Fusarium Mycotoxins in Maize Field Soils: Method Validation and Implications for Sampling Strategy

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Soil Samples for Method Validation
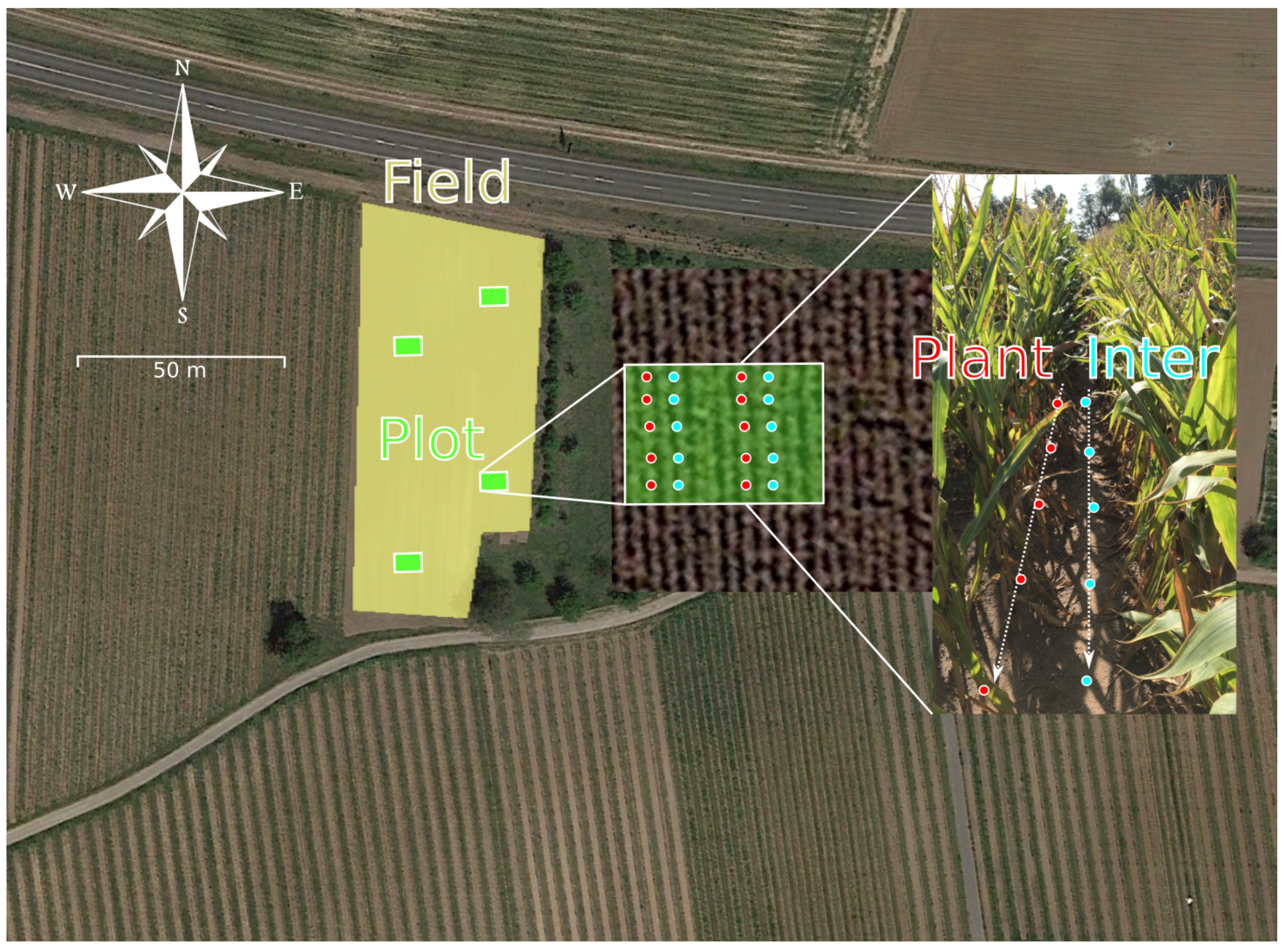
2.3. Environmental Monitoring Strategy
2.4. Soil Characterization
2.5. Soil Spiking and Extraction
2.6. LC–HRMS Analysis
2.7. Method Validation Criteria
2.8. Data Evaluation
3. Results
3.1. Soil Characterization
3.2. Linearity and Matrix Effect
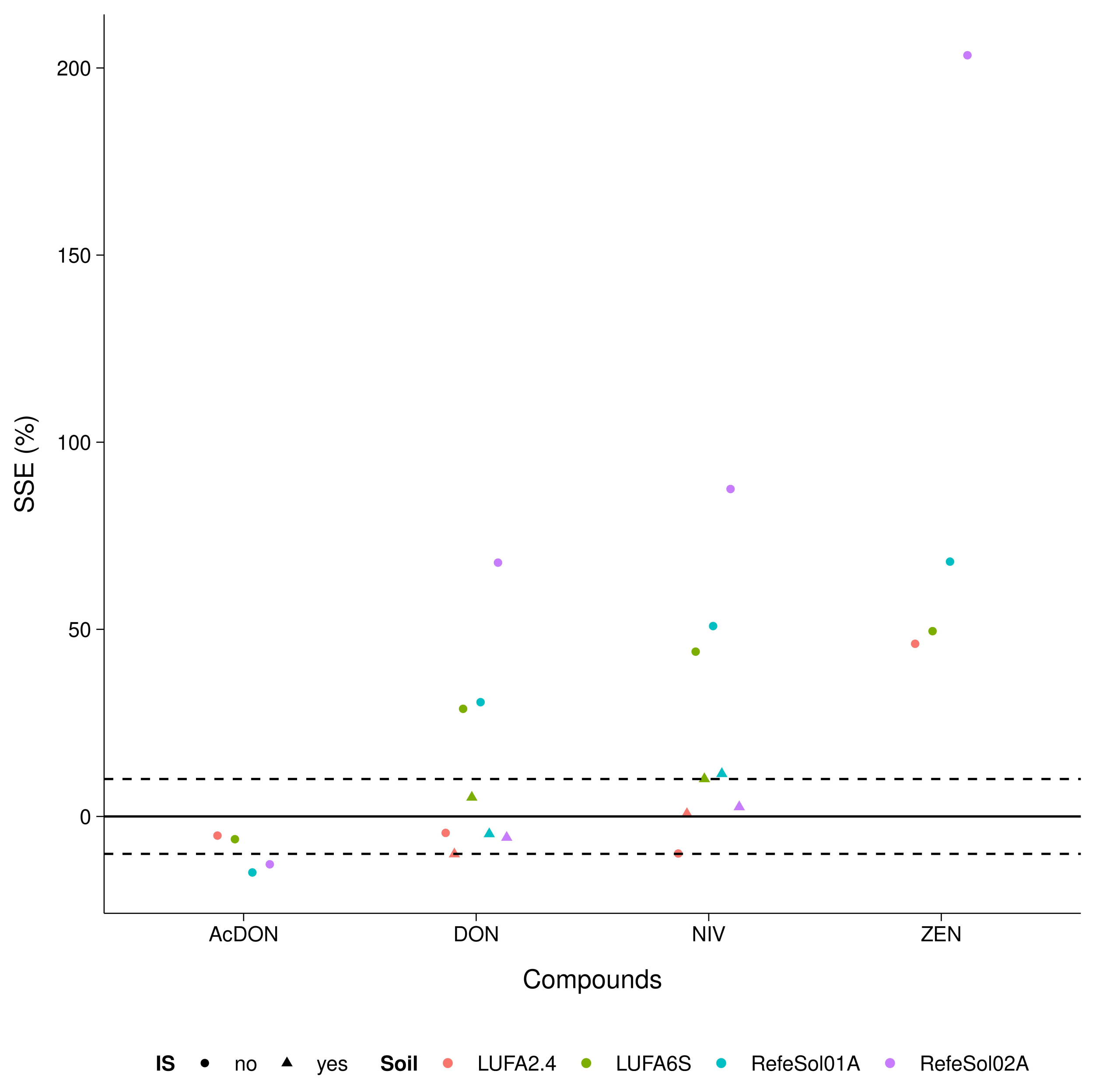
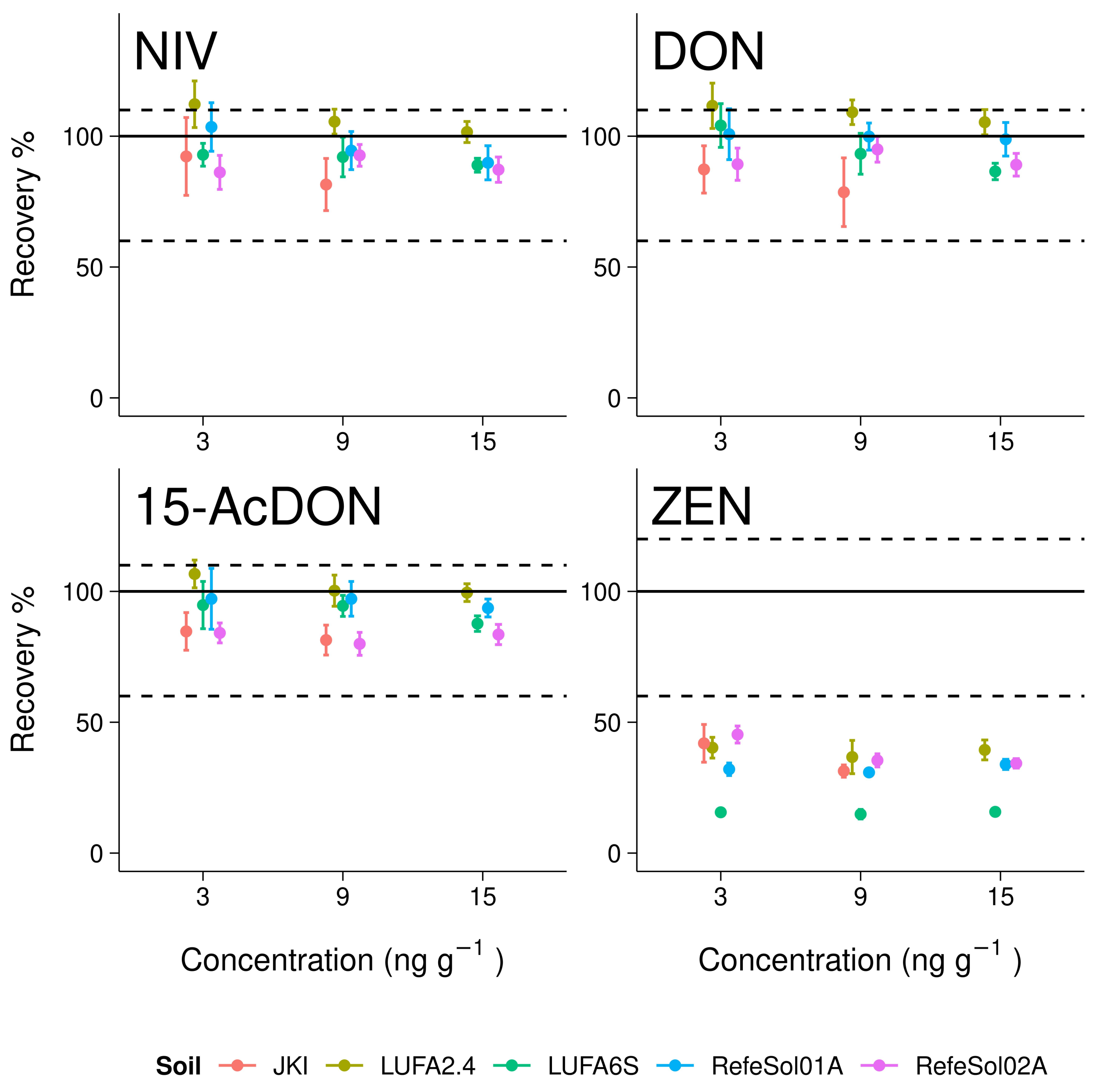
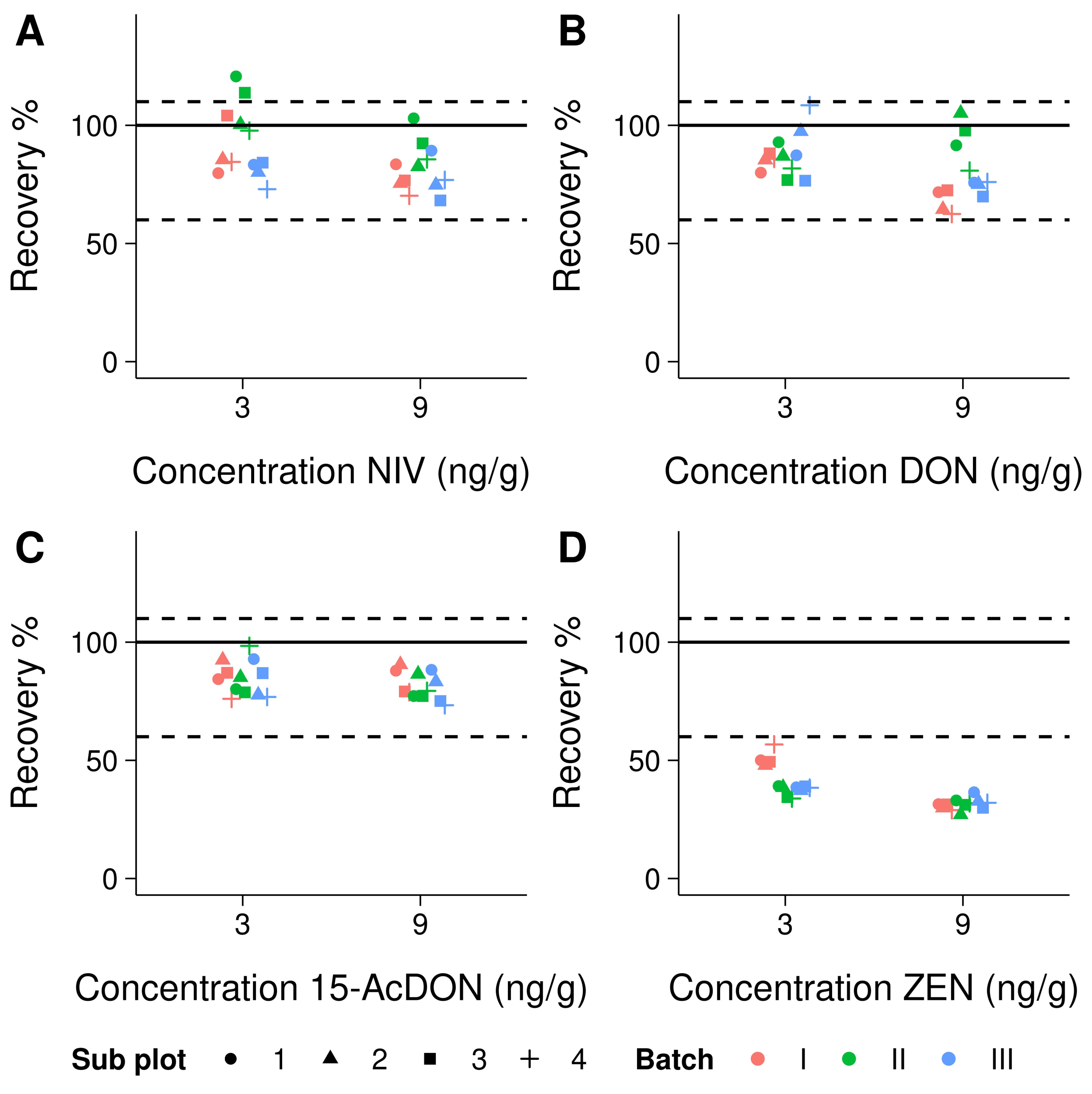
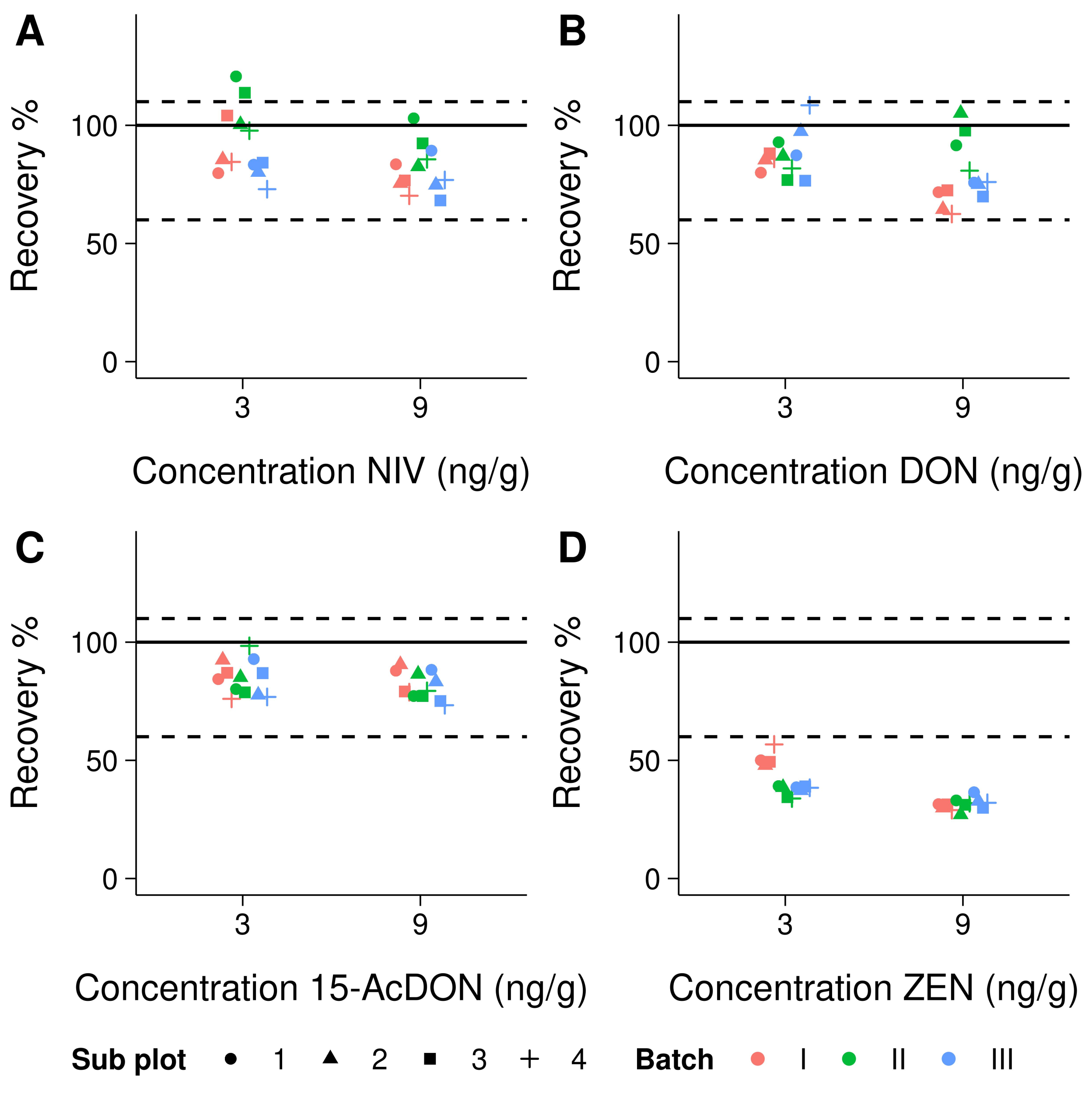
3.3. Trueness and Precision
3.4. Limit of Detection and Quantification
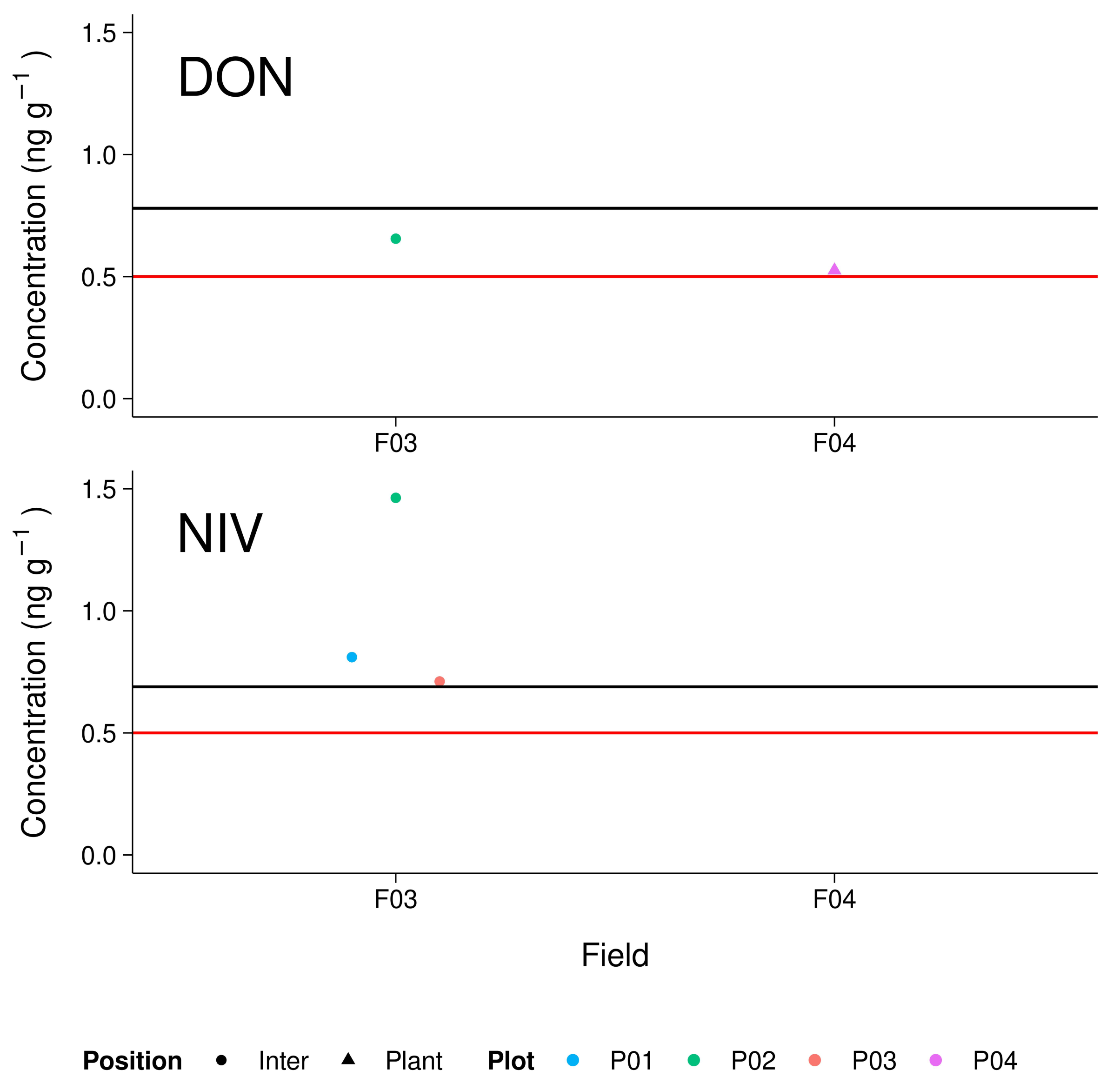
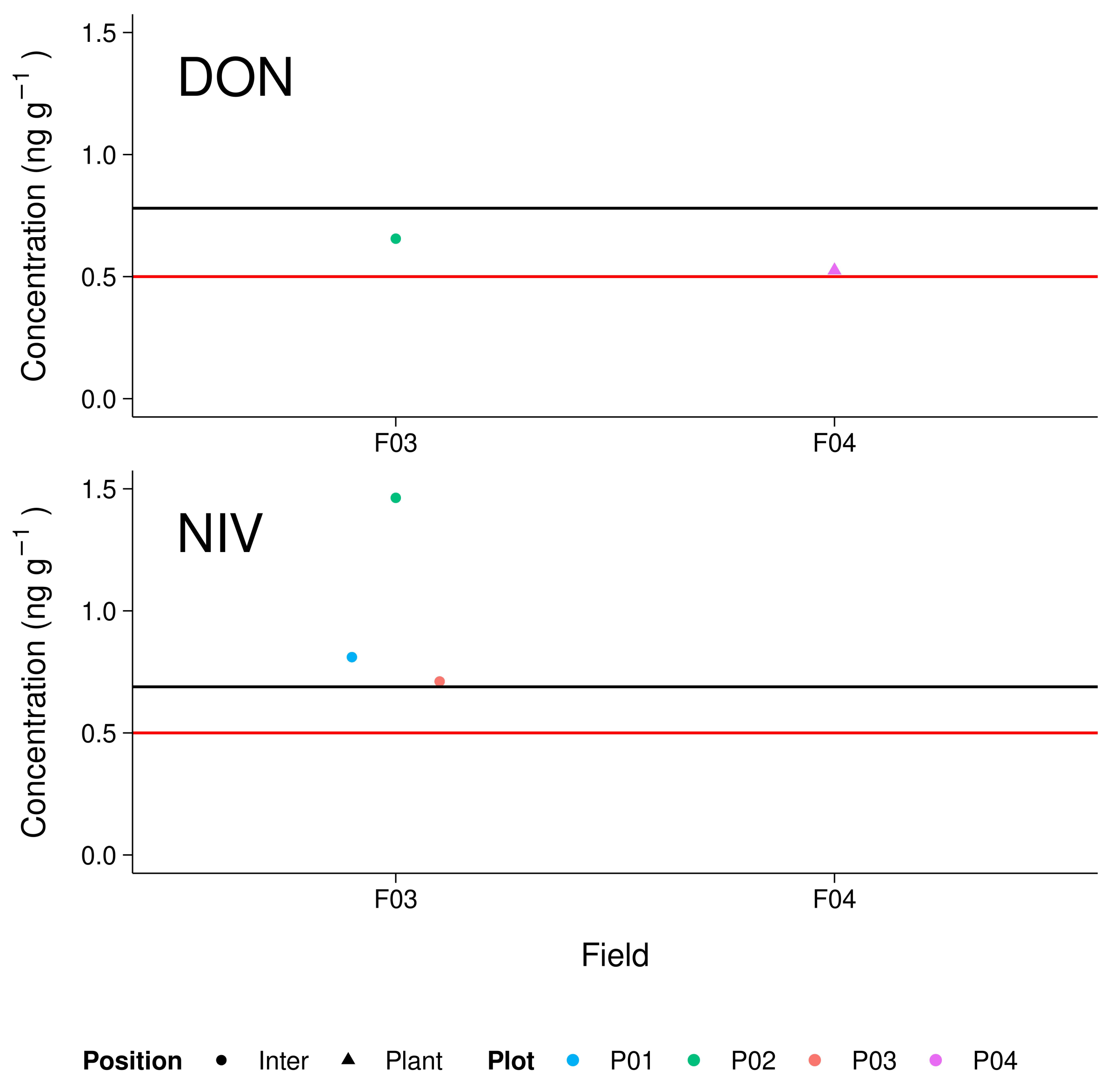
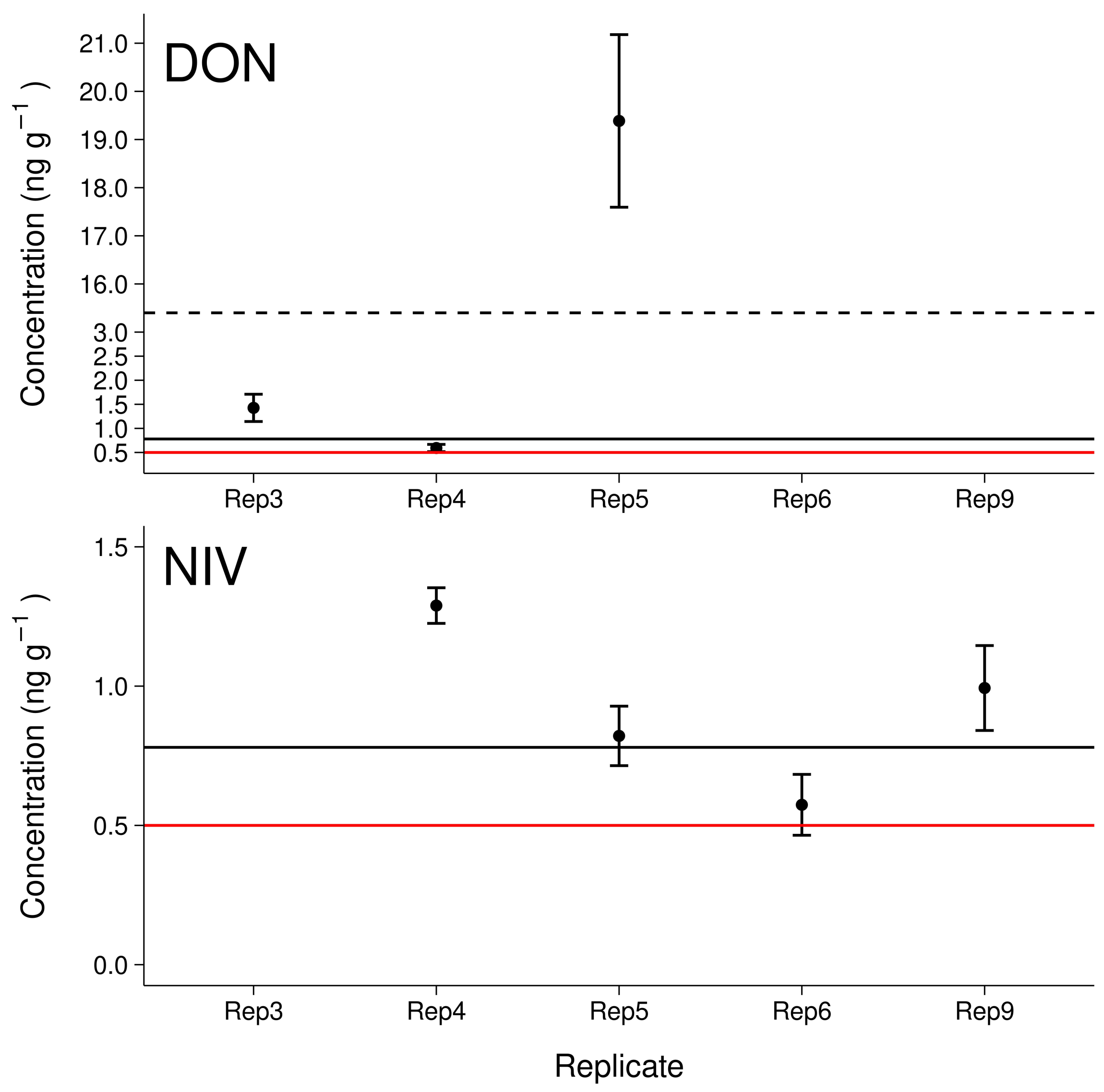
3.5. Maize Field Soil Samples
4. Discussion
4.1. Linearity and Matrix Effect
4.2. Trueness and Precision
4.3. Limit of Detection and Quantification
4.4. Environmental Samples and Implications for Further Investigations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| JKI | Julius Kühn Institute |
