Efflux at the Blood-Brain Barrier Reduces the Cerebral Exposure to Ochratoxin A, Ochratoxin α, Citrinin and Dihydrocitrinone


Abstract
1. Introduction
1.1. The Blood-Brain Barrier
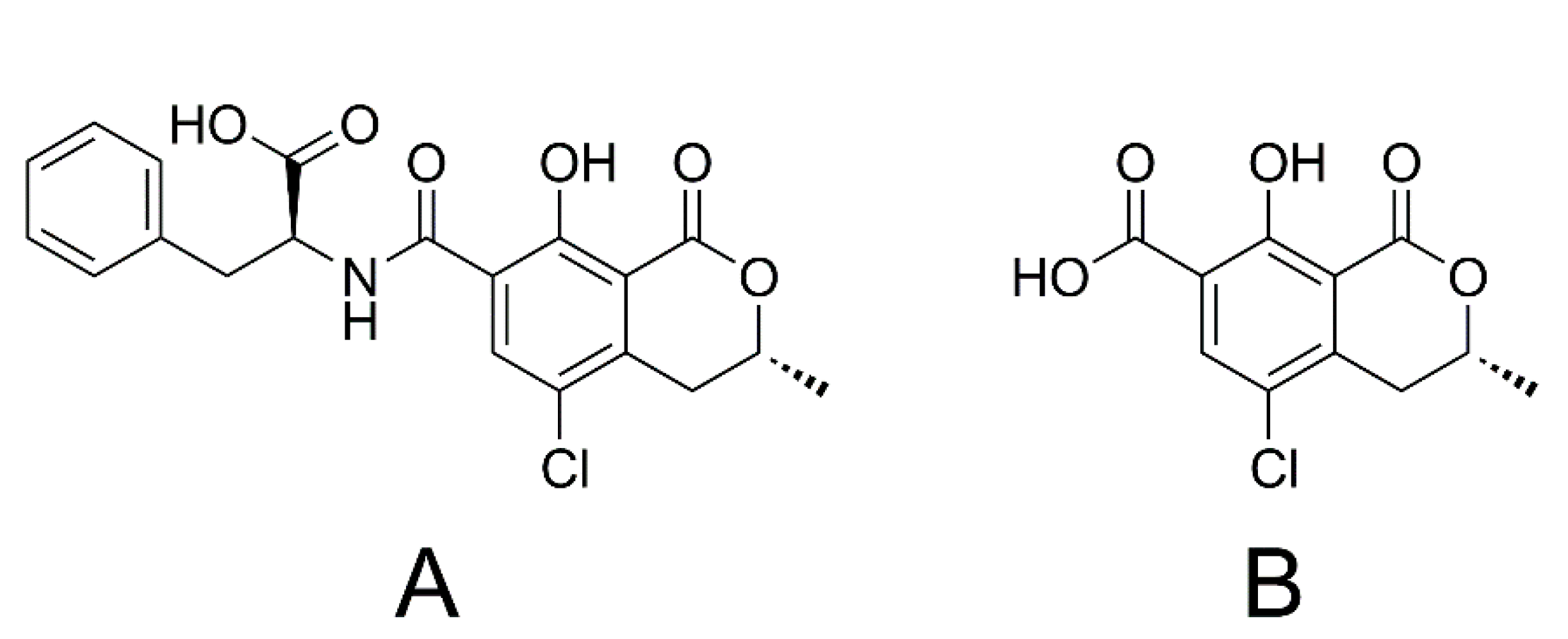
1.2. Ochratoxin A and Ochratoxin α
1.3. Citrinin and Dihydrocitrinone
1.4. Aim of the Study
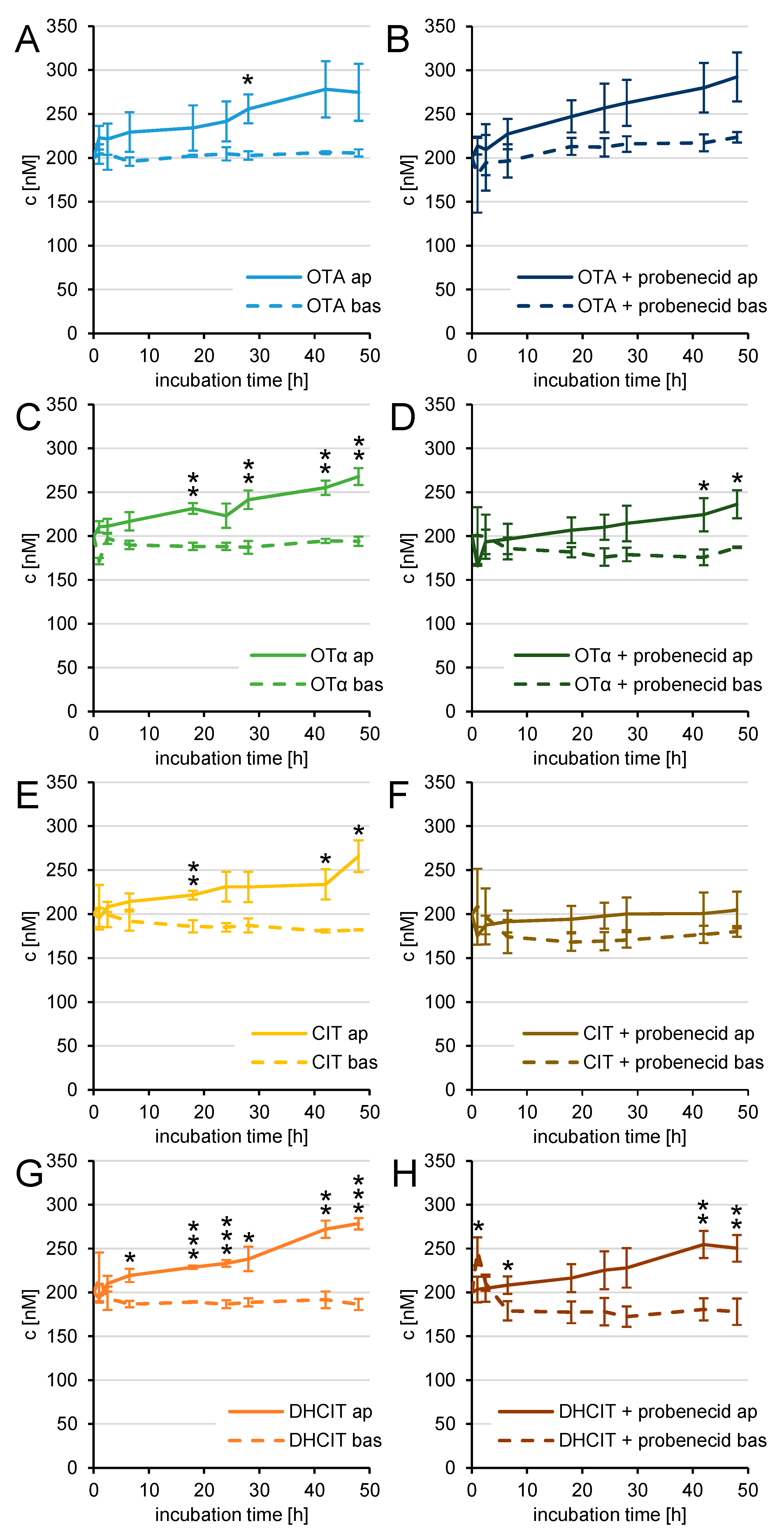
2. Results
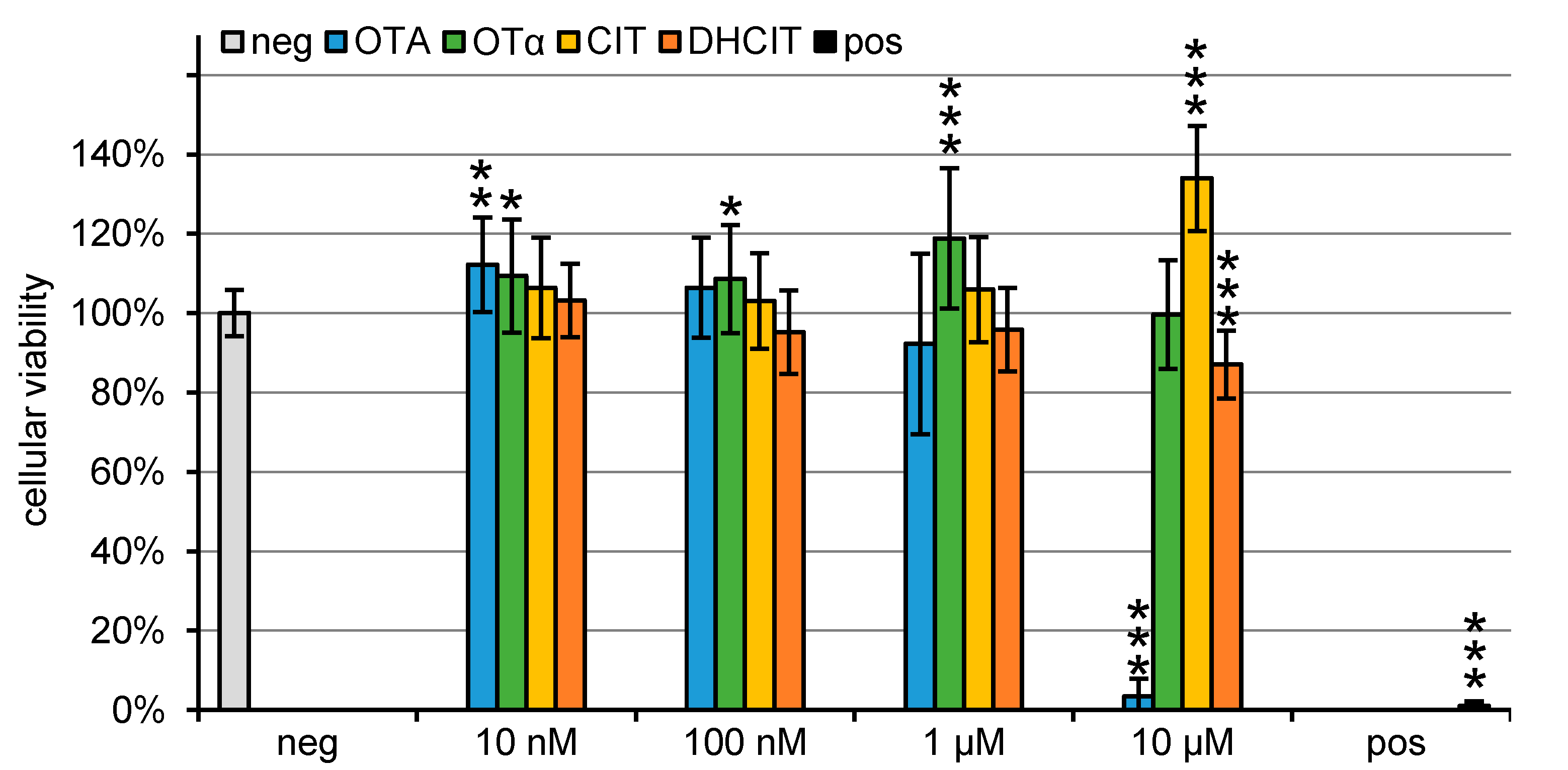
2.1. Viability Test
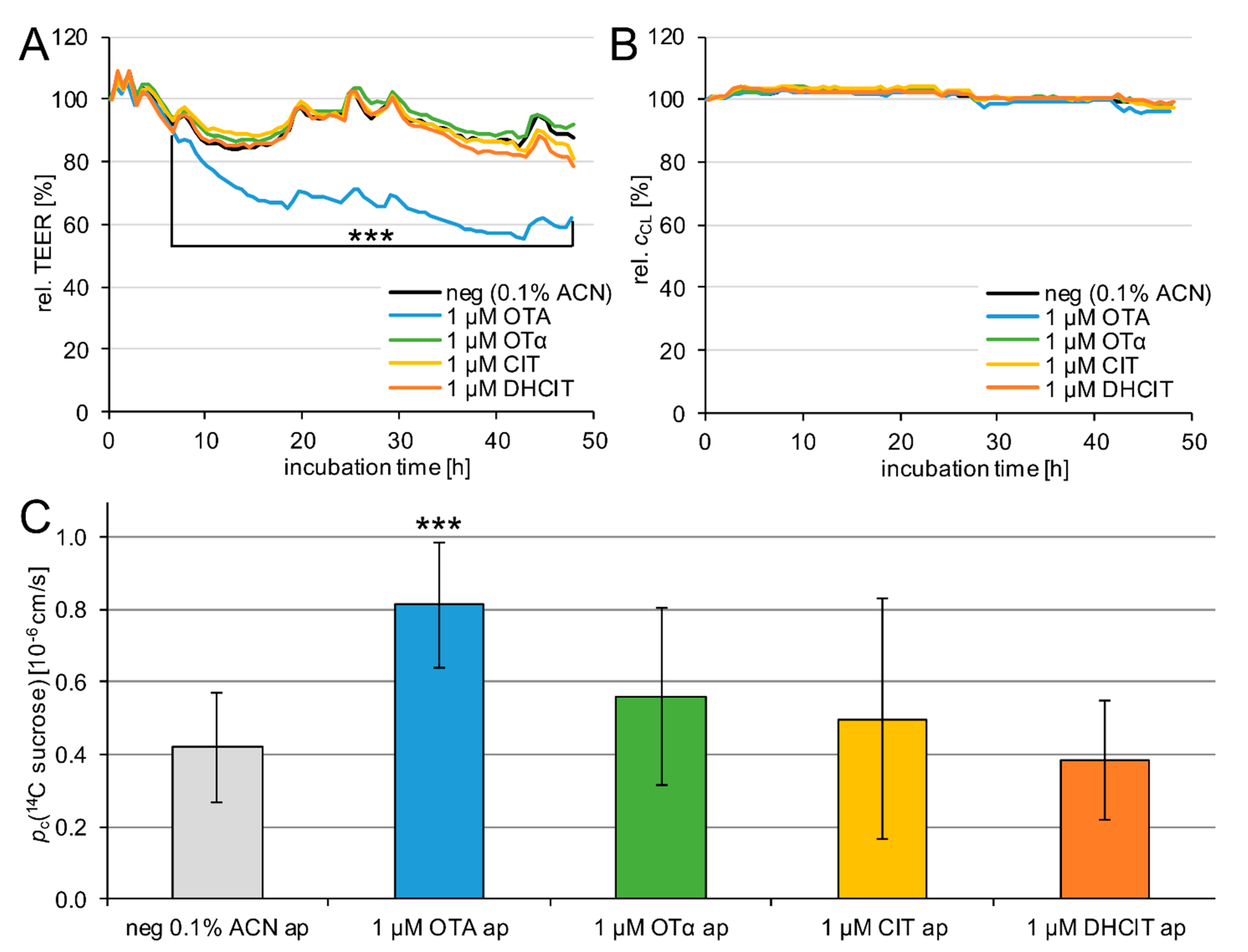
2.2. Barrier Integrity
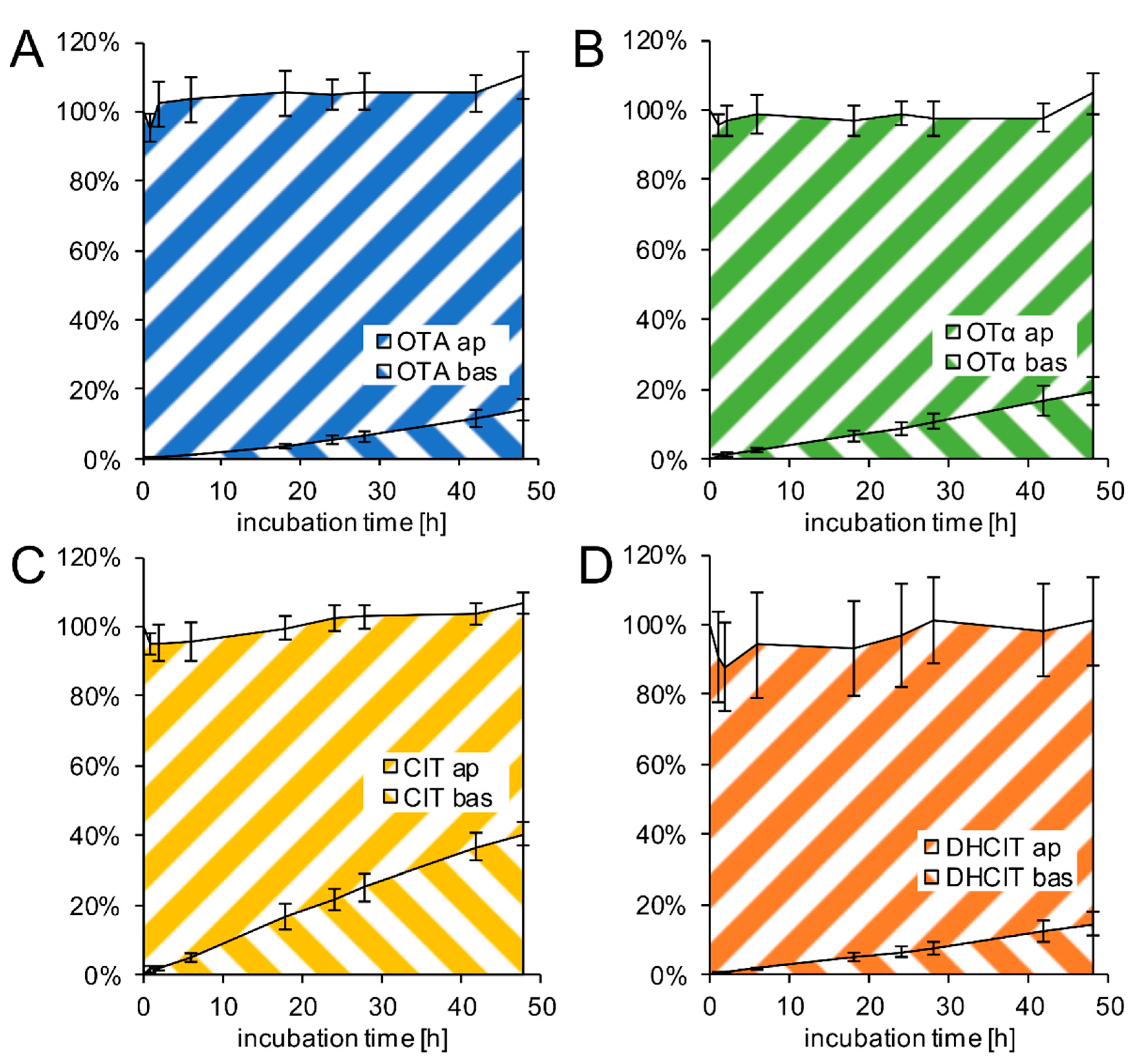
2.3. Transfer Studies
2.4. Active Transfer Studies
3. Discussion
3.1. Cellular Viability and Barrier Integrity
3.2. Transport Properties
4. Conclusions
5. Materials and Methods
