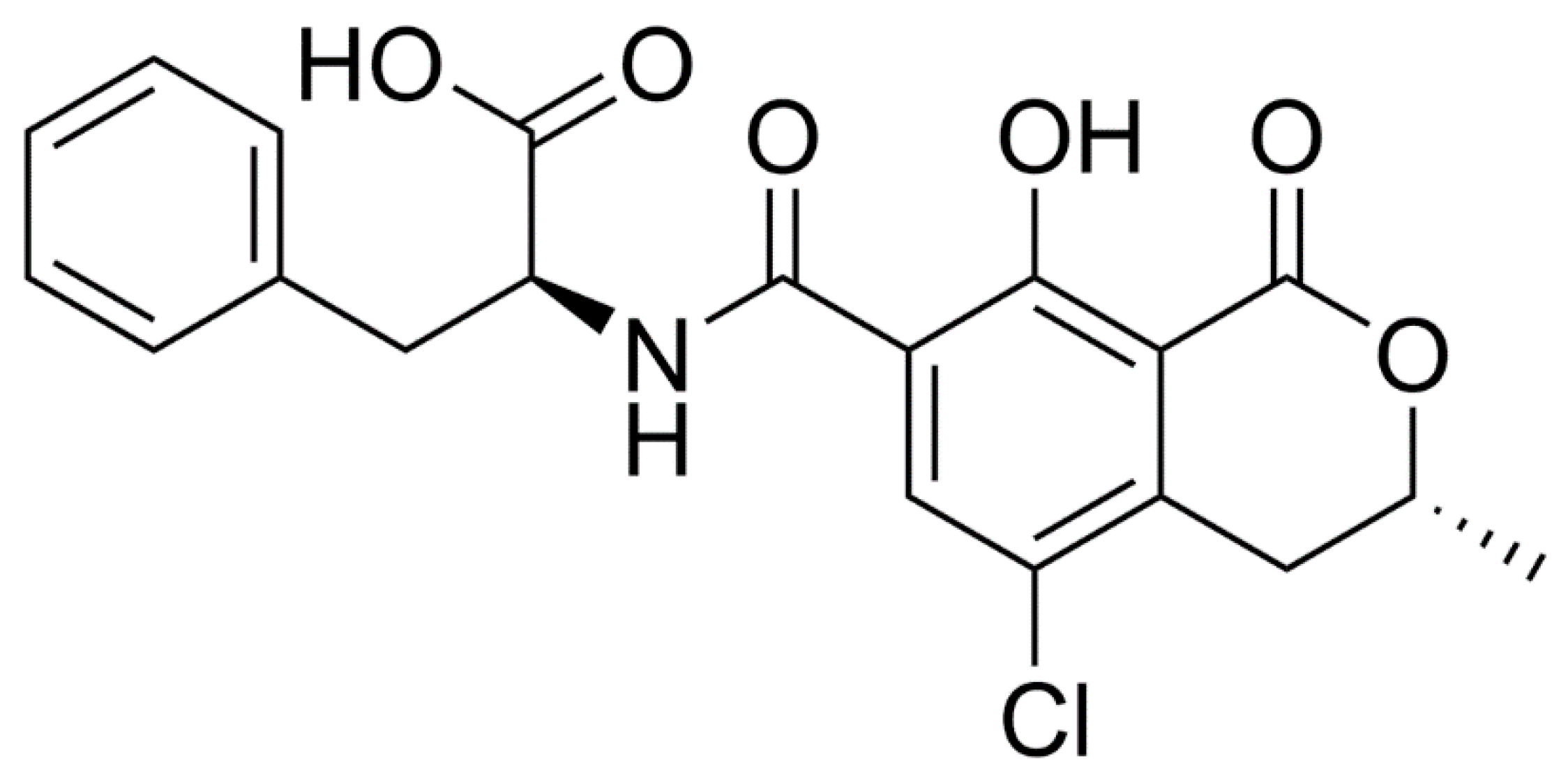
The Neurotoxic Effect of Ochratoxin-A on the Hippocampal Neurogenic Niche of Adult Mouse Brain

 , , , and
, , , and
Abstract
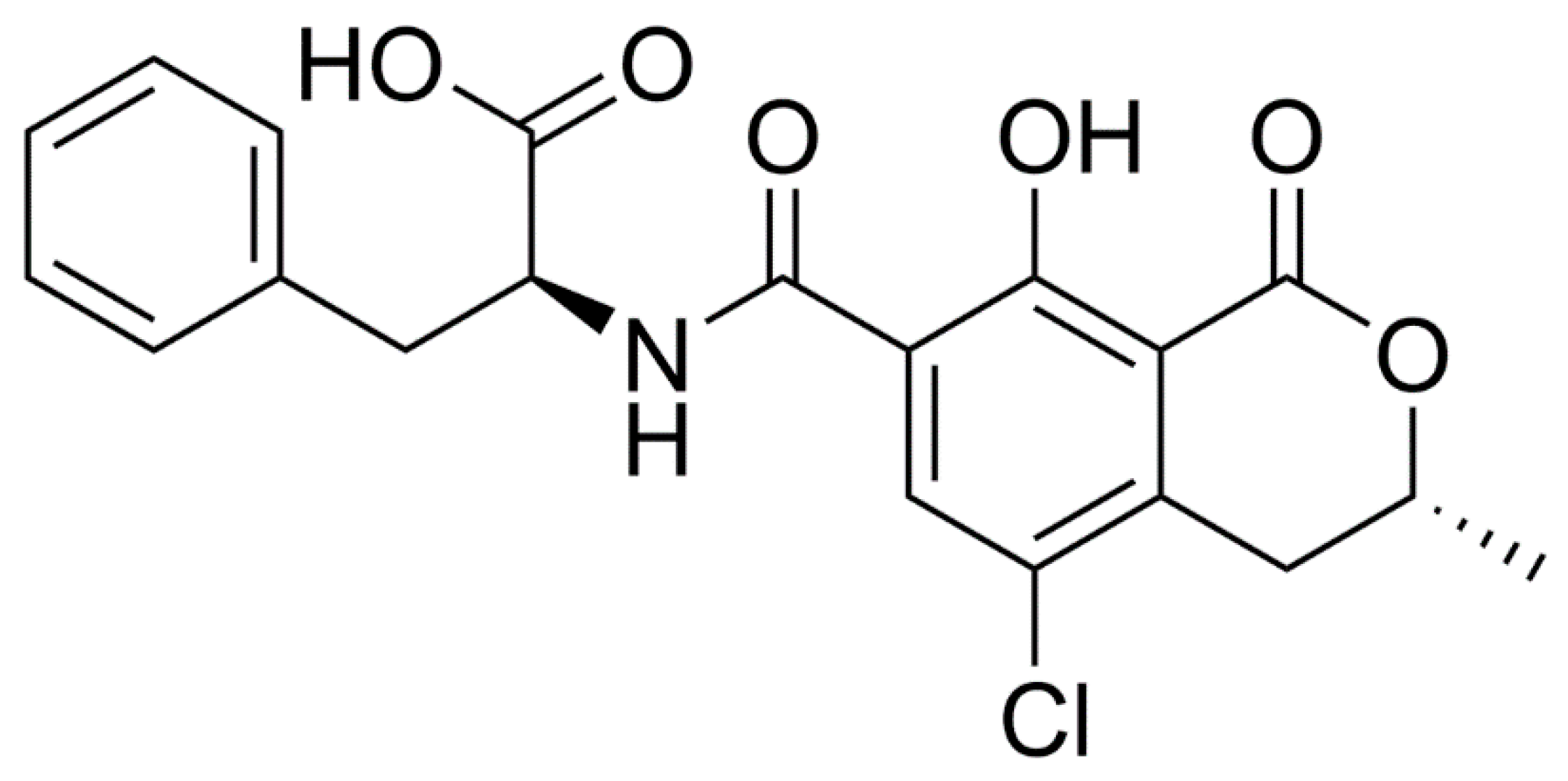
:1. Introduction
2. Results
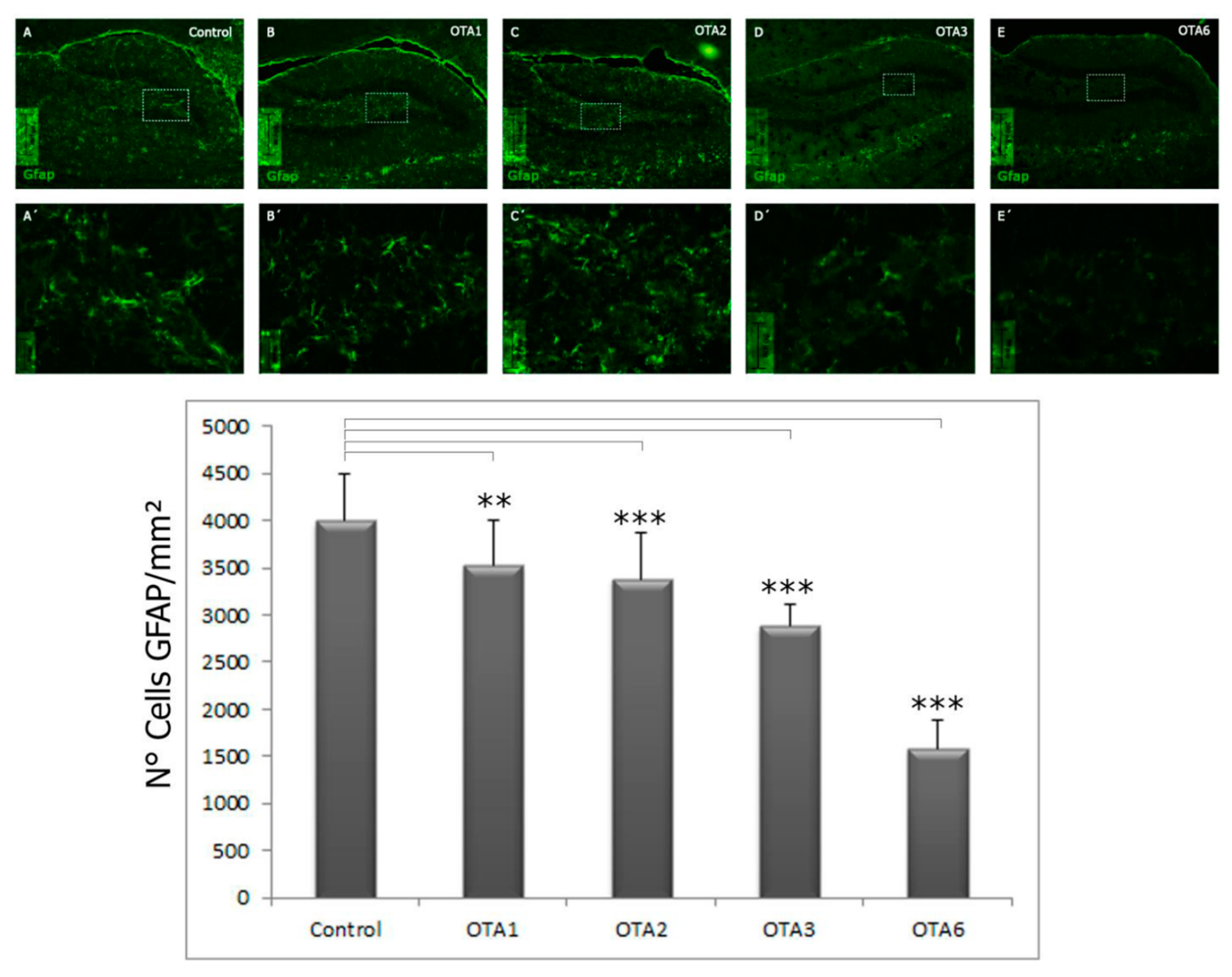
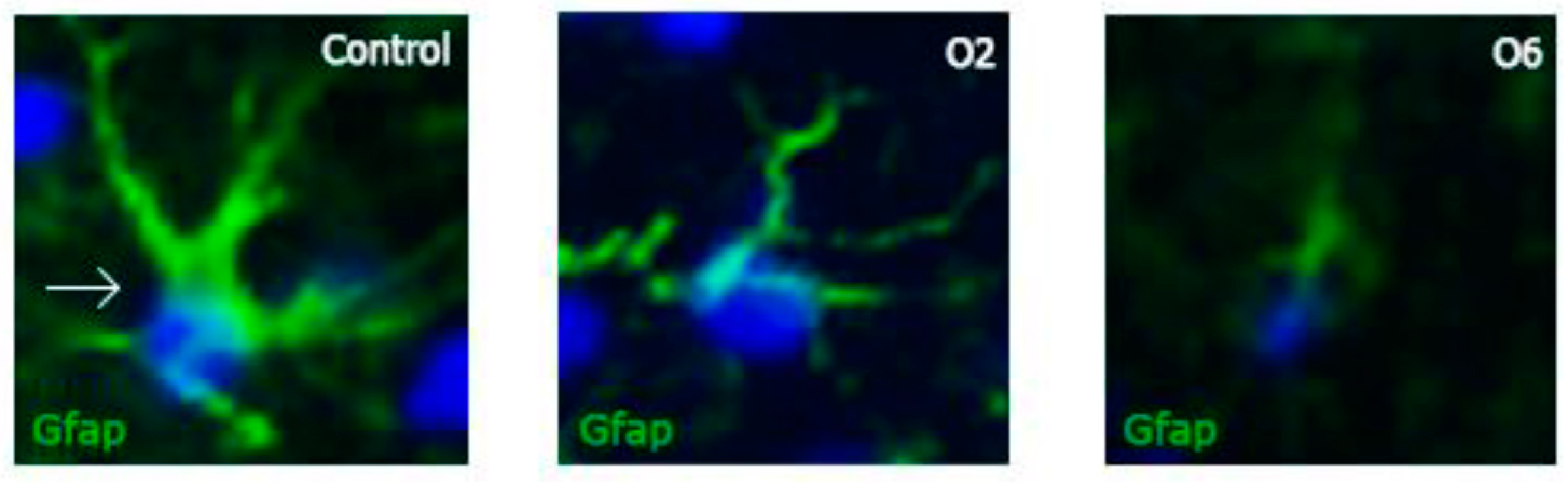
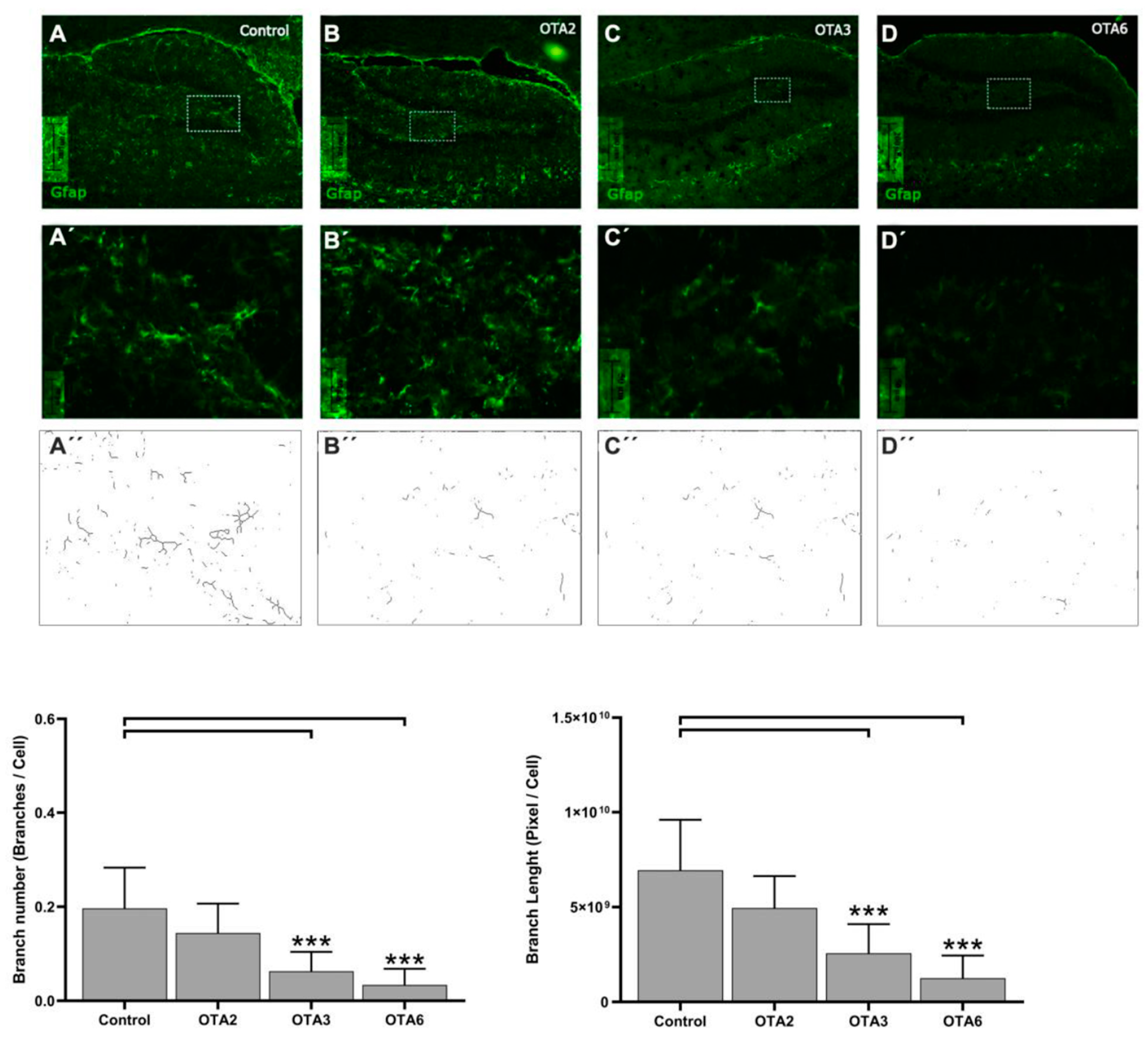
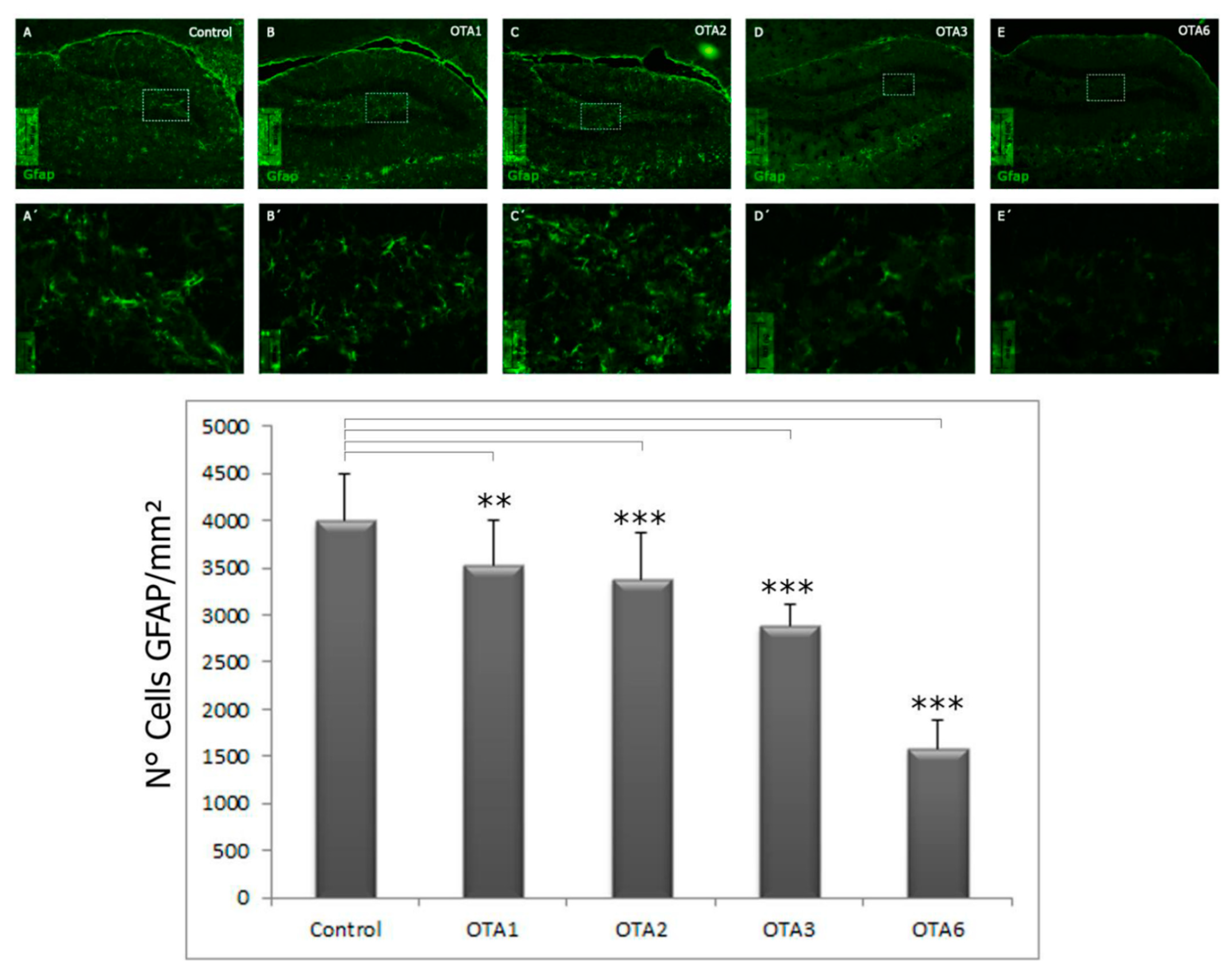
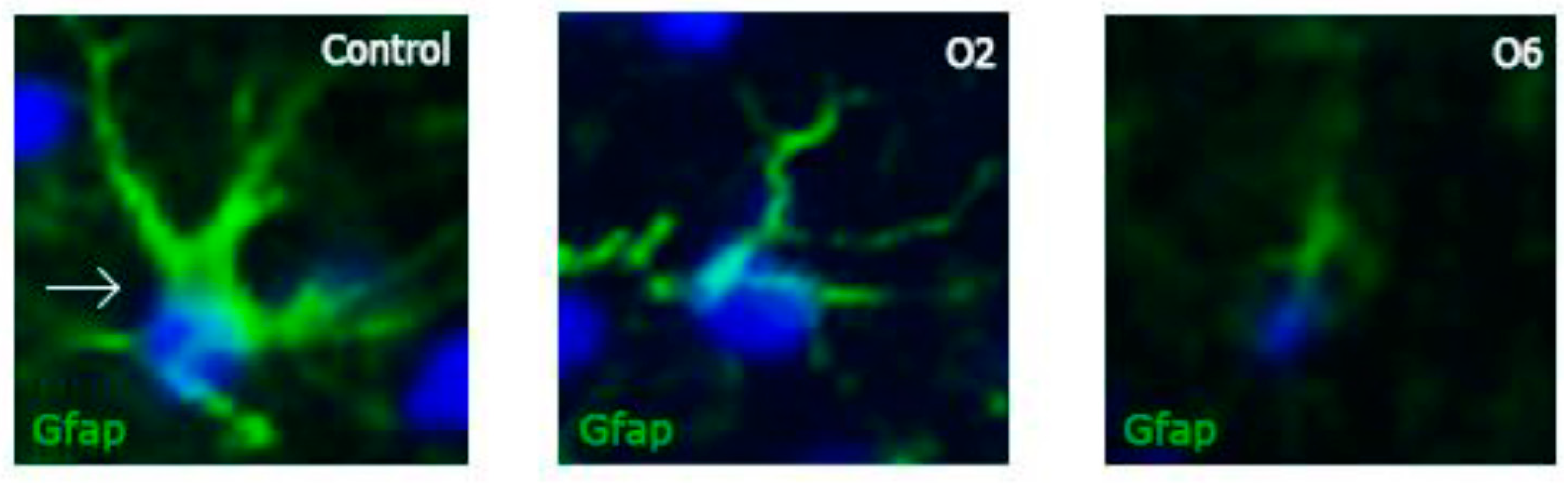
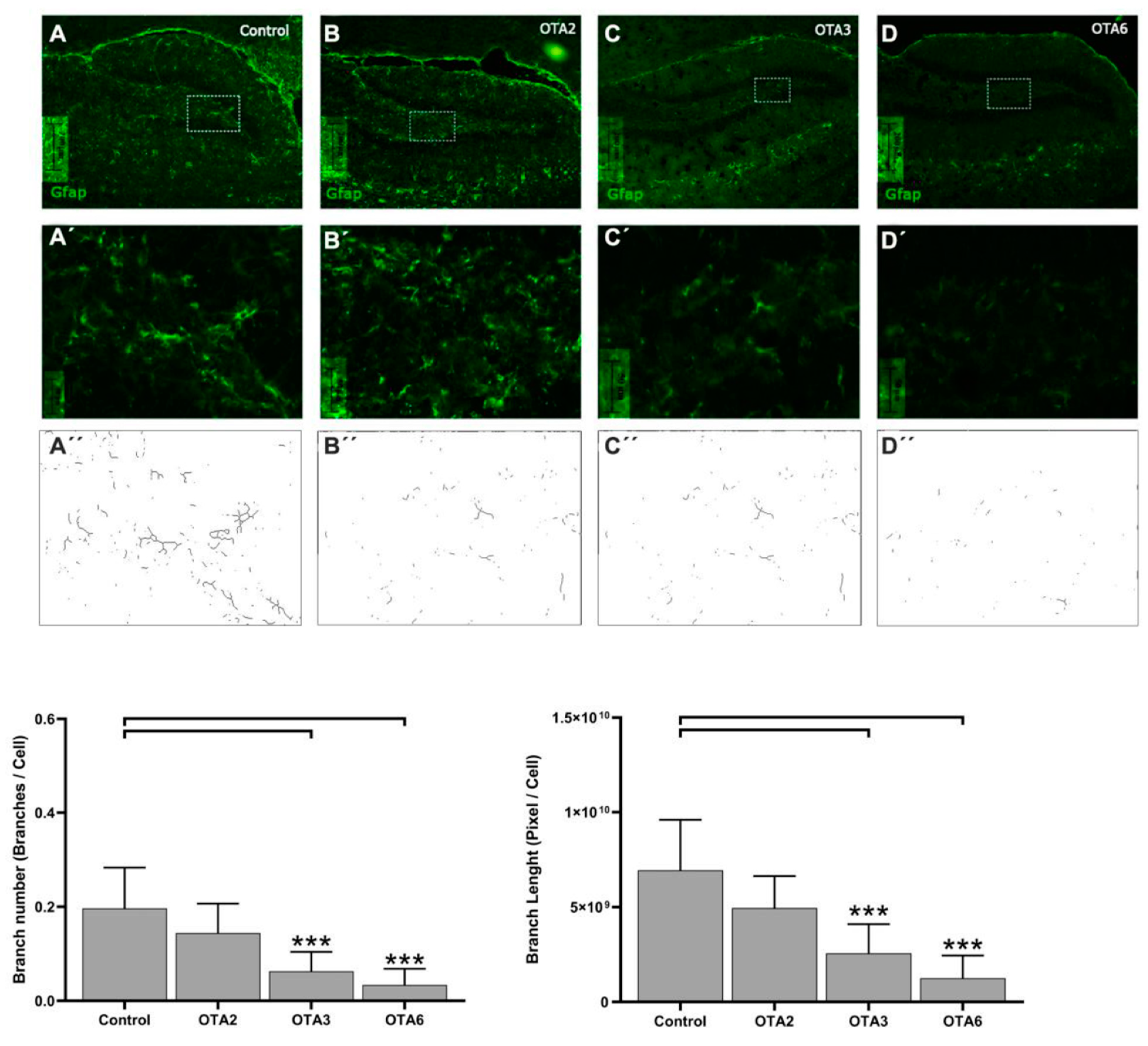
2.1. Ochratoxin A Reduces the Number of Astrocytes in the Dentate Gyrus
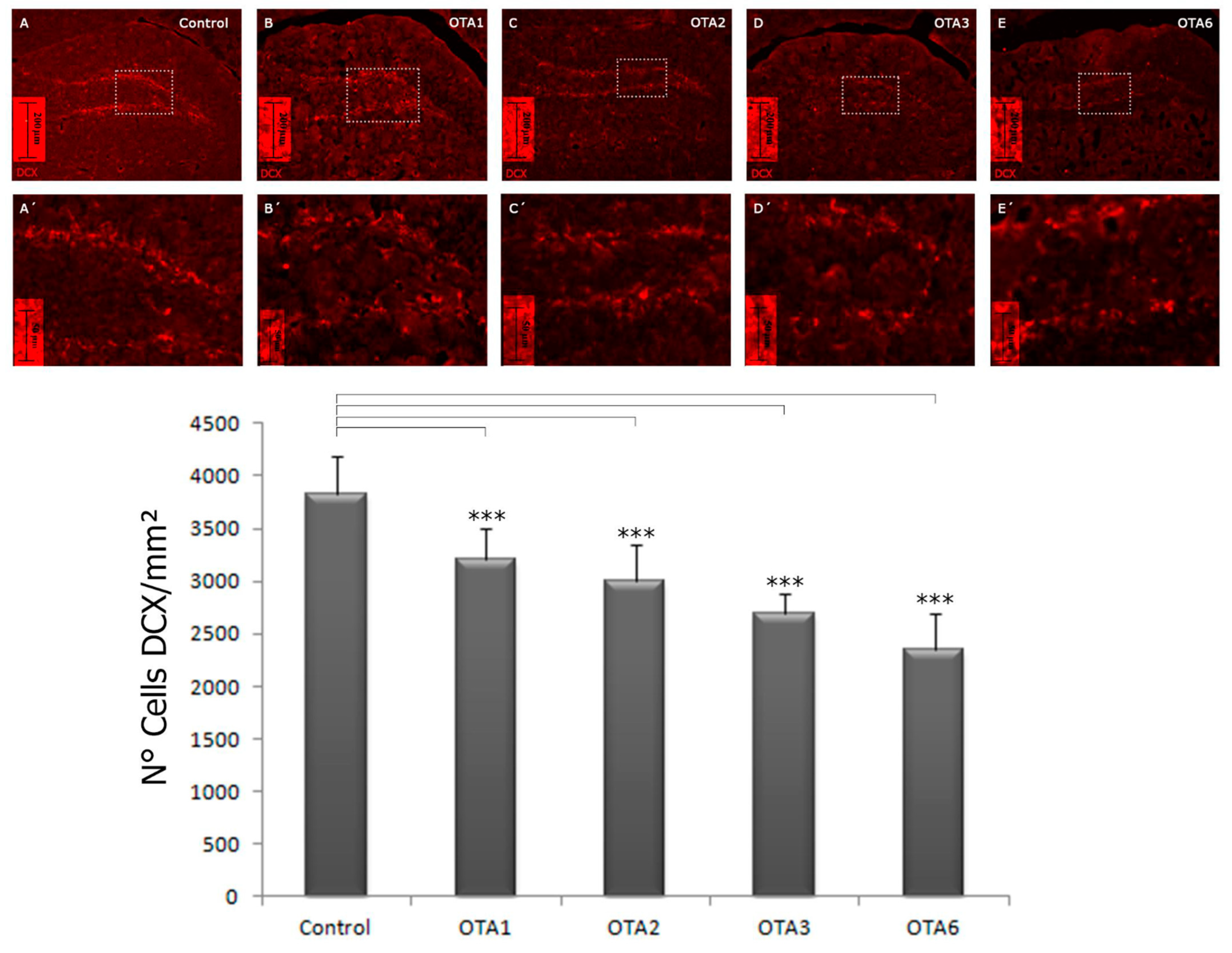
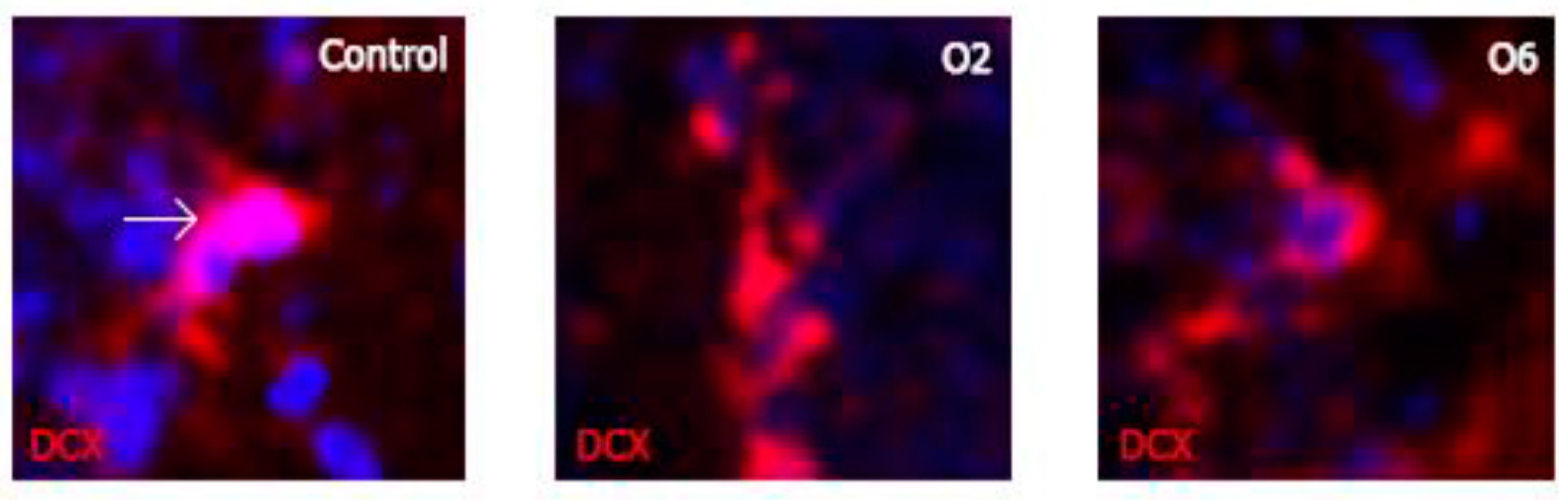
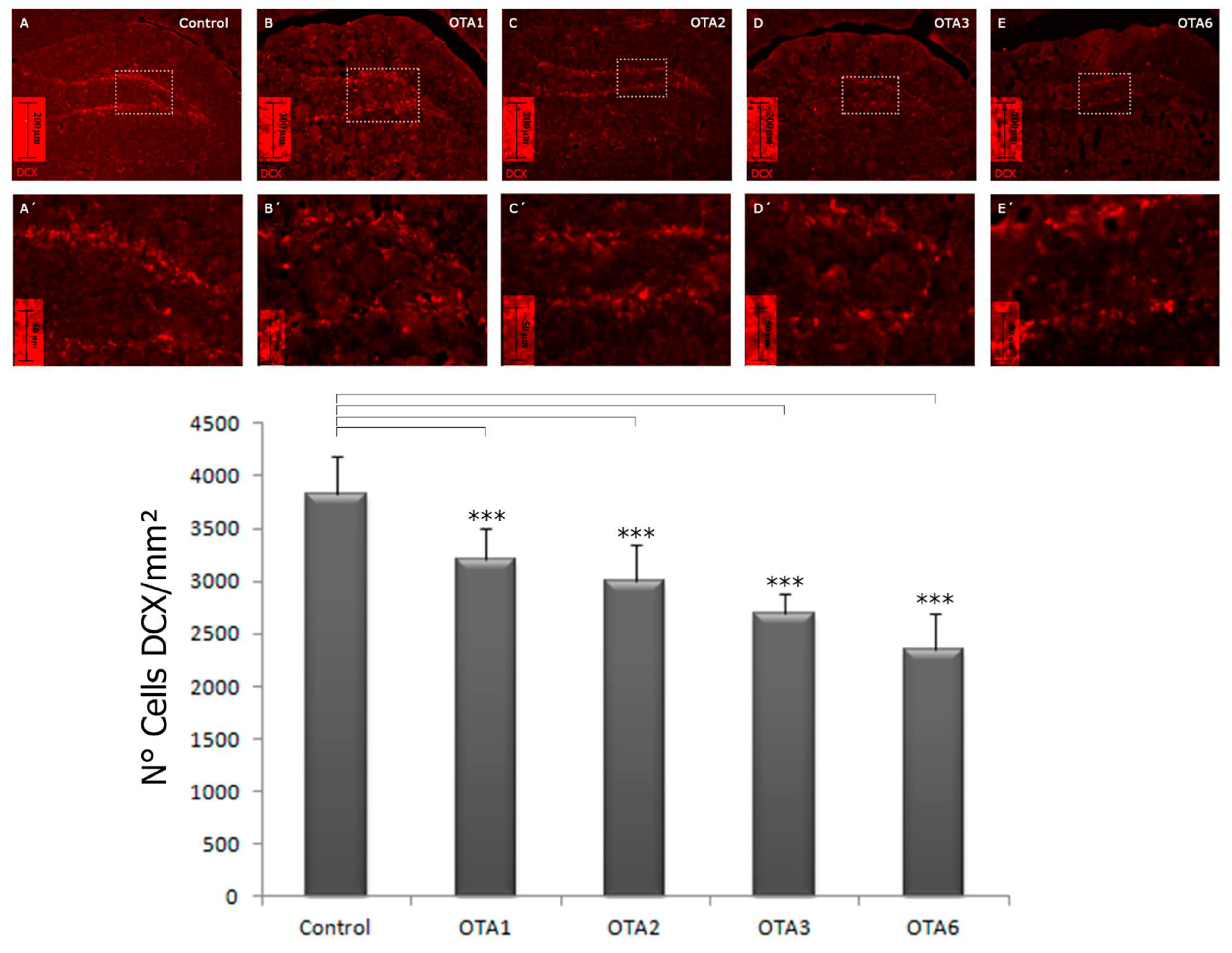
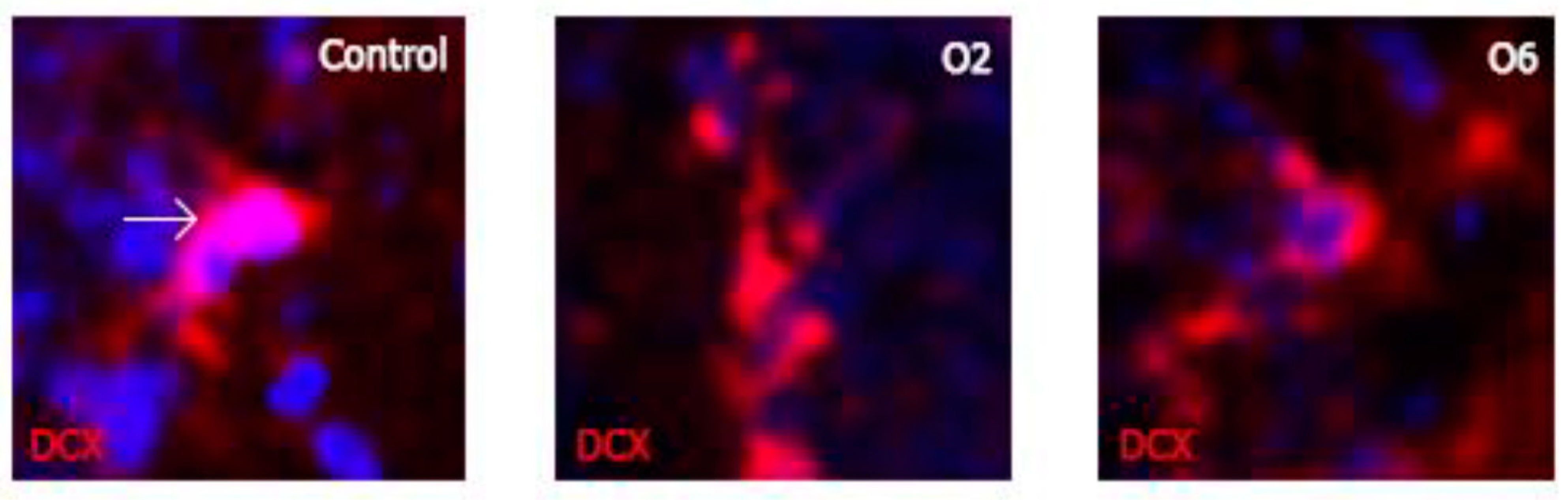
2.2. Ochratoxin A Reduces the Number of Young Neurons in the Dentate Gyrus
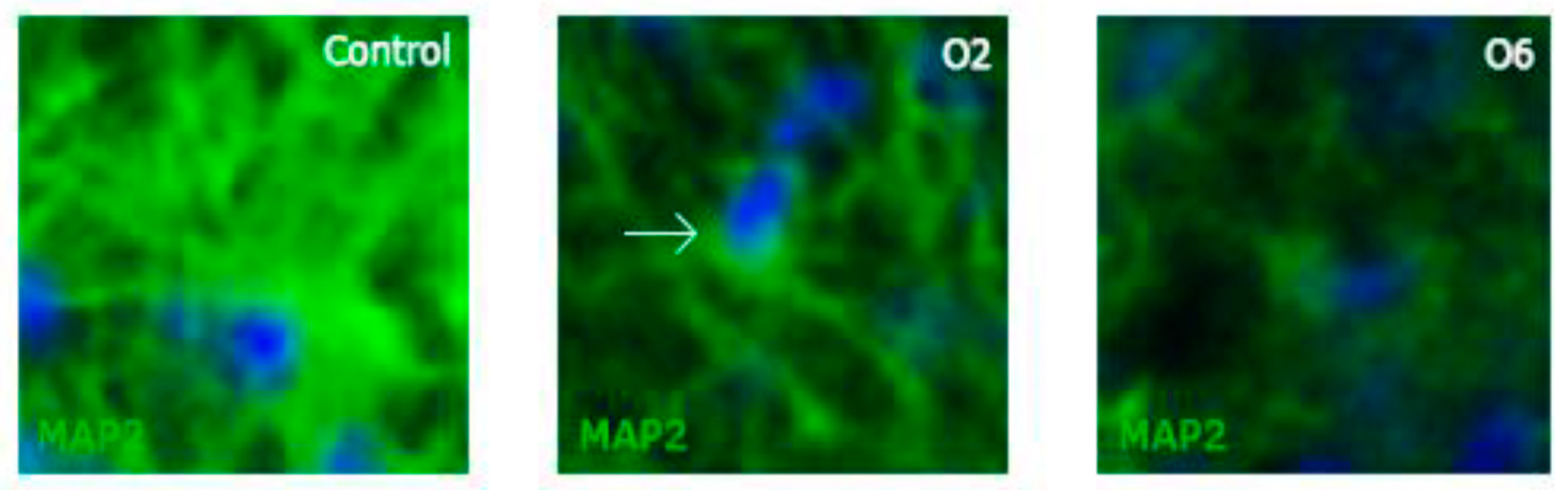
2.3. Ochratoxin A Reduces the Number of Mature Neurons in the Dentate Gyrus
3. Discussion
4. Materials and Methods
4.1. Experimental Animals
4.2. Ochratoxin A Administration in the In Vivo Assay
4.3. Immunohistochemistry
4.4. Cell Quantification and Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Walker, R. Risk assessment of ochratoxin: Current views of the european scientific committee on food, the JECFA and the CODEX committee on food additives and contaminants. Adv. Exp. Med. Biol. 2002, 504, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Galtier, P. Pharmacokinetics of ochratoxin A in animals. IARC Sci. Publ. 1991, 115, 187–200. [Google Scholar]
- Schrenk, D.; Bodin, L.; Chipman, J.K.; del Mazo, J.; Grasl-Kraupp, B.; Hogstrand, C.; Hoogenboom, L.; Leblanc, J.C.; Nebbia, C.S.; Nielsen, E.; et al. Risk assessment of ochratoxin A in food. EFSA J. 2020, 18, e06113. [Google Scholar] [CrossRef]
