska, B.; Wright, M.H.; Foyer, C.H. Catalase: A critical node in the regulation of cell fate. Free Rad. Biol. Med. 2023, 199, 56–66. [Google Scholar] [CrossRef]
- Thompson, J.L.; Thomas, P.M.; Schiller, K.A. Purification and properties of a glutathione peroxidase from Southern bluefin tuna (Thunnus maccoyii) liver. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2006, 144, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid Peroxidation: Production, Metabolism, and Signaling Mechanisms of Malondialdehyde and 4-Hydroxy-2-Nonenal. Oxidative Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef]
- Gaschler, M.M.; Stockwell, B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017, 482, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, D.; Meade, G.; Foley, V.M.; Dowd, C.A. Structure, function and evolution of glutathione transferases: Implications for classification of non-mammalian members of an ancient enzyme superfamily. Biochem. J. 2001, 360, 1–16. [Google Scholar] [CrossRef]
- Dalleau, S.; Baradat, M.; Guéraud, F.; Huc, L. Cell death and diseases related to oxidative stress: 4-hydroxynonenal (HNE) in the balance. Cell Death Differ. 2013, 20, 1615–1630. [Google Scholar] [CrossRef]
- Popescu, R.G.; Marinescu, G.C.; Rădulescu, A.L.; Marin, D.E.; Țăranu, I.; Dinischiotu, A. Natural Antioxidant By-Product Mixture Counteracts the Effects of Aflatoxin B1 and Ochratoxin A Exposure of Piglets after Weaning: A Proteomic Survey on Liver Microsomal Fraction. Toxins 2023, 15, 299. [Google Scholar] [CrossRef]
- Chen, L.; Zong, R.; Zhou, J.; Ma, J.X.; Liu, Z.; Zhou, Y. The oxidant role of 4-hydroxynonenal in corneal epithelium. Sci. Rep. 2015, 5, 10630. [Google Scholar] [CrossRef] [PubMed]
- Gryszczyńska, B.; Formanowicz, D.; Budzyń, M.; Wanic-Kossowska, M.; Pawliczak, E.; Formanowicz, P.; Majewski, W.; Strzyżewski, K.W.; Kasprzak, M.P.; Iskra, M. Advanced Oxidation Protein Products and Carbonylated Proteins as Biomarkers of Oxidative Stress in Selected Atherosclerosis-Mediated Diseases. Biomed. Res. Int. 2017, 2017, 4975264. [Google Scholar] [CrossRef]
- Davies, K.J.; Goldberg, A.L. Oxygen radicals stimulate intracellular proteolysis and lipid peroxidation by independent mechanisms in erythrocytes. J. Biol. Chem. 1987, 262, 8220–8226. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.J. Protein oxidation and peroxidation. Biochem. J. 2016, 473, 805–825. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of its protective roles, measurement, and biosynthesis. Mol. Asp. Med. 2009, 30, 1–12. [Google Scholar] [CrossRef]
- Schaaf, G.J.; Nijmeijer, S.M.; Maas, R.F.M.; Roestenberg, P.; de Groene, E.M.; Gremmels, F. The role of oxidative stress in the ochratoxin A-mediated toxicity in proximal tubular cells. Biochim. Biophys. Acta-Mol. Basis Dis. 2002, 1588, 149–158. [Google Scholar] [CrossRef]
- Garcia-Pćrez, E.; Ryu, D.; Lee, C.; Lee, H.J. Ochratoxin A Induces Oxidative Stress in HepG2 Cells by Impairing the Gene Expression of Antioxidant Enzymes. Toxins 2021, 13, 271. [Google Scholar] [CrossRef]
- Couto, N.; Wood, J.; Barber, J. The role of glutathione reductase and related enzymes on cellular redox homeostasis network. Free Rad. Biol. Med. 2016, 95, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Fan, Q.; Liang, Q.; Wu, Y.; Ye, Z.; Wu, H.; Sun, Q.; Tang, H.; Liu, Y.; Liu, Q.; et al. Human CYP1A1-activated aneugenicity of aflatoxin B1 in mammalian cells and its combined effect with benzo(a)pyrene. Chem. Biol. Interact. 2024, 392, 110923. [Google Scholar] [CrossRef]
- Lee, H.J.; Pyo, M.C.; Shin, H.G.; Ryu, D.; Lee, K.-W. Renal toxicity through AhR, PXR, and Nrf2 signaling pathway activation of ochratoxin A-induced oxidative stress in kidney cells. Food Chem. Toxicol. 2018, 122, 59–68. [Google Scholar] [CrossRef]
- Cremers, C.M.; Jakob, U. Oxidant sensing by reversible disulfide bond formation. J. Biol. Chem. 2013, 288, 26489–26496. [Google Scholar] [CrossRef]
