Transcriptional Stages of Conidia Germination and Associated Genes in Aspergillus flavus: An Essential Role for Redox Genes



Abstract
:1. Introduction
2. Results
2.1. Conidia Germination Rate
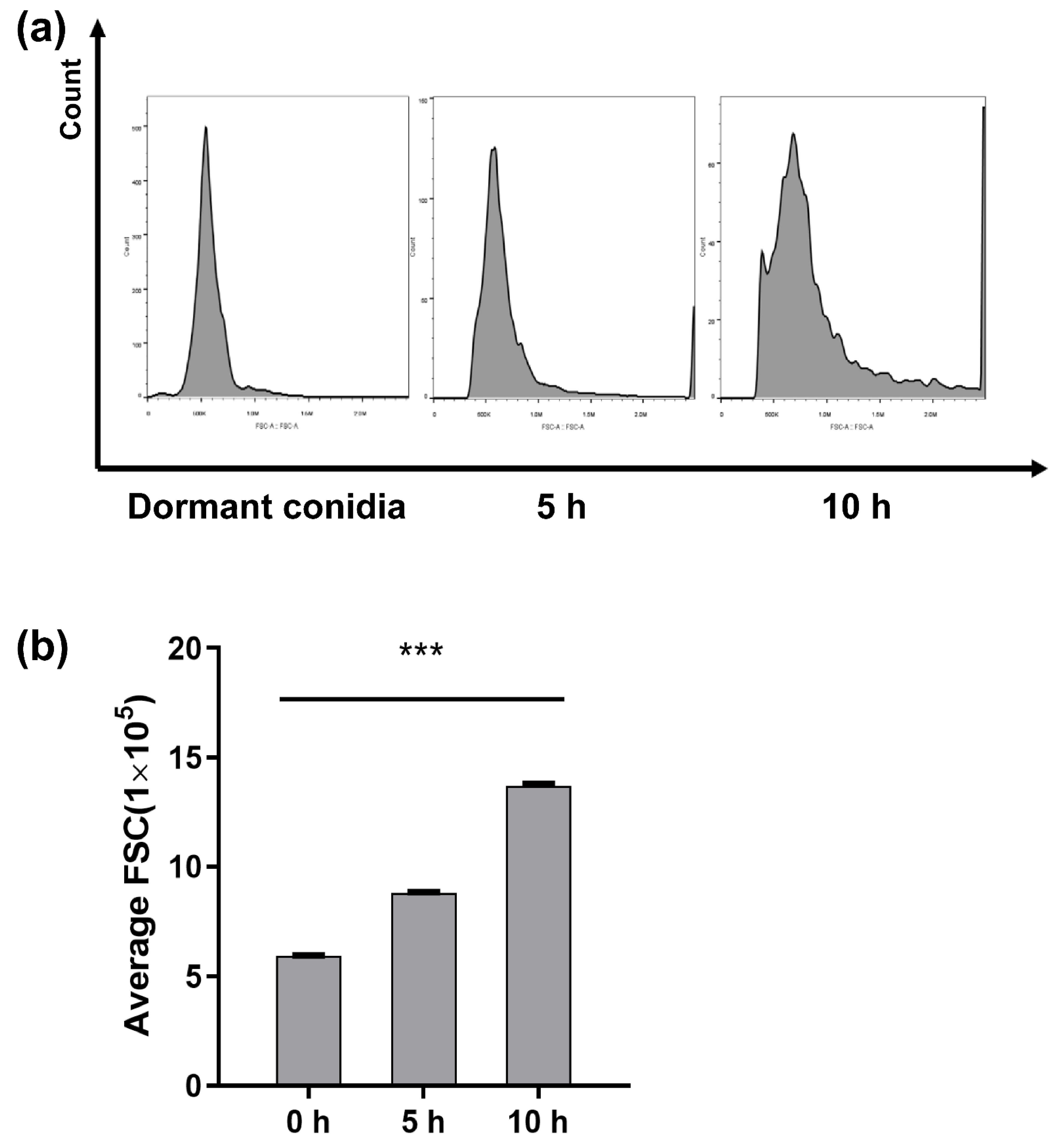
2.2. Flow Cytometry
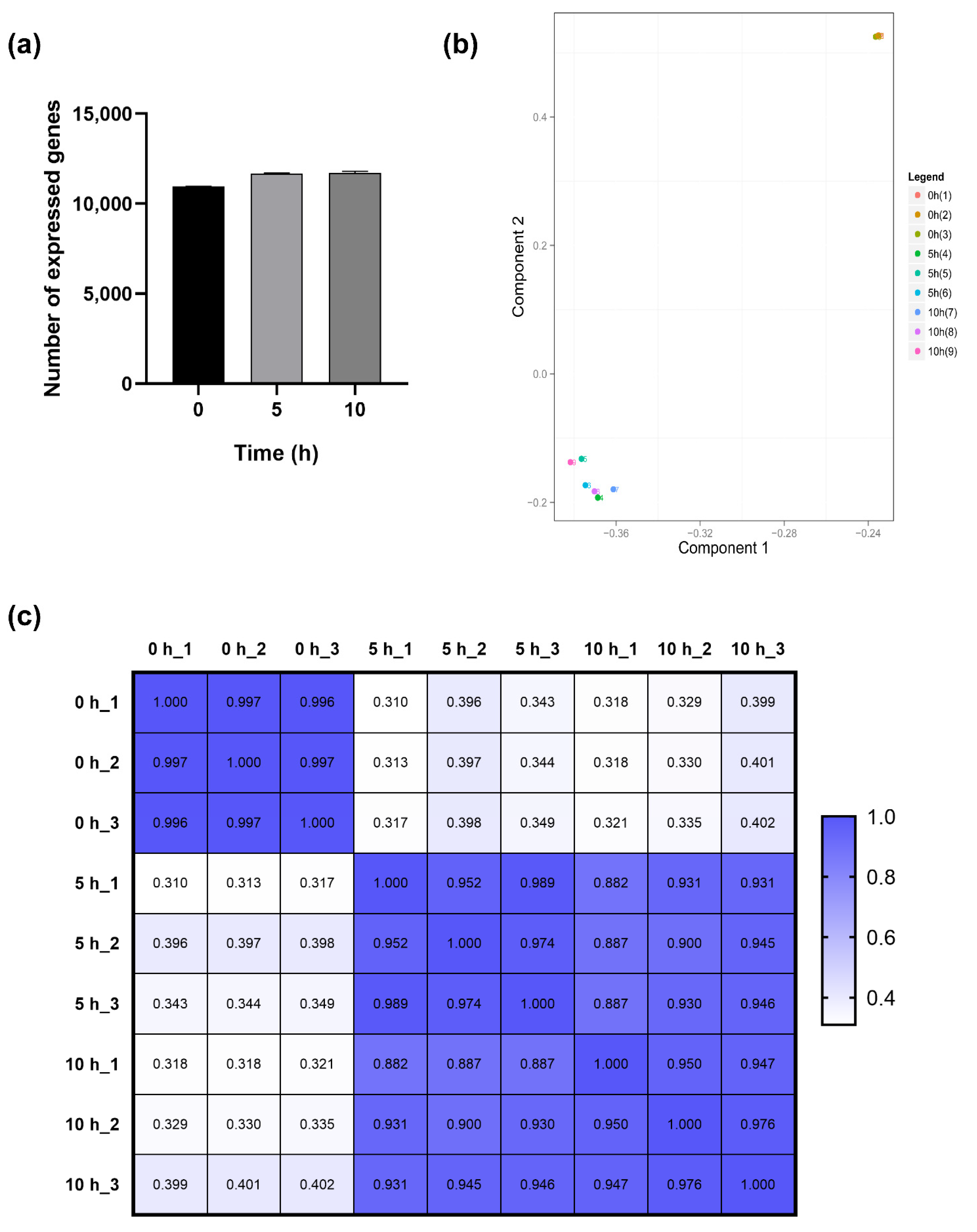
2.3. Transcriptional Profiling
2.4. Gene Expression
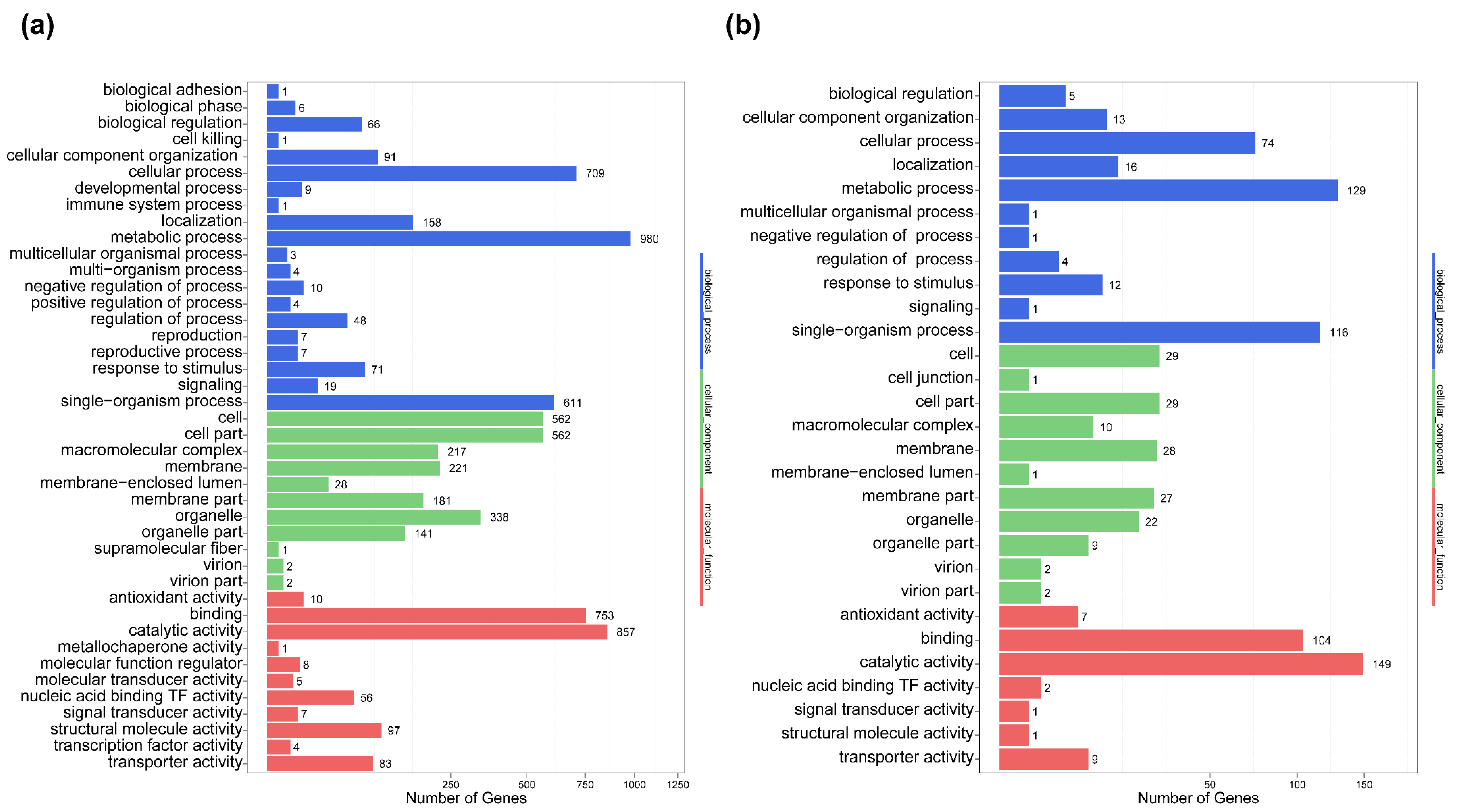
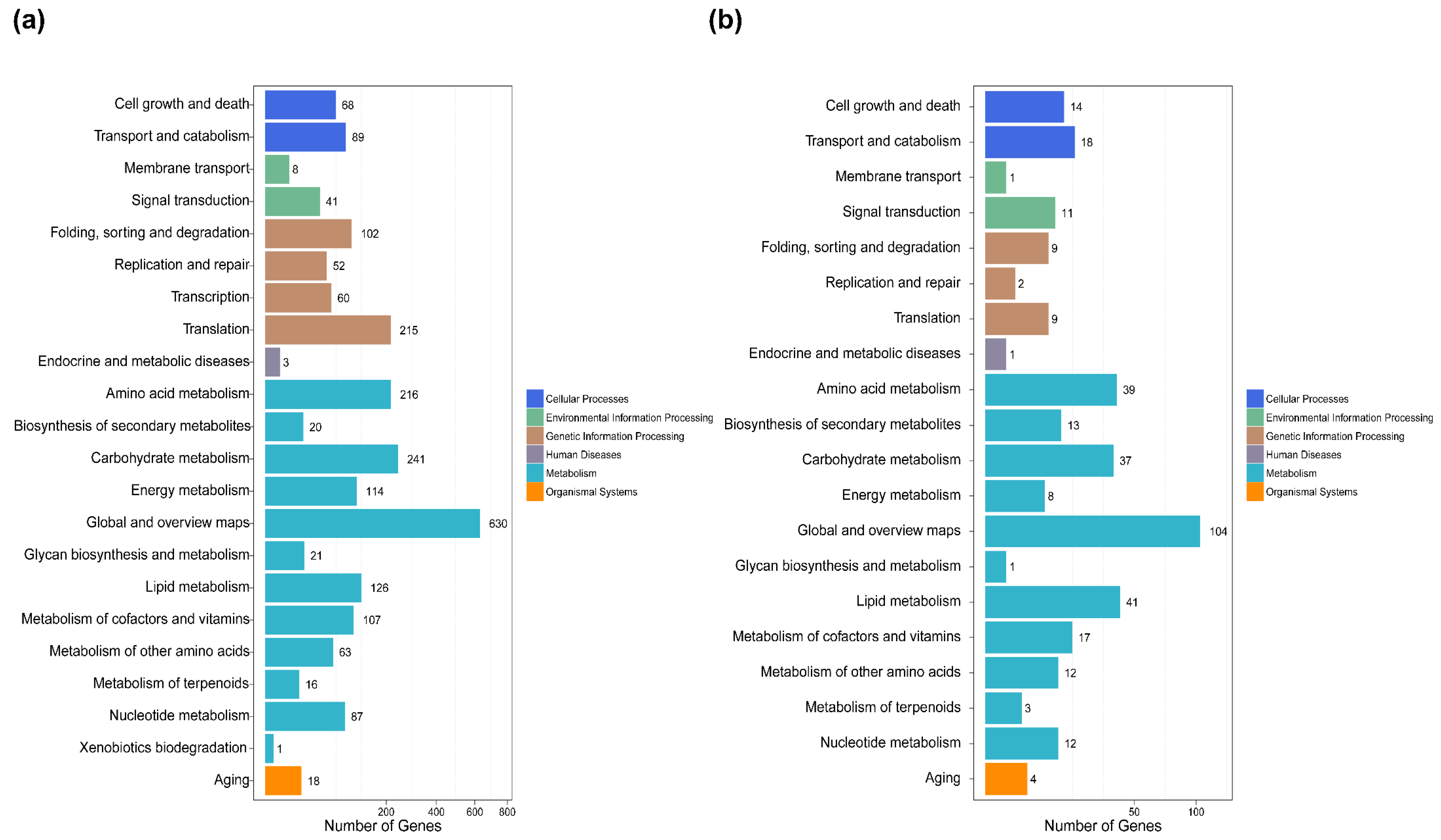
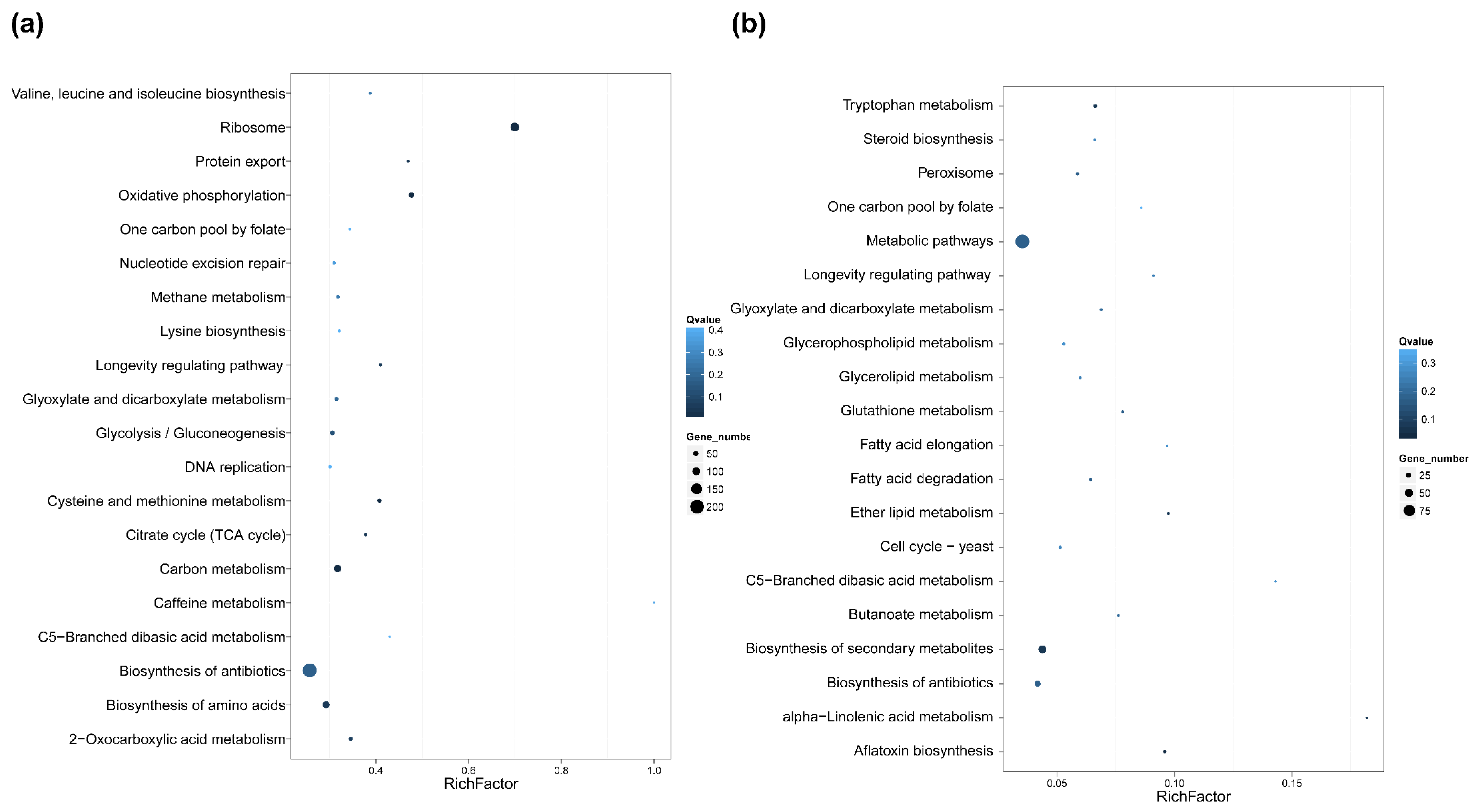
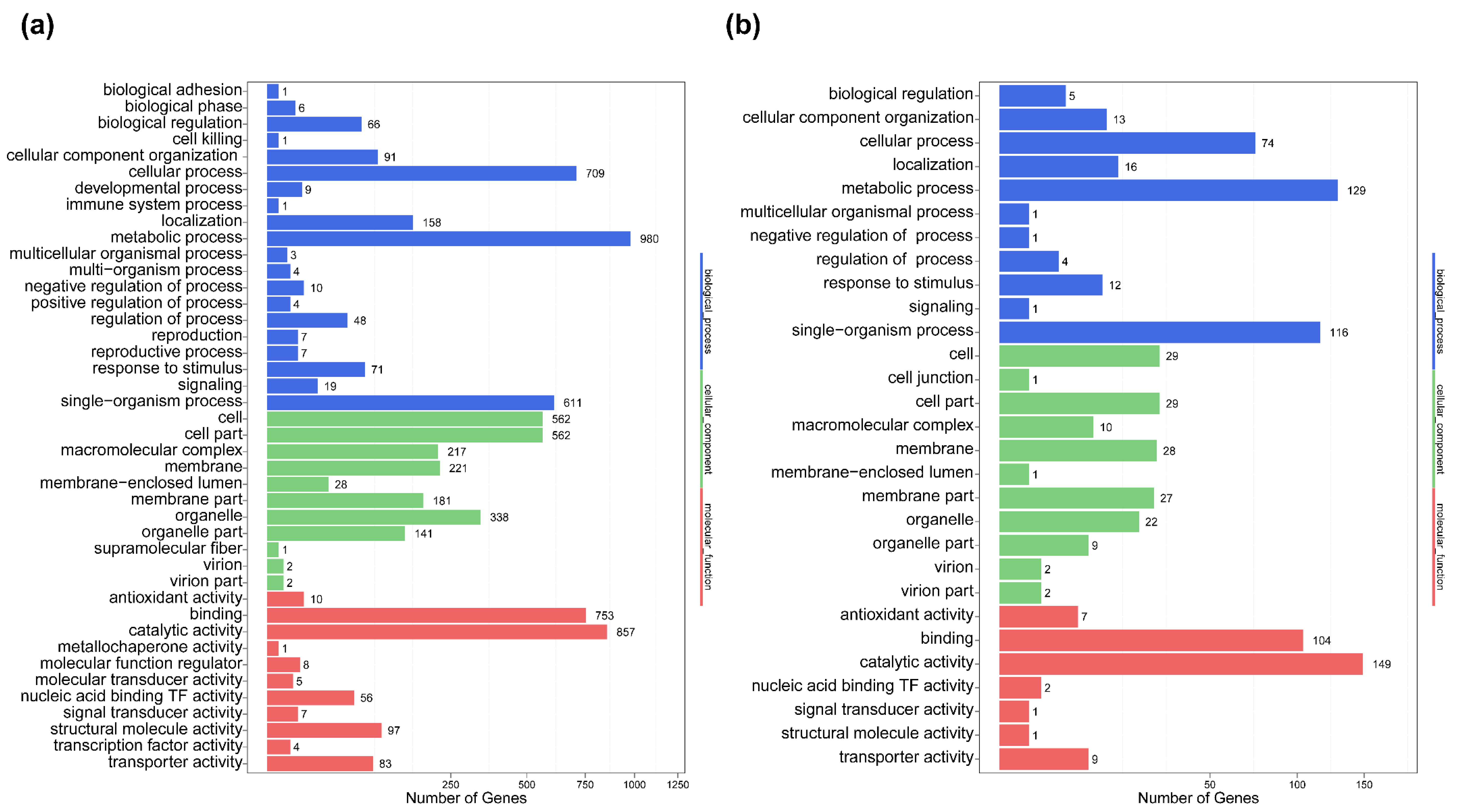
2.5. Differential Gene Expression and Functional Analysis
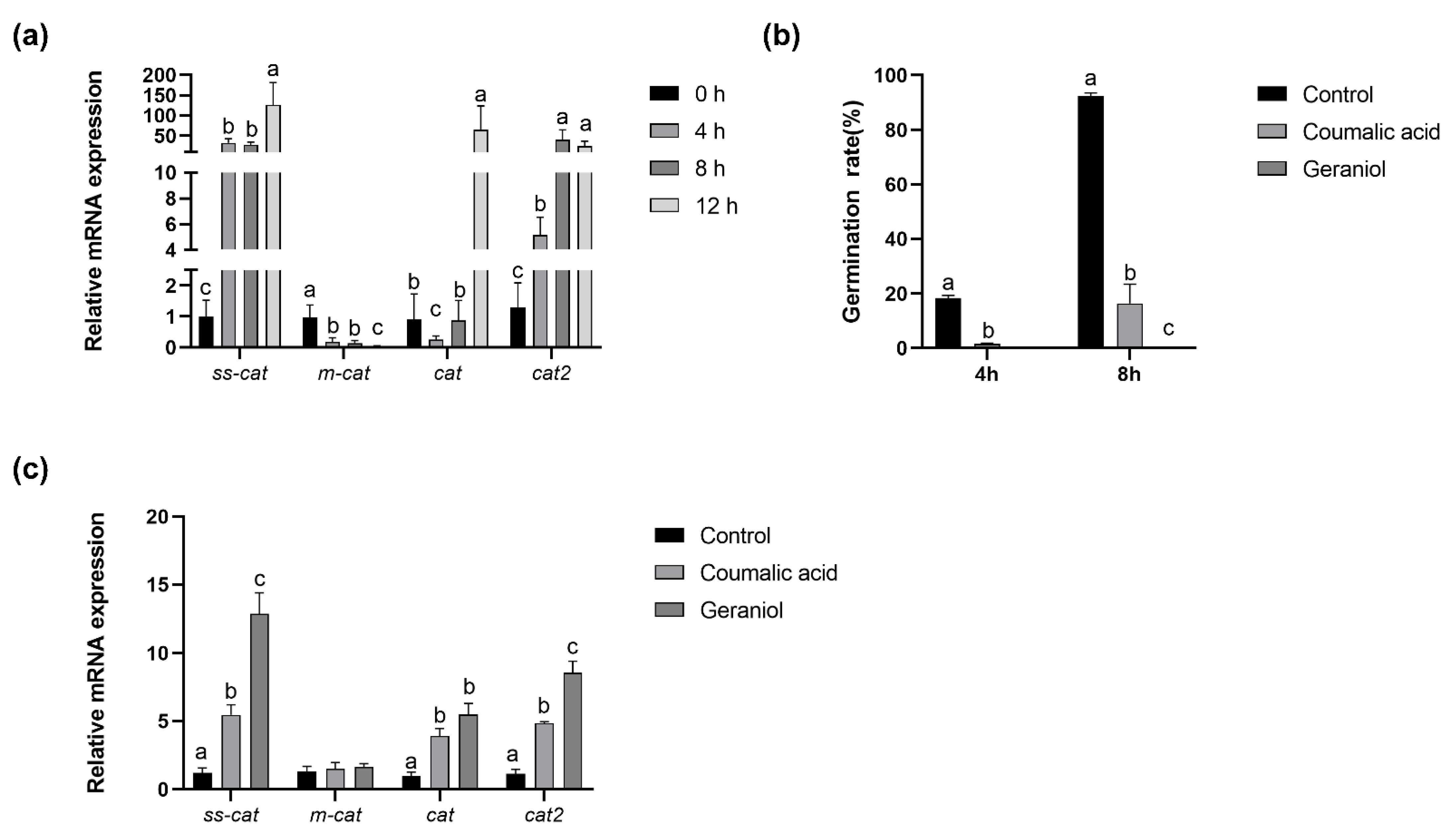
2.6. Antioxidant System during Conidia Germination
3. Discussion
4. Materials and Methods
4.1. Culturing Conditions and Sampling
4.2. Microscopy
4.3. Flow Cytometry of Spores
4.4. RNA Extraction and RNA-Seq
4.5. Real-Time Quantitative PCR
4.6. Statistical Analysis
4.7. Data Submission
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tian, J.; Ban, X.; Zeng, H.; He, J.; Huang, B.; Wang, Y. Chemical composition and antifungal activity of essential oil from Cicuta virosa L. var. latisecta Celak. Int. J. Food Microbiol. 2011, 145, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Sun, L.; Zhang, N.; Zhang, J.; Guo, J.; Li, C.; Rajput, S.A.; Qi, D. Effects of Nutrients in Substrates of Different Grains on Aflatoxin B1 Production by Aspergillus flavus. Biomed. Res. Int. 2016, 2016, 7232858. [Google Scholar] [CrossRef] [PubMed]
- WHO. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. Volume 56. Some Naturally Occurring Substances: Food Items and Constituents, Heterocyclic Aromatic Amines and Mycotoxins. Cancer Causes Control 1994, 5, 89–90. [Google Scholar]