- JECFA. Safety Evaluation of Certain Mycotoxins in Food; FAO/WHO JECFA: Geneva, Switzerland, 2001. [Google Scholar]
- Pickova, D.; Ostry, V.; Malir, J.; Toman, J.; Malir, F. A Review on Mycotoxins and Microfungi in Spices in the Light of the Last Five Years. Toxins 2020, 12, 789. [Google Scholar] [CrossRef] [PubMed]
- Pleadin, J.; Frece, J.; Markov, K. Mycotoxins in food and feed. Adv. Food Nutr. Res. 2019, 89, 297–345. [Google Scholar] [CrossRef]
- Dey, D.K.; Kang, J.I.; Bajpai, V.K.; Kim, K.; Lee, H.; Sonwal, S.; Simal-Gandara, J.; Xiao, J.; Ali, S.; Huh, Y.S.; et al. Mycotoxins in food and feed: Toxicity, preventive challenges, and advanced detection techniques for associated diseases. Crit. Rev. Food Sci. Nutr. 2022, 2022, 2059650. [Google Scholar] [CrossRef]
- Bui-Klimke, T.R.; Wu, F. Ochratoxin A and human health risk: A review of the evidence. Crit. Rev. Food Sci. Nutr. 2015, 55, 1860–1869. [Google Scholar] [CrossRef]
- Creppy, E.E.; Kane, A.; Dirheimer, G.; Lafarge-Frayssinet, C.; Mousset, S.; Frayssinet, C. Genotoxicity of ochratoxin A in mice: DNA single-strand break evaluation in spleen, liver and kidney. Toxicol. Lett. 1985, 28, 29–35. [Google Scholar] [CrossRef]
- Pfohl-Leszkowicz, A.; Manderville, R.A. Ochratoxin A: An overview on toxicity and carcinogenicity in animals and humans. Mol. Nutr. Food Res. 2007, 51, 61–99. [Google Scholar] [CrossRef]
- Kőszegi, T.; Poór, M. Ochratoxin A: Molecular Interactions, Mechanisms of Toxicity and Prevention at the Molecular Level. Toxins 2016, 8, 111. [Google Scholar] [CrossRef]