5.1. Chemicals and Reagents
5.2. Viability Test and PBCEC Cell Culture
5.3. Transfer Studies
5.4. Active Transport Studies
5.5. Barrier Integrity
5.6. Mycotoxin Quantification
5.7. Permeability Calculations
5.8. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood–brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.A.; Tran, N.D.; Li, Z.; Yang, F.; Zhou, W.; Fisher, M.J. Brain endothelial hemostasis regulation by pericytes. Br. J. Pharmacol. 2005, 26, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, F.L.; Brites, D.; Brito, M.A. Looking at the blood–brain barrier: Molecular anatomy and possible investigation approaches. Brain Res. Rev. 2010, 64, 328–363. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte–endothelial interactions at the blood–brain barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Pfrieger, F.W.; Ungerer, N. Cholesterol metabolism in neurons and astrocytes. Prog. Lipid Res. 2011, 50, 357–371. [Google Scholar] [CrossRef]
- Thanabalasundaram, G.; El-Gindi, J.; Lischper, M.; Galla, H.-J. Methods to assess pericyte-endothelial cell interactions in a coculture model. In the Blood-Brain and Other Neural Barriers; Nag, S., Ed.; Humana Press: Totowa, NJ, USA, 2011; pp. 379–399. [Google Scholar]
- Beuckmann, C.T.; Dernbach, K.; Hakvoort, A.; Galla, H.-J. A new astrocytic cell line which is able to induce a blood-brain barrier property in cultured brain capillary endothelial cells. Cytotechnology 1997, 24, 11–17. [Google Scholar] [CrossRef]