- Egal, S.; Hounsa, A.; Gong, Y.Y.; Turner, P.C.; Wild, C.P.; Hall, A.J.; Hell, K.; Cardwell, K.F. Dietary exposure to aflatoxin from maize and groundnut in young children from Benin and Togo, West Africa. Int. J. Food Microbiol. 2005, 104, 215–224. [Google Scholar] [CrossRef]
- Novodvorska, M.; Hayer, K.; Pullan, S.T.; Wilson, R.; Archer, D.B. Transcriptional landscape of Aspergillus niger at breaking of conidial dormancy revealed by RNA-Sequencing. BMC Genom. 2013, 14, 246. [Google Scholar] [CrossRef]
- Wang, B.; Lv, Y.; Li, X.; Lin, Y.; Deng, H.; Pan, L. Profiling of secondary metabolite gene clusters regulated by LaeA in Aspergillus niger FGSC A1279 based on genome sequencing and transcriptome analysis. Res. Microbiol. 2017, 169, 67–77. [Google Scholar] [CrossRef]
- Brown, G.D.; Denning, D.W.; Gow, N.A.R.; Levitz, S.M.; Netea, M.G.; White, T.C. Hidden Killers: Human Fungal Infections. Sci. Transl. Med. 2012, 4, 165rv113. [Google Scholar] [CrossRef]
- Verweij, P.P.E.; Zhang, J.; Debets, A.J.M.; Meis, J.F.; Melchers, W.J.G. In-host adaptation and acquired triazole resistance in Aspergillus fumigatus: A dilemma for clinical management. Lancet Infect. Dis. 2016, 16, e251–e260. [Google Scholar] [CrossRef]
- Nir, O.; May, G.S. The molecular mechanisms of conidial germination. FEMS Microbiol. Lett. 2001, 199, 153–160. [Google Scholar] [CrossRef]
- Lamarre, C.; Sokol, S.; Debeaupuis, J.P.; Henry, C.; Lacroix, C.; Glaser, P.; Coppée, J.-Y.; François, J.M.; Latgé, J.-P. Transcriptomic analysis of the exit from dormancy of Aspergillus fumigatus conidia. BMC Genom. 2008, 9, 417. [Google Scholar] [CrossRef]
- Baltussen, T.J.H.; Zoll, J.; Verweij, P.E.; Melchers, W.J.G. Molecular Mechanisms of Conidial Germination in Aspergillus spp. Microbiol. Mol. Biol. Rev. 2019, 84, e00049-19. [Google Scholar] [CrossRef]