- Malir, F.; Ostry, V.; Pfohl-Leszkowicz, A.; Novotna, E. Ochratoxin A: Developmental and reproductive toxicity-an overview. Birth Defects Res. B Dev. Reprod. Toxicol. 2013, 98, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Heussner, A.H.; Bingle, L.E.H. Comparative ochratoxin toxicity: A review of the available data. Toxins 2015, 7, 4253–4282. [Google Scholar] [CrossRef] [PubMed]
- Cramer, B.; Osteresch, B.; Muñoz, K.A.; Hillmann, H.; Sibrowski, W.; Humpf, H.U. Biomonitoring using dried blood spots: Detection of ochratoxin A and its degradation product 2’R-ochratoxin A in blood from coffee drinkers. Mol. Nutr. Food Res. 2015, 59, 1837–1843. [Google Scholar] [CrossRef] [PubMed]
- Coronel, M.B.; Sanchis, V.; Ramos, A.J.; Marin, S. Review. Ochratoxin A: Presence in human plasma and intake estimation. Food Sci. Technol. Int. 2010, 16, 5–18. [Google Scholar] [CrossRef]
- Paradells, S.; Rocamonde, B.; Llinares, C.; Herranz-Pérez, V.; Jimenez, M.; Garcia-Verdugo, J.M.; Zipancic, I.; Soria, J.M.; Garcia-Esparza, M.A. Neurotoxic effects of ochratoxin A on the subventricular zone of adult mouse brain. J. Appl. Toxicol. 2015, 35, 737–751. [Google Scholar] [CrossRef]
- Behrens, M.; Hüwel, S.; Galla, H.J.; Humpf, H.U. Efflux at the Blood-Brain Barrier Reduces the Cerebral Exposure to Ochratoxin A, Ochratoxin α, Citrinin and Dihydrocitrinone. Toxins 2021, 13, 327. [Google Scholar] [CrossRef]
- Belmadani, A.; Tramu, G.; Betbeder, A.M.; Creppy, E.E. Subchronic effects of ochratoxin A on young adult rat brain and partial prevention by aspartame, a sweetener. Hum. Exp. Toxicol. 1998, 17, 380–386. [Google Scholar] [CrossRef]
- Yoon, S.; Cong, W.T.; Bang, Y.; Lee, S.N.; Yoon, C.S.; Kwack, S.J.; Kang, T.S.; Lee, K.Y.; Choi, J.K.; Choi, H.J. Proteome response to ochratoxin A-induced apoptotic cell death in mouse hippocampal HT22 cells. Neurotoxicology 2009, 30, 666–676. [Google Scholar] [CrossRef]