| NIV | nivalenol |
| DON | deoxynivalenol |
| 15-AcDON | 15-acetyl-deoxynivalenol |
| ZEN | zearalenone |
| LC | liquid chromatography |
| HRMS | high-resolution mass spectrometry |
| RSDr | relative standard deviation |
| HPLC | high pressure liquid chromatography |
| PET | polyethylene |
| LOD | limit of detection |
| LOQ | limit of quantification |
| QQ plot | quantile–quantile plot |
| WLS | weighted least squares |
| F-ANOVA | analysis of variance based on F-test |
| LME | linear mixed effect |
| APCI | atmospheric pressure chemical ionization |
| EU | European Union |
| EC | European Commission |
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Soil | Recovery | LOD | LOQ | ||
|---|---|---|---|---|---|---|
| 3 | 9 | 15 | ||||
| NIV | LUFA2.4 | 112 (9) | 106 (5) | 102 (4) | 0.15 | 0.5 |
| LUFA6S | 93 (4) | 92 (8) | 89 (3) | 0.25 | 0.82 | |
| RefeSol01A | 104 (9) | 94 (7) | 90 (7) | 0.25 | 0.84 | |
| RefeSol02A | 86 (6) | 93 (4) | 87 (5) | 0.26 | 0.86 | |
| DON | LUFA2.4 | 112 (9) | 109 (5) | 105 (5) | 0.24 | 0.79 |
| LUFA6S | 104 (8) | 93 (8) | 87 (3) | 0.12 | 0.41 | |
