In Vitro Metabolism of Phenylspirodrimanes Derived from the Indoor Fungus Stachybotrys


Abstract
:1. Introduction
2. Results and Discussion
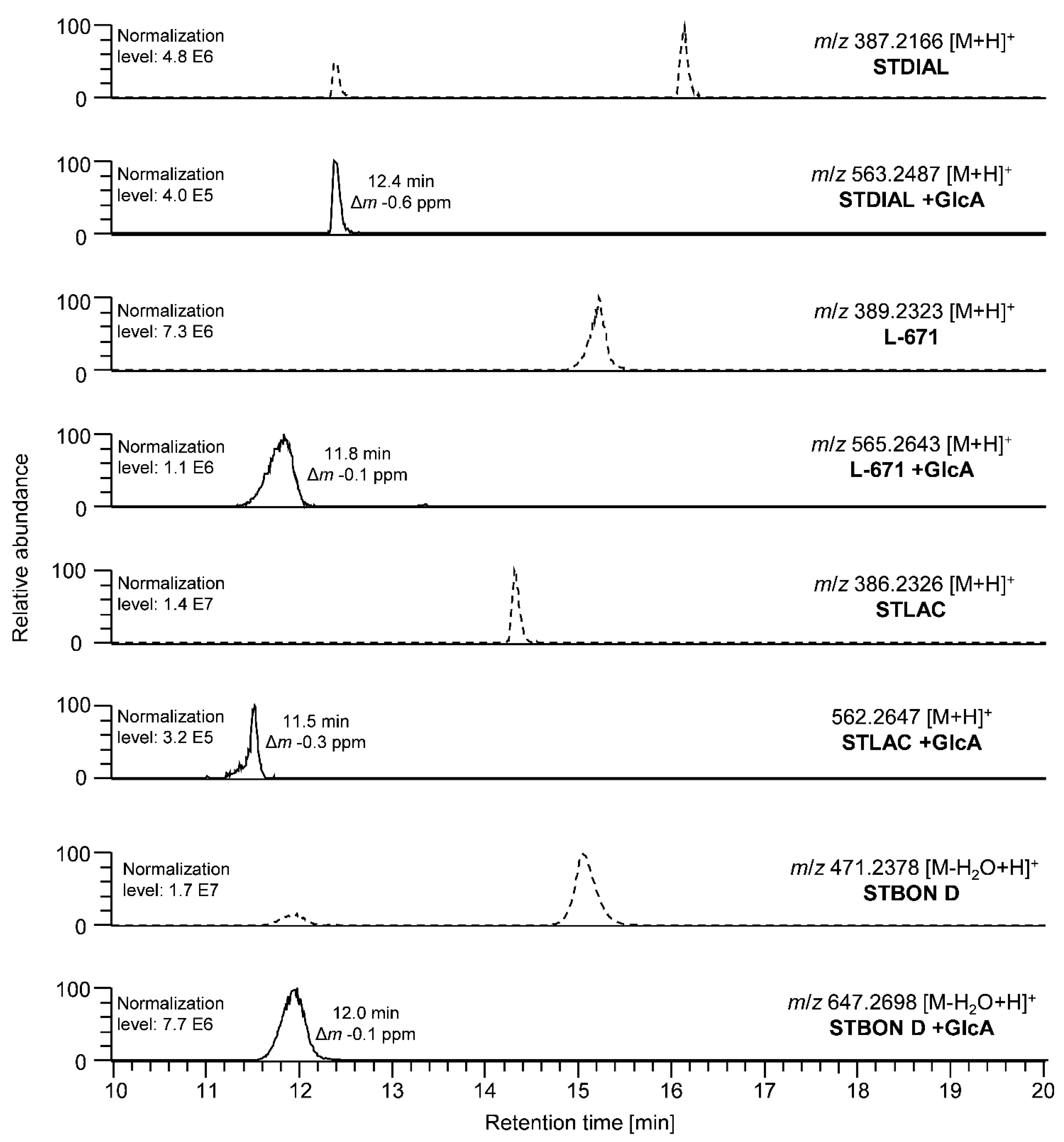
2.1. Metabolism Studies
2.1.1. Phase I Metabolism of PSDs in Human and Horse Liver Microsomes
2.1.2. Phase II Metabolism of PSDs in Human and Horse Liver Microsomes and Cytosol
3. Conclusions
4. Materials and Methods
4.1. Materials, Chemicals, and Reagents
4.2. Incubation with Liver Microsomes and Cytosol
4.2.1. Incubation with NADPH-Regenerating System
4.2.2. Incubation with UDPGA and PAPS
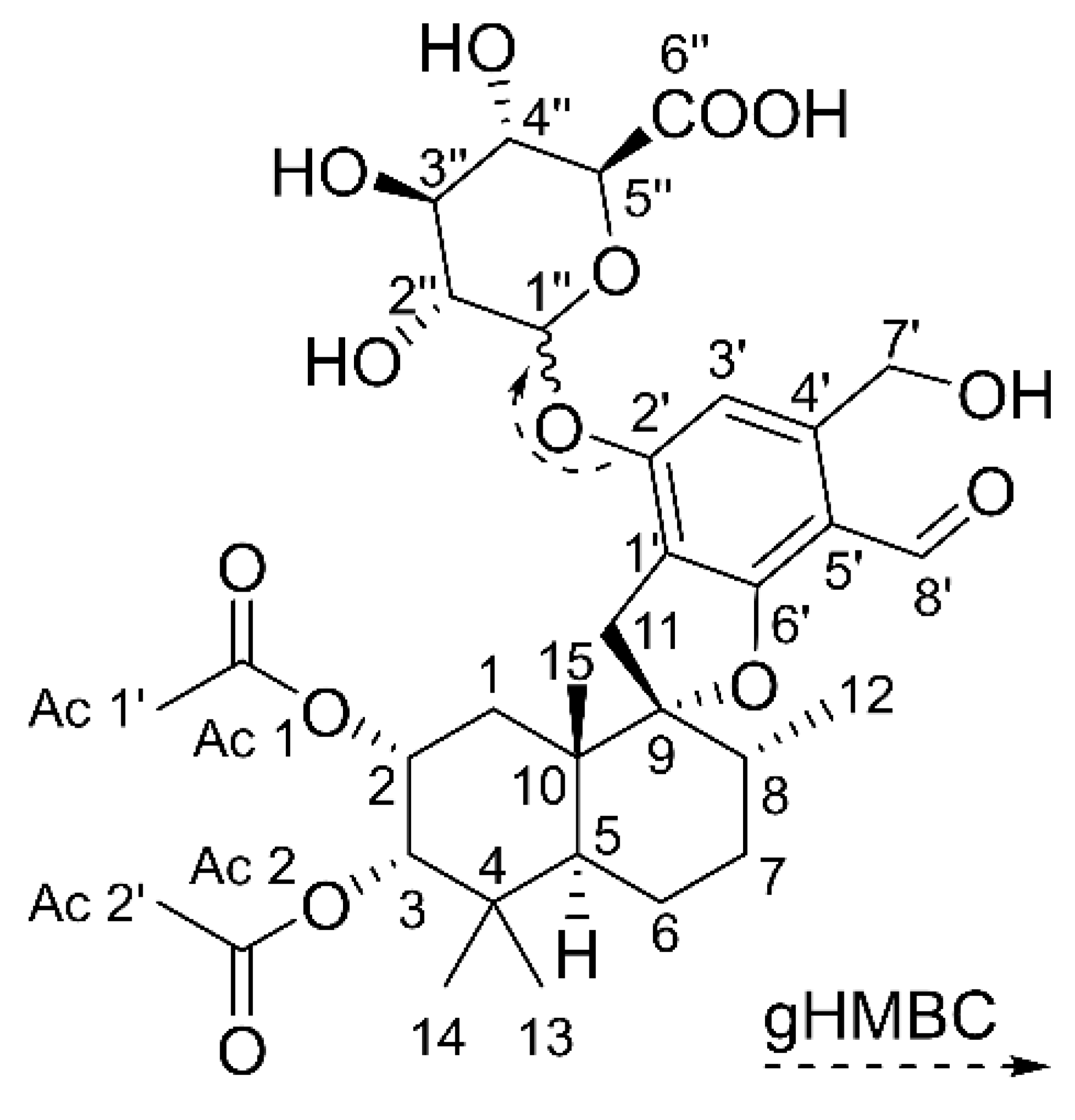
4.3. Isolation of STBON D +GlcA and Acquisition of Structural Data
4.4. Instruments, Chromatographic, and Mass Spectrometric Conditions
4.4.1. HPLC-Orbitrap-HRMS
4.4.2. HPLC-QTOF-HRMS
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Andersen, B.; Nielsen, K.F.; Jarvis, B.B. Characterization of Stachybotrys from water-damaged buildings based on morphology, growth, and metabolite production. Mycologia 2017, 94, 392–403. [Google Scholar] [CrossRef]
- Islam, Z.; Harkema, J.R.; Pestka, J.J. Satratoxin G from the black mold Stachybotrys chartarum evokes olfactory sensory neuron loss and inflammation in the murine nose and brain. Environ. Health Perspect 2006, 114, 1099–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corda, A.C.I. Icones Fungorum Hucusque Cognitorum; Tomus 1. CG Calve: Prague, Czechoslovakia, 1837. [Google Scholar]
- Harrach, B.; Bata, A.; Sándor, G.; Ványi, A. Isolation of macrocyclic and non-macrocyclic trichothecenes (Stachybotrys and Fusarium toxins) from the environment of 200 III sport horses. Mycotoxin. Res. 1987, 3, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Harrach, B.; Bata, A.; Bajmocy, E.; Benko, M. Isolation of satratoxins from the bedding straw of a sheep flock with fatal stachybotryotoxicosis. Appl. Environ. Microbiol. 1983, 45, 1419–1422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drobotko, V.G. Stachybotryotoxicosis: A new disease of horses and humans. Am. Rev. Sov. Med. 1945, 2, 238–242. [Google Scholar]
- Croft, W.A.; Jarvis, B.B.; Yatawara, C.S. Airborne outbreak of trichothecene toxicosis. Atmos Environ. 1986, 20, 549–552. [Google Scholar] [CrossRef]