- Erceg, S.; Mateo, E.M.; Zipancic, I.; Jiménez, F.J.R.; Aragó, M.A.P.; Jiménez, M.; Soria, J.M.; Garcia-Esparza, M.Á. Assessment of Toxic Effects of Ochratoxin A in Human Embryonic Stem Cells. Toxins 2019, 11, 217. [Google Scholar] [CrossRef]
- Pei, X.; Zhang, W.; Jiang, H.; Liu, D.; Liu, X.; Li, L.; Li, C.; Xiao, X.; Tang, S.; Li, D. Food-Origin Mycotoxin-Induced Neurotoxicity: Intend to Break the Rules of Neuroglia Cells. Oxid. Med. Cell. Longev. 2021, 2021, 9967334. [Google Scholar] [CrossRef]
- Zhang, X.; Boesch-Saadatmandi, C.; Lou, Y.; Wolffram, S.; Huebbe, P.; Rimbach, G. Ochratoxin A induces apoptosis in neuronal cells. Genes Nutr. 2009, 4, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sava, V.; Velasquez, A.; Song, S.; Sanchez-Ramos, J. Adult hippocampal neural stem/progenitor cells in vitro are vulnerable to the mycotoxin ochratoxin-A. Toxicol. Sci. 2007, 98, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Vukovic, J.; Borlikova, G.G.; Ruitenberg, M.J.; Robinson, G.J.; Sullivan, R.K.P.; Walker, T.L.; Bartlett, P.F. Immature doublecortin-positive hippocampal neurons are important for learning but not for remembering. J. Neurosci. 2013, 33, 6603–6613. [Google Scholar] [CrossRef] [PubMed]
- Sava, V.; Reunova, O.; Velasquez, A.; Harbison, R.; Sánchez-Ramos, J. Acute neurotoxic effects of the fungal metabolite ochratoxin-A. Neurotoxicology 2006, 27, 82–92. [Google Scholar] [CrossRef]
- Belmadani, A.; Tramu, G.; Betbeder, A.M.; Steyn, P.S.; Creppy, E.E. Regional selectivity to ochratoxin A, distribution and cytotoxicity in rat brain. Arch. Toxicol. 1998, 72, 656–662. [Google Scholar] [CrossRef]