| RefeSol01A | 100 (10) | 100 (5) | 99 (6) | 0.33 | 1.1 | |
| RefeSol02A | 89 (6) | 95 (5) | 89 (4) | 0.28 | 0.92 | |
| 15-AcDON | LUFA2.4 | 107 (5) | 100 (6) | 100 (3) | 0.26 | 0.88 |
| LUFA6S | 95 (9) | 94 (4) | 88 (3) | 0.11 | 0.36 | |
| RefeSol01A | 100 (10) | 97 (7) | 94 (3) | 0.18 | 0.58 | |
| RefeSol02A | 84 (4) | 80 (4) | 84 (4) | 0.18 | 0.62 | |
| ZEN | LUFA2.4 | 40 (4) | 37 (6) | 39 (4) | n.d. | n.d. |
| LUFA6S | 16 (1) | 15 (2) | 16 (1) | n.d. | n.d. | |
| RefeSol01A | 32 (2) | 31 (1) | 34 (2) | n.d. | n.d. | |
| RefeSol02A | 45 (3) | 35 (3) | 34 (2) | n.d. | n.d. | |


References
- Savary, S.; Willocquet, L.; Pethybridge, S.J.; Esker, P.; McRoberts, N.; Nelson, A. The Global Burden of Pathogens and Pests on Major Food Crops. Nat. Ecol. Evol. 2019, 3, 430–439. [Google Scholar] [CrossRef] [PubMed]
- Elmholt, S. Mycotoxins in the Soil Environment. In Secondary Metabolites in Soil Ecology; Springer: Berlin/Heidelberg, Germany, 2008; Volume 14, pp. 167–203. [Google Scholar] [CrossRef]