- Thanh, N.V.; Rombouts, F.M.; Nout, M.J.R. Effect of individual amino acids and glucose on activation and germination of Rhizopus oligosporus sporangiospores in tempe starter. J. Appl. Microbiol. 2005, 99, 1204–1214. [Google Scholar] [CrossRef] [PubMed]
- Taubitz, A.; Bauer, B.; Heesemann, J.; Ebel, F. Role of respiration in the germination process of the pathogenic mold Aspergillus fumigatus. Curr. Microbiol. 2007, 54, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Leeuwen, M.R.V.; Doorn, T.M.V.; Golovina, E.; Stark, J.; Dijksterhuis, J. Water- and Air-Distributed Conidia Differ in Sterol Content and Cytoplasmic Microviscosity. Appl. Environ. Microbiol. 2009, 76, 366–369. [Google Scholar] [CrossRef] [PubMed]
- Osherov, N.; May, G.S. Conidial germination in Aspergillus nidulans requires RAS signaling and protein synthesis. Genetics 2000, 155, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Van Leeuwen, M.R.; Smant, W.; De Boer, W.; Dijksterhuis, J. Filipin is a reliable in situ marker of ergosterol in the plasma membrane of germinating conidia (spores) of Penicillium discolor and stains intensively at the site of germ tube formation. J. Microbiol. Methods 2008, 74, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Mellon, J.E.; Dowd, M.K.; Cotty, P.J. Substrate Utilization by Aspergillus flavus in Inoculated Whole Corn Kernels and Isolated Tissues. J. Agric. Food Chem. 2005, 53, 2351–2357. [Google Scholar] [CrossRef]
- van Leeuwen, M.R.; Krijgsheld, P.; Bleichrodt, R.; Menke, H.; Stam, H.; Stark, J.; Wosten, H.A.; Dijksterhuis, J. Germination of conidia of Aspergillus niger is accompanied by major changes in RNA profiles. Stud. Mycol. 2013, 74, 59–70. [Google Scholar] [CrossRef]
- Szilagyi, M.; Miskei, M.; Karanyi, Z.; Lenkey, B.; Pocsi, I.; Emri, T. Transcriptome changes initiated by carbon starvation in Aspergillus nidulans. Microbiology 2013, 159, 176–190. [Google Scholar] [CrossRef]