- Sava, V.; Reunova, O.; Velasquez, A.; Sanchez-Ramos, J. Can low level exposure to ochratoxin-A cause parkinsonism? J. Neurol. Sci. 2006, 249, 68–75. [Google Scholar] [CrossRef]
- Kamp, H.G.; Eisenbrand, G.; Schlatter, J.; Würth, K.; Janzowski, C. Ochratoxin A: Induction of (oxidative) DNA damage, cytotoxicity and apoptosis in mammalian cell lines and primary cells. Toxicology 2005, 206, 413–425. [Google Scholar] [CrossRef]
- Damiano, S.; Longobardi, C.; Andretta, E.; Prisco, F.; Piegari, G.; Squillacioti, C.; Montagnaro, S.; Pagnini, F.; Badino, P.; Florio, S.; et al. Antioxidative effects of curcumin on the hepatotoxicity induced by ochratoxin a in rats. Antioxidants 2021, 10, 125. [Google Scholar] [CrossRef]
- Schwerdt, G.; Kopf, M.; Gekle, M. The impact of the nephrotoxin ochratoxin a on human renal cells studied by a novel co-culture model is influenced by the presence of fibroblasts. Toxins 2021, 13, 219. [Google Scholar] [CrossRef]
- Fukui, Y.; Hayasaka, S.; Itoh, M.; Takeuchi, Y. Development of neurons and synapses in ochratoxin A-induced microcephalic mice: A quantitative assessment of somatosensory cortex. Neurotoxicol. Teratol. 1992, 14, 191–196. [Google Scholar] [CrossRef]
- Wilk-Zasadna, I.; Minta, M. Developmental Toxicity of Ochratoxin A in Rat Embryo Midbrain Micromass Cultures. Int. J. Mol. Sci. 2009, 10, 37–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gage, F.H. Mammalian neural stem cells. Science 2000, 287, 1433–1438. [Google Scholar] [CrossRef] [PubMed]
- Doetsch, F.; Caille, I.; Lim, D.A.; Garcia-Verdugo, J.M.; Alvarez-Buylla, A. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell 1999, 97, 703–716. [Google Scholar] [CrossRef]