- Bryła, M.; Ksieniewicz-Woźniak, E.; Yoshinari, T.; Waśkiewicz, A.; Szymczyk, K. Contamination of Wheat Cultivated in Various Regions of Poland during 2017 and 2018 Agricultural Seasons with Selected Trichothecenes and Their Modified Forms. Toxins 2019, 11, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolpin, D.W.; Schenzel, J.; Meyer, M.T.; Phillips, P.J.; Hubbard, L.E.; Scott, T.M.; Bucheli, T.D. Mycotoxins: Diffuse and Point Source Contributions of Natural Contaminants of Emerging Concern to Streams. Sci. Total Environ. 2014, 470, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Vogelgsang, S.; Musa, T.; Bänziger, I.; Kägi, A.; Bucheli, T.D.; Wettstein, F.E.; Pasquali, M.; Forrer, H.R. Fusarium Mycotoxins in Swiss Wheat: A Survey of Growers’ Samples between 2007 and 2014 Shows Strong Year and Minor Geographic Effects. Toxins 2017, 9, 246. [Google Scholar] [CrossRef] [Green Version]
- Knutsen, H.K.; Alexander, J.; Barregård, L.; Bignami, M.; Brüschweiler, B.; Ceccatelli, S.; Cottrill, B.; Dinovi, M.; Grasl-Kraupp, B.; Hogstrand, C.; et al. Risks to Human and Animal Health Related to the Presence of Deoxynivalenol and Its Acetylated and Modified Forms in Food and Feed. EFSA J. 2017, 15, e04718. [Google Scholar] [CrossRef] [PubMed]