- Teixeira, J.; Gaspar, A.; Garrido, E.M.; Garrido, J.; Borges, F. Hydroxycinnamic acid antioxidants: An electrochemical overview. Biomed. Res. Int. 2013, 2013, 251754. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem 1976, 72, 248–254. [Google Scholar] [CrossRef] [PubMed]
- del Rio, D.; Pellegrini, N.; Colombi, B.; Bianchi, M.; Serafini, M.; Torta, F.; Tegoni, S.M.; Musci, M.; Brighenti, F. Rapid fluorimetric method to detect total plasmamalondialdehyde with mild derivatization conditions. Clin. Chem. 2003, 49, 690–692. [Google Scholar] [CrossRef] [PubMed]
- Witko-Sarsat, V.; Friedlander, M.; Capeillère-Blandin, C.; Nguyen-Khoa, T.; Nguyen, A.T.; Zingraff, J.; Jungers, P.; Descamps-Latscha, B. Advanced oxidation protein products as a novel marker of oxidative stress in uremia. Kidney Int. 1996, 49, 1304–1313. [Google Scholar] [CrossRef]
- Popescu, R.G.; Avramescu, S.; Marin, D.E.; Țăranu, I.; Georgescu, S.E.; Dinischiotu, A. The Reduction of the Combined Effects of Aflatoxin and Ochratoxin A in Piglet Livers and Kidneys by Dietary Antioxidants. Toxins 2021, 13, 648. [Google Scholar] [CrossRef]
- Ellman, G.L. Tissue sulfhydryl groups. Arch. Biochem. Biophys. 1959, 82, 70–77. [Google Scholar] [CrossRef]
- Riener, C.K.; Kada, G.; Gruber, H.J. Quick measurement of protein sulfhydryls with Ellman’s reagent and with 4,4′-dithiodipyridine. Anal. Bioanal. Chem. 2002, 373, 266–276. [Google Scholar] [CrossRef]
- Beers, R.F.; Sizer, I.W. A spectrophotometric method for measuring the breakdown of hydrogen peroxide by catalase. J. Biol. Chem. 1952, 195, 133–140. [Google Scholar] [CrossRef]
- Paoletti, F.; Aldinucci, D.; Mocali, A.; Caparrini, A. A sensitive spectrophotometric method for the determination of superoxide dismutase activity in tissue extracts. Anal. Biochem. 1986, 154, 536–541. [Google Scholar] [CrossRef]
- Beutler, E. Red Cell Metabolism: A Manual of Biochemical Methods, 3rd ed.; Grune and Stratton: Orlando, FL, USA, 1984; pp. 68–73. ISBN 978-0808916727. [Google Scholar]
- Habig, W.H.; Pabst, M.J.; Jakoby, W.B. Glutathione S-transferases. The first enzymatic step in mercapturic acid formation. J. Biol. Chem 1974, 249, 7130–7139. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, D.M.; Spooner, R.J. Glutathione reductase. In Methods of Enzymatic Analysis, 3rd ed.; Bergmeyer, H.U., Ed.; Verlag Chemie: Weinheim, Germany, 1986; Volume 3, pp. 258–265. ISBN 9780895732330. [Google Scholar]
- Lohr, G.W.; Waller, H.D. Glucose-6-phosphate dehydrogenase. In Methods of Enzymatic Analysis, 2nd ed.; Bergmeyer, H.U., Ed.; Academic Press: New York, NY, USA, 1974; Volume 4, pp. 744–751. ISBN 9780323161329. [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rădulescu, A.-L.; Popescu, R.G.; Balas, M.; Marinescu, G.C.; Dinischiotu, A. Modulation of the Antioxidant System of Caco-2 Cells in the Presence of Aflatoxin B1, Ochratoxin A, and Ferulic Acid. Toxins 2025, 17, 274. https://doi.org/10.3390/toxins17060274
Rădulescu A-L, Popescu RG, Balas M, Marinescu GC, Dinischiotu A. Modulation of the Antioxidant System of Caco-2 Cells in the Presence of Aflatoxin B1, Ochratoxin A, and Ferulic Acid. Toxins. 2025; 17(6):274. https://doi.org/10.3390/toxins17060274
Chicago/Turabian StyleRădulescu, Andreea-Luminița, Roua Gabriela Popescu, Mihaela Balas, George Cătălin Marinescu, and Anca Dinischiotu. 2025. "Modulation of the Antioxidant System of Caco-2 Cells in the Presence of Aflatoxin B1, Ochratoxin A, and Ferulic Acid" Toxins 17, no. 6: 274. https://doi.org/10.3390/toxins17060274
APA StyleRădulescu, A.-L., Popescu, R. G., Balas, M., Marinescu, G. C., & Dinischiotu, A. (2025). Modulation of the Antioxidant System of Caco-2 Cells in the Presence of Aflatoxin B1, Ochratoxin A, and Ferulic Acid. Toxins, 17(6), 274. https://doi.org/10.3390/toxins17060274