- Mirzadeh, Z.; Merkle, F.T.; Soriano-Navarro, M.; Garcia-Verdugo, J.M.; Alvarez-Buylla, A. Neural stem cells confer unique pinwheel architecture to the ventricular surface in neurogenic regions of the adult brain. Cell Stem Cell 2008, 3, 265–278. [Google Scholar] [CrossRef] [PubMed]
- Ponti, G.; Obernier, K.; Guinto, C.; Jose, L.; Bonfanti, L.; Alvarez-Buylla, A. Cell cycle and lineage progression of neural progenitors in the ventricular-subventricular zones of adult mice. Proc. Natl. Acad. Sci. USA 2013, 110, E1054. [Google Scholar] [CrossRef]
- Zurich, M.G.; Lengacher, S.; Braissant, O.; Monnet-Tschudi, F.; Pellerin, L.; Honegger, P. Unusual astrocyte reactivity caused by the food mycotoxin ochratoxin A in aggregating rat brain cell cultures. Neuroscience 2005, 134, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Janzer, R.C.; Raff, M.C. Astrocytes induce blood-brain barrier properties in endothelial cells. Nature 1987, 325, 253–257. [Google Scholar] [CrossRef]
- Barcia, C.; Sanderson, N.S.R.; Barrett, R.J.; Wawrowsky, K.; Kroeger, K.M.; Puntel, M.; Liu, C.; Castro, M.G.; Lowenstein, P.R. T Cells’ Immunological Synapses Induce Polarization of Brain Astrocytes In Vivo and In Vitro: A Novel Astrocyte Response Mechanism to Cellular Injury. PLoS ONE 2008, 3, e2977. [Google Scholar] [CrossRef]
- Acuña Castroviejo, E.; Martínez Arias de Reyna, L.; Andrés de la Calle, I.; Armengol y Butrón de Mújica, J.A.; Artigas Pérez, F. Manual de Neurociencias, 1st ed.; Sintesis: Madrid, Spain, 1998; ISBN 9788477386001. [Google Scholar]
- Sullivan, S.M.; Björkman, S.T.; Miller, S.M.; Colditz, P.B.; Pow, D.V. Structural remodeling of gray matter astrocytes in the neonatal pig brain after hypoxia/ischemia. Glia 2010, 58, 181–194. [Google Scholar] [CrossRef]