- Escrivá, L.; Font, G.; Manyes, L. In Vivo Toxicity Studies of Fusarium Mycotoxins in the Last Decade: A Review. Food Chem. Toxicol. 2015, 78, 185–206. [Google Scholar] [CrossRef]
- Kowalska, K.; Habrowska-Górczyńska, D.E.; Piastowska-Ciesielska, A.W. Zearalenone as an Endocrine Disruptor in Humans. Environ. Toxicol. Pharmacol. 2016, 48, 141–149. [Google Scholar] [CrossRef]
- Schwartz, P.; Thorpe, K.L.; Bucheli, T.D.; Wettstein, F.E.; Burkhardt-Holm, P. Short-Term Exposure to the Environmentally Relevant Estrogenic Mycotoxin Zearalenone Impairs Reproduction in Fish. Sci. Total Environ. 2010, 409, 326–333. [Google Scholar] [CrossRef]
- Commission of the European Communities. Commission Regulation (EC) No 1881/2006 of 19 December 2006 Setting Maximum Levels for Certain Contaminants in Foodstuffs (Text with EEA Relevance). Off. J. Eur. Union 2006, 364, 5–24. [Google Scholar]
- U.S. FDA. Guidance for Industry and FDA: Advisory Levels for Deoxynivalenol (DON) in Finished Wheat Products for Human Consumption and Grains and Grain By-Products Used for Animal Feed; U.S. FDA: Silver Spring, MD, USA, 2010.