- Dearborn, D.G.; Yike, I.; Sorenson, W.G.; Miller, M.J.; Etzel, R.A. Overview of investigations into pulmonary hemorrhage among infants in Cleveland, Ohio. Environ. Health Perspect. 1999, 107 (Suppl. 3), 495–499. [Google Scholar] [CrossRef] [Green Version]
- Croston, T.L.; Lemons, A.R.; Barnes, M.A.; Goldsmith, W.T.; Orandle, M.S.; Nayak, A.P.; Germolec, D.R.; Green, B.J.; Beezhold, D.H. Inhalation of Stachybotrys chartarum fragments induces pulmonary arterial remodeling. Am. J. Respir. Cell Mol. Biol. 2020, 62, 563–576. [Google Scholar] [CrossRef]
- Assouline-Dayan, Y.; Leong, A.; Shoenfeld, Y.; Gershwin, M.E. Studies of sick building syndrome. IV. Mycotoxicosis. J. Asthma 2002, 39, 191–201. [Google Scholar] [CrossRef]
- Brakhage, A.A. Regulation of fungal secondary metabolism. Nat. Rev. Microbiol. 2013, 11, 21–32. [Google Scholar] [CrossRef]
- Bennett, J.W.; Klich, M. Mycotoxins. Clin. Microbiol. Rev. 2003, 16, 497–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eppley, R.M.; Bailey, W.J. 12,13-epoxy-Δ9-trichothecenes as the probable mycotoxins responsible for stachybotryotoxicosis. Science 1973, 181, 758–760. [Google Scholar] [CrossRef] [PubMed]
- Ekruth, J.; Gottschalk, C.; Ulrich, S.; Gareis, M.; Schwaiger, K. Differentiation of S. chartarum (Ehrenb.) S. Hughes chemotypes A and S via FT-IR spectroscopy. Mycopathologia 2020, 185, 993–1004. [Google Scholar] [CrossRef] [PubMed]
- El-Maghraby, O.M.O.; Bean, G.A.; Jarvis, B.B.; Aboul-Nasr, M.B. Macrocyclic trichothecenes produced by Stachybotrys isolated from Egypt and Eastern Europe. Mycopathologia 1991, 113, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Bata, A.; Harrach, B.; Ujszászi, K.; Kis-Tamás, A.; Lásztity, R. Macrocyclic trichothecene toxins produced by Stachybotrys atra strains isolated in Middle Europe. Appl. Environ. Microbiol. 1985, 49, 678. [Google Scholar] [CrossRef] [Green Version]
- Jarvis, B.B. Stachybotrys chartarum: A fungus for our time. Phytochemistry 2003, 64, 53–60. [Google Scholar] [CrossRef]
- Nielsen, C.; Casteel, M.; Didier, A.; Dietrich, R.; Märtlbauer, E. Trichothecene-induced cytotoxicity on human cell lines. Mycotoxin Res. 2009, 25, 77–84. [Google Scholar] [CrossRef]