- Morel, G.R.; Andersen, T.; Pardo, J.; Zuccolilli, G.O.; Cambiaggi, V.L.; Hereñú, C.B.; Goya, R.G. Cognitive impairment and morphological changes in the dorsal hippocampus of very old female rats. Neuroscience 2015, 303, 189–199. [Google Scholar] [CrossRef]
- Le, G.; Yuan, X.; Hou, L.; Ge, L.; Liu, S.; Muhmood, A.; Liu, K.; Lin, Z.; Liu, D.; Gan, F.; et al. Ochratoxin A induces glomerular injury through activating the ERK/NF-κB signaling pathway. Food Chem. Toxicol. 2020, 143, 111516. [Google Scholar] [CrossRef] [PubMed]
- Franklin, K.B.J.; George, P. Paxinos and Franklin’s the Mouse Brain in Stereotaxic Coordinates, 7th ed.; Academic Press Inc.: Cambridge, MA, USA, 2019; ISBN 9780128161593. [Google Scholar]
- Young, K.; Morrison, H. Quantifying Microglia Morphology from Photomicrographs of Immunohistochemistry Prepared Tissue Using ImageJ. J. Vis. Exp. 2018, 2018, e57648. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | OTA1 | OTA2 | OTA3 | OTA6 | |
|---|---|---|---|---|---|
| N = cells/mm2 | 4 | 4 | 4 | 4 | 5 |
| 3825 ± 182 | 3217 ± 1445 | 2999 ± 175 | 2686 ± 99 | 2343 ± 154 | |
| Significance compared to the control | - | <0.001 | <0.001 | <0.001 | <0.001 |
| Control | OTA1 | OTA2 | OTA3 | OTA6 | |
|---|---|---|---|---|---|
| N = cells/mm2 | 4 | 4 | 4 | 4 | 5 |
| 7990 ± 426 | 5466 ± 488 | 5247 ± 297 | 4095 ± 617 | 3006 ± 694 | |
| Significance compared to the control | - | <0.001 | <0.001 | <0.001 | <0.001 |
| Product | Antibody | Dilution | Specificity | Wavelength | Reference | Company |
|---|---|---|---|---|---|---|
| H-1200 | 4′,6-diamidino-2-fenylindool (DAPI) | - | Binds with chromatin- DNA/RNA | 461 nm (blue) | Chazotte 2011 | Vector Labs, UK |
| Z0334 | Glial fibrillary acidic protein (GFAP) | 1:500 | Astroglial lineage | - | Eng et al., 2000 | Dako, Denmark |