- Kralj Cigić, I.; Prosen, H. An Overview of Conventional and Emerging Analytical Methods for the Determination of Mycotoxins. Int. J. Mol. Sci. 2009, 10, 62–115. [Google Scholar] [CrossRef]
- Pereira, V.L.; Fernandes, J.O.; Cunha, S.C. Mycotoxins in Cereals and Related Foodstuffs: A Review on Occurrence and Recent Methods of Analysis. Trends Food Sci. Technol. 2014, 36, 96–136. [Google Scholar] [CrossRef]
- Schenzel, J.; Hungerbühler, K.; Bucheli, T.D. Mycotoxins in the Environment: II. Occurrence and Origin in Swiss River Waters. Environ. Sci. Technol. 2012, 46, 13076–13084. [Google Scholar] [CrossRef] [PubMed]
- Bucheli, T.D. Phytotoxins: Environmental Micropollutants of Concern? Environ. Sci. Technol. 2014, 48, 13027–13033. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, N.; Erbs, M.; Wettstein, F.E.; Hoerger, C.C.; Schwarzenbach, R.P.; Bucheli, T.D. Quantification of Zearalenone in Various Solid Agroenvironmental Samples Using D6-Zearalenone as the Internal Standard. J. Agric. Food Chem. 2008, 56, 2926–2932. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, K.; Schmidt-Heydt, M.; Stoll, D.; Diehl, D.; Ziegler, J.; Geisen, R.; Schaumann, G.E. Effect of Plastic Mulching on Mycotoxin Occurrence and Mycobiome Abundance in Soil Samples from Asparagus Crops. Mycotoxin Res. 2015, 31, 191–201. [Google Scholar] [CrossRef]
- Muñoz, K.; Buchmann, C.; Meyer, M.; Schmidt-Heydt, M.; Steinmetz, Z.; Diehl, D.; Thiele-Bruhn, S.; Schaumann, G.E. Physicochemical and Microbial Soil Quality Indicators as Affected by the Agricultural Management System in Strawberry Cultivation Using Straw or Black Polyethylene Mulching. Appl. Soil Ecol. 2017, 113, 36–44. [Google Scholar] [CrossRef]
- Meyer, M.; Diehl, D.; Schaumann, G.E.; Muñoz, K. Agricultural Mulching and Fungicides—Impacts on Fungal Biomass, Mycotoxin Occurrence, and Soil Organic Matter Decomposition. Environ. Sci. Pollut. Res. 2021, 28, 36535–36550. [Google Scholar] [CrossRef]
- Bucheli, T.D.; Wettstein, F.E.; Hartmann, N.; Erbs, M.; Vogelgsang, S.; Forrer, H.R.; Schwarzenbach, R.P. Fusarium Mycotoxins: Overlooked Aquatic Micropollutants? J. Agric. Food Chem. 2008, 56, 1029–1034. [Google Scholar] [CrossRef]
- Schenzel, J.; Forrer, H.R.; Vogelgsang, S.; Hungerbühler, K.; Bucheli, T.D. Mycotoxins in the Environment: I. Production and Emission from an Agricultural Test Field. Environ. Sci. Technol. 2012, 46, 13067–13075. [Google Scholar] [CrossRef]
- Meyer-Wolfarth, F.; Oldenburg, E.; Meiners, T.; Muñoz, K.; Schrader, S. Effects of Temperature and Soil Fauna on the Reduction and Leaching of Deoxynivalenol and Zearalenone from Fusarium Graminearum-Infected Maize Stubbles. Mycotoxin Res. 2021, 37, 249–263. [Google Scholar] [CrossRef]
- Chen, P.; Xiang, B.; Shi, H.; Yu, P.; Song, Y.; Li, S. Recent Advances on Type A Trichothecenes in Food and Feed: Analysis, Prevalence, Toxicity, and Decontamination Techniques. Food Control 2020, 118, 107371. [Google Scholar] [CrossRef]
- Malachová, A.; Stránská, M.; Václavíková, M.; Elliott, C.T.; Black, C.; Meneely, J.; Hajšlová, J.; Ezekiel, C.N.; Schuhmacher, R.; Krska, R. Advanced LC–MS-based Methods to Study the Co-Occurrence and Metabolization of Multiple Mycotoxins in Cereals and Cereal-Based Food. Anal. Bioanal. Chem. 2018, 410, 801–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schenzel, J.; Schwarzenbach, R.P.; Bucheli, T.D. Multi-Residue Screening Method To Quantify Mycotoxins in Aqueous Environmental Samples. J. Agric. Food Chem. 2010, 58, 11207–11217. [Google Scholar] [CrossRef]
- De Santis, B.; Debegnach, F.; Gregori, E.; Russo, S.; Marchegiani, F.; Moracci, G.; Brera, C. Development of a LC–MS/MS Method for the Multi-Mycotoxin Determination in Composite Cereal-Based Samples. Toxins 2017, 9, 169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patzold, S.; Mertens, F.M.; Bornemann, L.; Koleczek, B.; Franke, J.; Feilhauer, H.; Welp, G. Soil Heterogeneity at the Field Scale: A Challenge for Precision Crop Protection. Precis. Agric. 2008, 9, 367–390. [Google Scholar] [CrossRef]
- Franklin, R.B.; Mills, A.L. Importance of Spatially Structured Environmental Heterogeneity in Controlling Microbial Community Composition at Small Spatial Scales in an Agricultural Field. Soil Biol. Biochem. 2009, 41, 1833–1840. [Google Scholar] [CrossRef]
- Albert, J.; More, C.A.; Dahlke, N.R.P.; Steinmetz, Z.; Schaumann, G.E.; Muñoz, K. Validation of a Simple and Reliable Method for the Determination of Aflatoxins in Soil and Food Matrices. ACS Omega 2021, 6, 18684–18693. [Google Scholar] [CrossRef]
- Wettstein, F.E.; Bucheli, T.D. Poor Elimination Rates in Waste Water Treatment Plants Lead to Continuous Emission of Deoxynivalenol into the Aquatic Environment. Water Res. 2010, 44, 4137–4142. [Google Scholar] [CrossRef]
- Fabregat-Cabello, N.; Zomer, P.; Sancho, J.; Roig-Navarro, A.; Mol, H. Comparison of Approaches to Deal with Matrix Effects in LC–MS/MS Based Determinations of Mycotoxins in Food and Feed. World Mycotoxin J. 2016, 9, 149–161. [Google Scholar] [CrossRef] [Green Version]
- Mortensen, G.K.; Strobel, B.W.; Hansen, H.C.B. Determination of Zearalenone and Ochratoxin A in Soil. Anal. Bioanal. Chem. 2003, 376, 98–101. [Google Scholar] [CrossRef]
- Klein, T.A.; Burgess, L.W.; Ellison, F.W. The Incidence and Spatial Patterns of Wheat Plants Infected by Fusarium Graminearum Group 1 and the Effect of Crown Rot on Yield. Aust. J. Agric. Res. 1991, 42, 399–407. [Google Scholar] [CrossRef]
- Rolli, E.; Righetti, L.; Galaverna, G.; Suman, M.; Dall’Asta, C.; Bruni, R. Zearalenone Uptake and Biotransformation in Micropropagated Triticum Durum Desf. Plants: A Xenobolomic Approach. J. Agric. Food Chem. 2018, 66, 1523–1532. [Google Scholar] [CrossRef] [PubMed]