- Rand, T.G.; Flemming, J.; David Miller, J.; Womiloju, T.O. Comparison of inflammatory responses in mouse lungs exposed to atranones A and C from Stachybotrys chartarum. J. Toxicol. Environ. Health Part A 2006, 69, 1239–1251. [Google Scholar] [CrossRef]
- Nielsen, K.F.; Huttunen, K.; Hyvärinen, A.; Andersen, B.; Jarvis, B.B.; Hirvonen, M.-R. Metabolite profiles of Stachybotrys isolates from water-damaged buildings and their induction of inflammatory mediators and cytotoxicity in macrophages. Mycopathologia 2002, 154, 201. [Google Scholar] [CrossRef]
- Jarvis, B.B.; Salemme, J.; Morals, A. Stachybotrys toxins. 1. Nat. Toxins 1995, 3, 10–16. [Google Scholar] [CrossRef]
- Hinkley, S.F.; Jiang, J.; Mazzola, E.P.; Jarvis, B.B. Atranones: Novel diterpenoids from the toxigenic mold Stachybotrys atra. Tetrahedron Lett. 1999, 40, 2725–2728. [Google Scholar] [CrossRef]
- Matsuda, Y.; Abe, I. Biosynthesis of fungal meroterpenoids. Nat. Prod. Rep. 2016, 33, 26–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagels, A.; Stephan, F.; Ernst, S.; Lindemann, V.; Cramer, B.; Hübner, F.; Humpf, H.-U. Artificial vs. natural Stachybotrys infestation—Comparison of mycotoxin production on various building materials. Indoor Air 2020, 30, 1268–1282. [Google Scholar] [CrossRef] [PubMed]
- Ayer, W.A.; Miao, S. Secondary metabolites of the aspen fungus Stachybotrys cylindrospora. Can. J. Chem. 1993, 71, 487–493. [Google Scholar] [CrossRef]
- Kaise, H.; Shinohara, M.; Miyazaki, W.; Izawa, T.; Nakano, Y.; Sugawara, M.; Sugiura, K.; Sasaki, K. Structure of K-76, a complement inhibitor produced by Stachybotrys complementi nov. Sp. K-76. J. Chem. Soc. Chem. Commun. 1979, 16, 726–727. [Google Scholar] [CrossRef]
- Zhao, J.; Feng, J.; Tan, Z.; Liu, J.; Zhao, J.; Chen, R.; Xie, K.; Zhang, D.; Li, Y.; Yu, L.; et al. Stachybotrysins A-G, phenylspirodrimane derivatives from the fungus Stachybotrys chartarum. J. Nat. Prod. 2017, 80, 1819–1826. [Google Scholar] [CrossRef]