| AB18723 | Doublecortin (DCX) | 1:300 | Neuronal precursor cells and immature neurons | - | Gleave et al. | Abcam, UK |
| AB5622 | Microtubule-associated protein 2 (MAP2) | 1:200 | Mature neurons | - | Lyck et al. | Millipore, CA, USA |
| A11008 | Alexa Fluor™ 488 goat anti-rabbit | 1:200 | Secondary antibody | 495–519 (green) | Borg et al. | Invitrogen, Spain |
| A1102 | Alexa Fluor™ 594-conjugated donkey anti-rabbit | 1:200 | Secondary antibody | 590–617 (red) | Purkartova et al. | Invitrogen, Spain |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mateo, E.; Tonino, R.P.B.; Canto, A.; Monroy Noyola, A.; Miranda, M.; Soria, J.M.; Garcia Esparza, M.A. The Neurotoxic Effect of Ochratoxin-A on the Hippocampal Neurogenic Niche of Adult Mouse Brain. Toxins 2022, 14, 624. https://doi.org/10.3390/toxins14090624
Mateo E, Tonino RPB, Canto A, Monroy Noyola A, Miranda M, Soria JM, Garcia Esparza MA. The Neurotoxic Effect of Ochratoxin-A on the Hippocampal Neurogenic Niche of Adult Mouse Brain. Toxins. 2022; 14(9):624. https://doi.org/10.3390/toxins14090624
Chicago/Turabian StyleMateo, Eva, Rik Paulus Bernardus Tonino, Antolin Canto, Antonio Monroy Noyola, Maria Miranda, Jose Miguel Soria, and María Angeles Garcia Esparza. 2022. "The Neurotoxic Effect of Ochratoxin-A on the Hippocampal Neurogenic Niche of Adult Mouse Brain" Toxins 14, no. 9: 624. https://doi.org/10.3390/toxins14090624
APA StyleMateo, E., Tonino, R. P. B., Canto, A., Monroy Noyola, A., Miranda, M., Soria, J. M., & Garcia Esparza, M. A. (2022). The Neurotoxic Effect of Ochratoxin-A on the Hippocampal Neurogenic Niche of Adult Mouse Brain. Toxins, 14(9), 624. https://doi.org/10.3390/toxins14090624