- Mantle, P.G. Uptake of Radiolabelled Ochratoxin A from Soil by Coffee Plants. Phytochemistry 2000, 53, 377–378. [Google Scholar] [CrossRef]
- Righetti, L.; Damiani, T.; Rolli, E.; Galaverna, G.; Suman, M.; Bruni, R.; Dall’Asta, C. Exploiting the Potential of Micropropagated Durum Wheat Organs as Modified Mycotoxin Biofactories: The Case of Deoxynivalenol. Phytochemistry 2020, 170, 112194. [Google Scholar] [CrossRef] [PubMed]
- Snigdha, M.; Hariprasad, P.; Venkateswaran, G. Mechanism of Aflatoxin Uptake in Roots of Intact Groundnut (Arachis hypogaea L.) Seedlings. Environ. Sci. Pollut. Res. 2013, 20, 8502–8510. [Google Scholar] [CrossRef]
- Goertz, A.; Zuehlke, S.; Spiteller, M.; Steiner, U.; Dehne, H.W.; Waalwijk, C.; de Vries, I.; Oerke, E.C. Fusarium Species and Mycotoxin Profiles on Commercial Maize Hybrids in Germany. Eur. J. Plant Pathol. 2010, 128, 101–111. [Google Scholar] [CrossRef]
- Schollenberger, M.; Müller, H.M.; Ernst, K.; Sondermann, S.; Liebscher, M.; Schlecker, C.; Wischer, G.; Drochner, W.; Hartung, K.; Piepho, H.P. Occurrence and Distribution of 13 Trichothecene Toxins in Naturally Contaminated Maize Plants in Germany. Toxins 2012, 4, 778–787. [Google Scholar] [CrossRef] [PubMed]
- Schenzel, J.; Goss, K.U.; Schwarzenbach, R.P.; Bucheli, T.D.; Droge, S.T.J. Experimentally Determined Soil Organic Matter–Water Sorption Coefficients for Different Classes of Natural Toxins and Comparison with Estimated Numbers. Environ. Sci. Technol. 2012, 46, 6118–6126. [Google Scholar] [CrossRef]
- Zachariášová, M. Multi-Mycotoxin Screening and Quantitation Using UHPLC, High Resolution and Accurate Mass. 2011. Available online: https://www.thermofisher.com/document-connect/document-connect.html?url=https://assets.thermofisher.com/TFS-Assets/LSG/Application-Notes/Article-Food-Science-Standout-plus-AN-51961-Multi-mycotoxin-Screening-Quantitation.pdf (accessed on 17 December 2018).
- McCullough, B.J.; Hopley, C.J. A Validated LC–HRMS Method for the Detection of T-2 and HT-2 Toxins in Cereals. J. Assoc. Public Anal. 2017, 45, 41–73. [Google Scholar]
- Tolosa, J.; Graziani, G.; Gaspari, A.; Chianese, D.; Ferrer, E.; Mañes, J.; Ritieni, A. Multi-Mycotoxin Analysis in Durum Wheat Pasta by Liquid Chromatography Coupled to Quadrupole Orbitrap Mass Spectrometry. Toxins 2017, 9, 59. [Google Scholar] [CrossRef] [Green Version]
- Slobodchikova, I.; Sivakumar, R.; Rahman, M.S.; Vuckovic, D. Characterization of Phase I and Glucuronide Phase II Metabolites of 17 Mycotoxins Using Liquid Chromatography—High-Resolution Mass Spectrometry. Toxins 2019, 11, 433. [Google Scholar] [CrossRef] [Green Version]
- Magnusson, B.; Örnemark, U. (Eds.) Eurachem Guide: The Fitness for Purpose of Analytical Methods—A Laboratory Guide to Method Validation and Related Topics, 2nd ed.; 2014; ISBN 978-91-87461-59-0. Available online: www.eurachem.org (accessed on 17 December 2018).
- Zuur, A.F.; Ieno, E.N.; Elphick, C.S. A Protocol for Data Exploration to Avoid Common Statistical Problems. Methods Ecol. Evol. 2010, 1, 3–14. [Google Scholar] [CrossRef]
- Almeida, A.M.; Castel-Branco, M.M.; Falcão, A.C. Linear Regression for Calibration Lines Revisited: Weighting Schemes for Bioanalytical Methods. J. Chromatogr. B 2002, 774, 215–222. [Google Scholar] [CrossRef]
- Commission of the European Communities. Commission Regulation (EC) No 401/2006 of 23 February 2006 Laying down the Methods of Sampling and Analysis for the Official Control of the Levels of Mycotoxins in Foodstuffs (Text with EEA Relevance). Off. J. Eur. Union 2006, 70, 12–34. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Dowle, M.; Srinivasan, A. Extension of ‘data.Frame’ [R Package Data.Table Version 1.14.0]. 2021. Available online: https://CRAN.R-project.org/package=data.table (accessed on 14 May 2021).
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis, 2nd ed.; Springer International Publishing: Berlin/Heidelberg, Germany, 2016. [Google Scholar] [CrossRef]
- Steinmetz, Z. Envalysis: Miscellaneous Functions for Environmental Analyses. 2021. Available online: https://cran.r-project.org/package=envalysis (accessed on 14 June 2021).
- Kuznetsova, A.; Brockhoff, P.B.; Christensen, R.H.B. lmerTest Package: Tests in Linear Mixed Effects Models. J. Stat. Softw. 2017, 82, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Halekoh, U.; Højsgaard, S. A Kenward–Roger Approximation and Parametric Bootstrap Methods for Tests in Linear Mixed Models - The R Package Pbkrtest. J. Stat. Softw. 2014, 59, 1–32. [Google Scholar] [CrossRef] [Green Version]
- Kappenberg, A.; Juraschek, L.M. Development of a LC–MS/MS Method for the Simultaneous Determination of the Mycotoxins Deoxynivalenol (DON) and Zearalenone (ZEA) in Soil Matrix. Toxins 2021, 13, 470. [Google Scholar] [CrossRef] [PubMed]
- Salvia, M.V.; Cren-Olivé, C.; Vulliet, E. Statistical Evaluation of the Influence of Soil Properties on Recoveries and Matrix Effects during the Analysis of Pharmaceutical Compounds and Steroids by Quick, Easy, Cheap, Effective, Rugged and Safe Extraction Followed by Liquid Chromatography–Tandem Mass Spectrometry. J. Chromatogr. A 2013, 1315, 53–60. [Google Scholar] [CrossRef] [PubMed]
- European Commission. SANCO/12571/2013: Guidance Document on Analytical Quality Control and Validation Procedures for Pesticides Residues Analysis in Food and Feed. 2013. Available online: https://www.eurl-pesticides.eu/docs/public/tmplt_article.asp?CntID=727 (accessed on 14 June 2021).