- Zhang, F.; Guo, Z.; Zhong, H.; Wang, S.; Yang, W.; Liu, Y.; Wang, S. RNA-Seq-based transcriptome analysis of aflatoxigenic Aspergillus flavus in response to water activity. Toxins 2014, 6, 3187–3207. [Google Scholar] [CrossRef]
- Baltussen, T.J.H.; Coolen, J.P.M.; Zoll, J.; Verweij, P.E.; Melchers, W.J.G. Gene co-expression analysis identifies gene clusters associated with isotropic and polarized growth in Aspergillus fumigatus conidia. Fungal Genet. Biol. 2018, 116, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Borkovich, K.A.; Ebbole, D.J. Hyphal Growth and Polarity. In Cellular and Molecular Biology of Filamentous Fungi; ASM Press: Washington, DC, USA, 2010; pp. 238–259. [Google Scholar] [CrossRef]
- Lestrade, P.P.A.; Meis, J.F.; Melchers, W.J.G.; Verweij, P.E. Triazole resistance in Aspergillus fumigatus: Recent insights and challenges for patient management. Clin. Microbiol. Infect. 2018, 25, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Dijksterhuis, J. Fungal spores: Highly variable and stress-resistant vehicles for distribution and spoilage. Food Microbiol. 2019, 81, 2–11. [Google Scholar] [CrossRef]
- Pereira de Souza, C.C.; Moreira Prado, G.; da Conceicao Freitas, R.C.; Silva Santos Guimaraes, P.; Calegario de Oliveira, L.; Eustaquio Alvim Brito-Melo, G.; de Figueiredo Conte Vanzela, A.P. Analysis of Aspergillus nidulans germination, initial growth and carbon source response by flow cytometry. J. Basic Microbiol. 2011, 51, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Krijgsheld, P.; Bleichrodt, R.; Veluw, G.J.V.; Wang, F.; Müller, W.H.; Dijksterhuis, J.; Wsten, H.A.B. Development in Aspergillus. Stud. Mycol. 2013, 74, 1–29. [Google Scholar] [CrossRef]
- Battaglia, M.; Olvera-Carrillo, Y.; Garciarrubio, A.; Campos, F.; Covarrubias, A.A. The Enigmatic LEA Proteins and Other Hydrophilins. Plant Physiol. 2008, 148, 6–24. [Google Scholar] [CrossRef]