- Ma, X.-H.; Zheng, W.-M.; Sun, K.-H.; Gu, X.-F.; Zeng, X.-M.; Zhang, H.-T.; Zhong, T.-H.; Shao, Z.-Z.; Zhang, Y.-H. Two new phenylspirodrimanes from the deep-sea derived fungus Stachybotrys sp. MCCC 3A00409. Nat. Prod. Res. 2018, 33, 386–392. [Google Scholar] [CrossRef]
- Williams, R.T. Detoxication mechanisms. In The Metabolism and Detoxication of Drugs, Toxic Substances, and Other Organic Compounds; Wiley: New York, NY, USA, 1959. [Google Scholar]
- Amuzie, C.J.; Islam, Z.; Kim, J.K.; Seo, J.-H.; Pestka, J.J. Kinetics of satratoxin G tissue distribution and excretion following intranasal exposure in the mouse. Toxicol. Sci. 2010, 116, 433–440. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, H.S.; Schulz, M.; Focke, C.; Becker, S.; Cramer, B.; Humpf, H.-U. Glucosylation of T-2 and HT-2 toxins using biotransformation and chemical synthesis: Preparation, stereochemistry, and stability. Mycotoxin. Res. 2018, 34, 159–172. [Google Scholar] [CrossRef]
- Knowles, B.B.; Howe, C.C.; Aden, D.P. Human hepatocellular carcinoma cell lines secrete the major plasma proteins and hepatitis B surface antigen. Science 1980, 209, 497–499. [Google Scholar] [CrossRef]
- O’Brien, J.; Wilson, I.; Orton, T.; Pognan, F. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur. J. Biochem. 2000, 267, 5421–5426. [Google Scholar] [CrossRef] [PubMed]
- Jagels, A.; Lindemann, V.; Ulrich, S.; Gottschalk, C.; Cramer, B.; Hübner, F.; Gareis, M.; Humpf, H.-U. Exploring secondary metabolite profiles of Stachybotrys spp. by LC-MS/MS. Toxins 2019, 11, 133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lake, B.G. Preparation and characterisation of microsomal fractions for studies on xenobiotic metabolism. In Biochemical Toxicology: A Practical Approach; Snell, K., Mullock, B., Eds.; IRL Press: Oxford, UK, 1987; pp. 183–215. [Google Scholar]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Li, S.; Liu, W.; Teng, L.; Cheng, X.; Wang, Z.; Wang, C. Metabolites identification of harmane in vitro/in vivo in rats by ultra-performance liquid chromatography combined with electrospray ionization quadrupole time-of-flight tandem mass spectrometry. J. Pharm. Biomed. Anal. 2014, 92, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Jia, R.; Ye, C.; Garcia, M.; Li, J.; Hidalgo, I.J. Glucuronidation and sulfation of 7-hydroxycoumarin in liver matrices from human, dog, monkey, rat, and mouse. Vitr. Cell. Dev. Biol. Anim. 2005, 41, 97–103. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | δC (ppm), Type | δH (ppm), Type (J in Hz) | Position | δC (ppm), Type | δH (ppm), Type (J in Hz) |