- Zachariasova, M.; Cajka, T.; Godula, M.; Malachova, A.; Veprikova, Z.; Hajslova, J. Analysis of Multiple Mycotoxins in Beer Employing (Ultra)-High-Resolution Mass Spectrometry. Rapid Commun. Mass Spectrom. 2010, 24, 3357–3367. [Google Scholar] [CrossRef]
- Rahmani, A.; Jinap, S.; Soleimany, F. Qualitative and Quantitative Analysis of Mycotoxins. Compr. Rev. Food Sci. Food Saf. 2009, 8, 202–251. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Van der Fels-Klerx, H.J. Quantitative Modeling of Climate Change Impacts on Mycotoxins in Cereals: A Review. Toxins 2021, 13, 276. [Google Scholar] [CrossRef]
- Moretti, A.; Pascale, M.; Logrieco, A.F. Mycotoxin Risks under a Climate Change Scenario in Europe. Trends Food Sci. Technol. 2019, 84, 38–40. [Google Scholar] [CrossRef]
- Jensen, T.; de Boevre, M.; Preußke, N.; de Saeger, S.; Birr, T.; Verreet, J.A.; Sönnichsen, F.D. Evaluation of High-Resolution Mass Spectrometry for the Quantitative Analysis of Mycotoxins in Complex Feed Matrices. Toxins 2019, 11, 531. [Google Scholar] [CrossRef] [Green Version]
- Winter, M.; Samuels, P.L.; Dong, Y.; Dill-Macky, R. Trichothecene Production Is Detrimental to Early Root Colonization by Fusarium Culmorum and F. Graminearum in Fusarium Crown and Root Rot of Wheat. Plant Pathol. 2019, 68, 185–195. [Google Scholar] [CrossRef] [Green Version]
- Asran, M.R.; Buchenauer, H. Pathogenicity of Fusarium Graminearum Isolates on Maize (Zea mays L.) Cultivars and Relation with Deoxynivalenol and Ergosterol Contents/Pathogenität von Fusarium Graminearum Isolaten an Mais- (Zea mays L.) Sorten Und Beziehung Zu Deoxynivalenol- Und Ergosterol-Gehalten. Z. Pflanzenkrankh. Pflanzenschutz J. Plant Dis. Prot. 2003, 110, 209–219. [Google Scholar]
- Abid, M.; Fayolle, L.; Edel-Hermann, V.; Gautheron, N.; Héraud, C.; Leplat, J.; Steinberg, C. Fate of Deoxynivalenol (DON) and Impact on the Soil Microflora and Soil Fauna. Appl. Soil Ecol. 2021, 162, 103898. [Google Scholar] [CrossRef]
- Ahad, R.; Zhou, T.; Lepp, D.; Pauls, K.P. Microbial Detoxification of Eleven Food and Feed Contaminating Trichothecene Mycotoxins. BMC Biotechnol. 2017, 17, 30. [Google Scholar] [CrossRef] [Green Version]
- Islam, R.; Zhou, T.; Christopher Young, J.; Goodwin, P.H.; Peter Pauls, K. Aerobic and Anaerobic De-Epoxydation of Mycotoxin Deoxynivalenol by Bacteria Originating from Agricultural Soil. World J. Microbiol. Biotechnol. 2012, 28, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Vanhoutte, I.; De Mets, L.; De Boevre, M.; Uka, V.; Di Mavungu, J.; De Saeger, S.; De Gelder, L.; Audenaert, K. Microbial Detoxification of Deoxynivalenol (DON), Assessed via a Lemna Minor L. Bioassay, through Biotransformation to 3-Epi-DON and 3-Epi-DOM-1. Toxins 2017, 9, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, J.W.; Hassan, Y.I.; Perilla, N.; Li, X.Z.; Boland, G.J.; Zhou, T. Bacterial Epimerization as a Route for Deoxynivalenol Detoxification: The Influence of Growth and Environmental Conditions. Front. Microbiol. 2016, 7, 572. [Google Scholar] [CrossRef] [Green Version]
- Zhai, Y.; Zhong, L.; Gao, H.; Lu, Z.; Bie, X.; Zhao, H.; Zhang, C.; Lu, F. Detoxification of Deoxynivalenol by a Mixed Culture of Soil Bacteria With 3-Epi-Deoxynivalenol as the Main Intermediate. Front. Microbiol. 2019, 10, 2172. [Google Scholar] [CrossRef] [PubMed]

| Soil | Sand | Silt | Clay | Soil Texture | Carbon Content | Soil pH |
|---|---|---|---|---|---|---|
| % | % | % | % | |||
| LUFA2.4 | 32.1 | 41.6 | 26.3 | Lt2 | 1.78 | 7.4 |
| LUFA6S | 23.8 | 35.3 | 40.9 | Lt3 | 1.99 | 7.2 |
| RefeSol01A | 74 | 19.8 | 6.2 | Sl2 | 0.89 | 5.3 |
| RefeSol02A | 5.7 | 78.3 | 16 | Ut3 | 1.04 | 6.6 |
| JKI soil | 63.5 | 32.5 | 4 | Su3 | 0.75 | Na |
| Field 1 | 92.5 | 5 | 2.5 | Ss | 1.42 | 6.6 |
| Field 2 | 63.5 | 32.5 | 4 | Su3 | 0.68 | 5.9 |
| Field 3 | 31 | 65 | 4 | Us | 0.89 | 7.1 |
| Field 4 | 80 | 17.5 | 2.5 | Su2 | 0.54 | 5.7 |
| Field 5 | 19 | 57.5 | 23.5 | Lu | 1.41 | 7.4 |
| Field 6 | 19 | 57.5 | 23.5 | Lu | 2.22 | 7.2 |
| Field 7 | 22.5 | 22.5 | 55 | Tl | 2.4 | 7.5 |
| Field 8 | Na | 17.5 | 82.5 | Tt | 1.2 | 6.8 |
| Analyte | Retention Time | Molecular Weight | Adduct | Target Mass |
|---|---|---|---|---|
| (min) | (Da) | (m/z) | ||
| NIV | 2.2 | 312.32 | [M + COOH] | 357.1195 |
| DON | 3.4 | 296.16 | [M + COOH] | 341.1242 |
| 15-AcDON | 6.45 | 338.35 | [M + Na]+ | 361.1257 |
| [M + NH4]+ | 356.1705 | |||
| ZEN | 8.4 | 318.36 | [M − H] | 317.1389 |
| 13C15-NIV | 2.2 | 327.32 | [M + COOH] | 372.1701 |
| 13C15-DON | 3.4 | 311.21 | [M + COOH] | 356.1750 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kenngott, K.G.J.; Albert, J.; Meyer-Wolfarth, F.; Schaumann, G.E.; Muñoz, K. Fusarium Mycotoxins in Maize Field Soils: Method Validation and Implications for Sampling Strategy. Toxins 2022, 14, 130. https://doi.org/10.3390/toxins14020130
Kenngott KGJ, Albert J, Meyer-Wolfarth F, Schaumann GE, Muñoz K. Fusarium Mycotoxins in Maize Field Soils: Method Validation and Implications for Sampling Strategy. Toxins. 2022; 14(2):130. https://doi.org/10.3390/toxins14020130
Chicago/Turabian StyleKenngott, Kilian G. J., Julius Albert, Friederike Meyer-Wolfarth, Gabriele E. Schaumann, and Katherine Muñoz. 2022. "Fusarium Mycotoxins in Maize Field Soils: Method Validation and Implications for Sampling Strategy" Toxins 14, no. 2: 130. https://doi.org/10.3390/toxins14020130
APA StyleKenngott, K. G. J., Albert, J., Meyer-Wolfarth, F., Schaumann, G. E., & Muñoz, K. (2022). Fusarium Mycotoxins in Maize Field Soils: Method Validation and Implications for Sampling Strategy. Toxins, 14(2), 130. https://doi.org/10.3390/toxins14020130