- Sasse, C.; Bignell, E.M.; Hasenberg, M.; Haynes, K.; Gunzer, M.; Braus, G.H.; Krappmann, S. Basal expression of the Aspergillus fumigatus transcriptional activator CpcA is sufficient to support pulmonary aspergillosis. Fungal Genet. Biol. 2008, 45, 693–704. [Google Scholar] [CrossRef]
- Ramakrishnan, V. Ribosome Structure and the Mechanism of Translation. Cell 2002, 108, 557–572. [Google Scholar] [CrossRef]
- Pearson, C.L.; Xu, K.; Sharpless, K.E.; Harris, S.D. MesA, a Novel Fungal Protein Required for the Stabilization of Polarity Axes in Aspergillus nidulans. Mol. Biol. Cell 2004, 15, 3658. [Google Scholar] [CrossRef]
- Yan, S.; Liang, Y.; Zhang, J.; Chen, Z.; Liu, C.-M. Autoxidated linolenic acid inhibits aflatoxin biosynthesis in Aspergillus flavus via oxylipin species. Fungal Genet. Biol. 2015, 81, 229–237. [Google Scholar] [CrossRef]
- Chitarra, G.S.; Abee, T.; Rombouts, F.M.; Dijksterhuis, J. 1-Octen-3-ol inhibits conidia germination of Penicillium paneum despite of mild effects on membrane permeability, respiration, intracellular pH, and changes the protein composition. FEMS Microbiol. Ecol. 2005, 54, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Yang, M.; Bai, Y.; Ge, F.; Wang, S. Antioxidant-related catalase CTA1 regulates development, aflatoxin biosynthesis, and virulence in pathogenic fungus Aspergillus flavus. Environ. Microbiol. 2020, 22, 2792–2810. [Google Scholar] [CrossRef] [PubMed]
- Paris, S.; Wysong, D.; Debeaupuis, J.P.; Shibuya, K.; Philippe, B.; Diamond, R.D.; Latgé, J.P. Catalases of Aspergillus fumigatus. Infect. Immun. 2003, 71, 3551–3562. [Google Scholar] [CrossRef] [PubMed]