| 1 1 | 31.5, CH2 | 1.43, 1.52, m | 1′ | 116.5, C | - |
| 2 | 69.7, CH | 5.23, m | 2′ | 156.8, C | - |
| 3 | 78.4, CH | 4.92, m | 3′ | 100.3, CH | 6.57, s |
| 4 | 39.2, C | - | 4′ | 147.6, C | - |
| 5 | 42.0, CH | 2.17, m | 5′ | 112.9, C | - |
| 6 1 | 21.5, CH2 | 1.52, 1.60, m | 6′ | 157.7, C | - |
| 7 1 | 32.1, CH2 | 1.52, 1.60, m | 7′ | 63.8, CH2 | 4.87, m |
| 8 | 37.7, CH | 1.94, m | 8′ | 189.7, CH | 10.35, s |
| 9 | 100.9, C | - | 1′′ | 102.1, CH | 4.96, m |
| 10 | 45.0, C | - | 2′′ 2 | 74.7, CH | 3.50, m |
| 11 | 33.1, CH2 | 2.94, 3.34, m | 3′′ 2 | 77.8, CH | 3.50, m |
| 12 | 15.8, CH3 | 0.81, d (6.5) | 4′′ | 73.6, CH | 3.53, m |
| 13 | 22.0, CH3 | 1.04, s | 5′′ | 76.6, CH | 3.76, m |
| 14 | 28.4, CH3 | 0.93, s | 6′′ | n.d. 3, C | - |
| 15 | 17.2, CH3 | 1.16, s | |||
| Ac 1 | 172.3, C | - | |||
| Ac 1′ | 21.0, CH3 | 1.86, s | |||
| Ac 2 | 172.4, C | - | |||
| Ac 2′ | 20.8, CH3 | 2.00, s |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lindemann, V.; Jagels, A.; Behrens, M.; Hübner, F.; Humpf, H.-U. In Vitro Metabolism of Phenylspirodrimanes Derived from the Indoor Fungus Stachybotrys. Toxins 2022, 14, 395. https://doi.org/10.3390/toxins14060395
Lindemann V, Jagels A, Behrens M, Hübner F, Humpf H-U. In Vitro Metabolism of Phenylspirodrimanes Derived from the Indoor Fungus Stachybotrys. Toxins. 2022; 14(6):395. https://doi.org/10.3390/toxins14060395
Chicago/Turabian StyleLindemann, Viktoria, Annika Jagels, Matthias Behrens, Florian Hübner, and Hans-Ulrich Humpf. 2022. "In Vitro Metabolism of Phenylspirodrimanes Derived from the Indoor Fungus Stachybotrys" Toxins 14, no. 6: 395. https://doi.org/10.3390/toxins14060395
APA StyleLindemann, V., Jagels, A., Behrens, M., Hübner, F., & Humpf, H.-U. (2022). In Vitro Metabolism of Phenylspirodrimanes Derived from the Indoor Fungus Stachybotrys. Toxins, 14(6), 395. https://doi.org/10.3390/toxins14060395