- Ohi, M.; Kitamura, T.; Hata, S. Stimulation by caffeic acid, coumalic acid, and corilagin of the germination of resting spores of the clubroot pathogen Plasmodiophora brassicae. Biosci. Biotechnol. Biochem. 2003, 67, 170–173. [Google Scholar] [CrossRef]
- Geraci, A.; Di Stefano, V.; Di Martino, E.; Schillaci, D.; Schicchi, R. Essential oil components of orange peels and antimicrobial activity. Nat. Prod. Res. 2017, 31, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Rao, R.; Kumar, S.; Mahant, S.; Khatkar, S. Therapeutic Potential of Citronella Essential Oil: A Review. Curr. Drug Discov. Technol. 2019, 16, 330–339. [Google Scholar] [CrossRef]
- Nierman, W.C.; Yu, J.; Fedorova-Abrams, N.D.; Losada, L.; Payne, G.A. Genome Sequence of Aspergillus flavus NRRL 3357, a Strain That Causes Aflatoxin Contamination of Food and Feed. Genome Announc. 2014, 3, e00168-15. [Google Scholar] [CrossRef]
- Shishodia, S.K.; Tiwari, S.; Hoda, S.; Vijayaraghavan, P.; Shankar, J. SEM and qRT-PCR revealed quercetin inhibits morphogenesis of Aspergillus flavus conidia via modulating calcineurin-Crz1 signalling pathway. Mycology 2020, 11, 118–125. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Drai, D.; Elmer, G.; Kafkafi, N.; Golani, I. Controlling the false discovery rate in behavior genetics research. Behav. Brain Res. 2001, 125, 279–284. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Data Size (bp) | Raw Reads Number | Clean Data Size (bp) | Clean Reads Number | Clean Data Rate 1 (%) |
|---|---|---|---|---|---|
| 0 h | 1,206,821,150 | 24,136,423 | 1,206,488,600 | 24,129,772 | 99.97 |
| 0 h | 1,206,804,050 | 24,136,081 | 1,206,255,800 | 24,125,116 | 99.95 |
| 0 h | 1,206,810,000 | 24,136,200 | 1,206,384,900 | 24,127,698 | 99.96 |
| 5 h | 1,206,793,700 | 24,135,874 | 1,206,071,500 | 24,121,430 | 99.94 |
| 5 h | 1,206,826,500 | 24,136,530 | 1,205,452,250 | 24,109,045 | 99.88 |
| 5 h | 1,206,836,250 | 24,136,725 | 1,206,384,850 | 24,127,697 | 99.96 |
| 10 h | 1,206,827,750 | 24,136,555 | 1,206,450,400 | 24,129,008 | 99.96 |
| 10 h | 1,206,830,400 | 24,136,608 | 1,206,429,400 | 24,128,588 | 99.96 |
| 10 h | 1,206,829,750 | 24,136,595 | 1,206,415,650 | 24,128,313 | 99.96 |
| Pathway | Gene ID | log2Ratio a | Up/Down | Probability | Description |
|---|---|---|---|---|---|
| Alpha-Linolenic acid metabolism | 7917785 | −1.02 | Down | 0.80373 | 3-ketoacyl-coA thiolase peroxisomal A precursor, mRNA |
| 7915336 | −1.69 | Down | 0.80566 | FMN binding oxidoreductase, putative, mRNA | |
| 7912988 | −1.95 | Down | 0.86628 | NADH-dependent flavin oxidoreductase, putative, mRNA | |
| 7912986 | −2.05 | Down | 0.89858 | Fatty-acyl coenzyme A oxidase (Pox1), putative, mRNA | |
| 7910815 | 2.87 | Up | 0.929795 | 40S ribosomal protein S22, partial mRNA | |
| Aflatoxin biosynthesis | 7909985 | −1.08 | Down | 0.810561786 | PKS-like enzyme, putative, mRNA |
| 7910374 | 3.32 | Up | 0.863160112 | short chain type dehydrogenase, putative, mRNA | |
| 7912783 | 1.45 | Up | 0.842033907 | toxin biosynthesis ketoreductase, putative, mRNA | |
| 7914380 | 7.38 | Up | 0.83854578 | benzoate 4-monooxygenase cytochrome P450, mRNA | |
| 7911412 | 5.07 | Up | 0.865160849 | cytochrome P450, putative, mRNA | |
| 7911415 | 6.16 | Up | 0.941794345 | short-chain dehydrogenase, putative, mRNA | |
| 7911112 | 3.25 | Up | 0.826041173 | cytochrome P450, putative, mRNA | |
| 7915318 | 5.90 | Up | 0.834952351 | O-methyltransferase family protein, mRNA | |
| 7912683 | 8.03 | Up | 0.80137419 | O-methyltransferase, putative, mRNA | |
| 7915318 | 5.90 | Up | 0.834952351 | O-methyltransferase family protein, mRNA | |
| 7911961 | 1.56 | Up | 0.801874375 | O-methyltransferase family protein, mRNA |
| Pathway | Gene ID | log2Ratio a | Up/Down | Probability | Description |
|---|---|---|---|---|---|
| Ribosome | 7913639 | 3.02 | Up | 0.905636 | ribosomal protein YmL41, partial mRNA |
| 7910217 | 2.97 | Up | 0.929728 | 60S ribosomal protein L31, partial mRNA | |
| 7921558 | 2.87 | Up | 0.929755 | 60S ribosomal protein L1, partial mRNA | |
| 7913089 | 2.87 | Up | 0.929914 | 40S ribosomal protein S20, partial mRNA | |
| 7910815 | 2.87 | Up | 0.929795 | 40S ribosomal protein S22, partial mRNA | |
| Redox genes | 7912906 | 7.00 | Up | 0.974886 | catalase Cat, mRNA |
| 7920700 | −1.68 | Down | 0.904521 | spore-specific catalase CatA, mRNA | |
| 7917068 | 6.11 | Up | 0.950608 | mycelial catalase Cat1, mRNA | |
| 7918464 | 1.84 | Up | 0.850788 | bifunctional catalase-peroxidase Cat2, mRNA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, C.; Jia, S.; Rajput, S.A.; Qi, D.; Wang, S. Transcriptional Stages of Conidia Germination and Associated Genes in Aspergillus flavus: An Essential Role for Redox Genes. Toxins 2022, 14, 560. https://doi.org/10.3390/toxins14080560
Li C, Jia S, Rajput SA, Qi D, Wang S. Transcriptional Stages of Conidia Germination and Associated Genes in Aspergillus flavus: An Essential Role for Redox Genes. Toxins. 2022; 14(8):560. https://doi.org/10.3390/toxins14080560
Chicago/Turabian StyleLi, Chong, Sifan Jia, Shahid Ali Rajput, Desheng Qi, and Shuai Wang. 2022. "Transcriptional Stages of Conidia Germination and Associated Genes in Aspergillus flavus: An Essential Role for Redox Genes" Toxins 14, no. 8: 560. https://doi.org/10.3390/toxins14080560
APA StyleLi, C., Jia, S., Rajput, S. A., Qi, D., & Wang, S. (2022). Transcriptional Stages of Conidia Germination and Associated Genes in Aspergillus flavus: An Essential Role for Redox Genes. Toxins, 14(8), 560. https://doi.org/10.3390/toxins14080560