- Franke, H.; Galla, H.-J.; Beuckmann, C.T. An improved low-permeability in vitro-model of the blood–brain barrier: Transport studies on retinoids, sucrose, haloperidol, caffeine and mannitol. Brain Res. 1999, 818, 65–71. [Google Scholar] [CrossRef]
- Franke, H.; Galla, H.-J.; Beuckmann, C.T. Primary cultures of brain microvessel endothelial cells: A valid and flexible model to study drug transport through the blood–brain barrier in vitro. Brain Res. Protoc. 2000, 5, 248–256. [Google Scholar] [CrossRef]
- Hoheisel, D.; Nitz, T.; Franke, H.; Wegener, J.; Hakvoort, A.; Tilling, T.; Galla, H.-J. Hydrocortisone reinforces the blood–brain barrier properties in a serum free cell culture system. Biochem. Biophys. Res. Commun. 1998, 244, 312–316. [Google Scholar] [CrossRef]
- Schrot, S.; Weidenfeller, C.; Schäffer, T.E.; Robenek, H.; Galla, H.-J. Influence of hydrocortisone on the mechanical properties of the cerebral endothelium in vitro. Biophys. J. 2005, 89, 3904–3910. [Google Scholar] [CrossRef] [PubMed]
- Naik, P.; Cucullo, L. In vitro blood–brain barrier models: Current and perspective technologies. J. Pharm. Sci. 2012, 101, 1337–1354. [Google Scholar] [CrossRef]
- Mulac, D.; Hüwel, S.; Galla, H.-J.; Humpf, H.-U. Permeability of ergot alkaloids across the blood-brain barrier in vitro and influence on the barrier integrity. Mol. Nutr. Food Res. 2011, 56, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Weidner, M.; Hüwel, S.; Ebert, F.; Schwerdtle, T.; Galla, H.-J.; Humpf, H.-U. Influence of T-2 and HT-2 toxin on the blood-brain barrier in vitro: New experimental hints for neurotoxic effects. PLoS ONE 2013, 8, e60484. [Google Scholar] [CrossRef] [PubMed]
- Behrens, M.; Hüwel, S.; Galla, H.-J.; Humpf, H.-U. Blood-brain barrier effects of the fusarium mycotoxins deoxynivalenol, 3 acetyldeoxynivalenol, and moniliformin and their transfer to the brain. PLoS ONE 2015, 10, e0143640. [Google Scholar] [CrossRef]
- Ostry, V.; Malir, F.; Ruprich, J. Producers and important dietary sources of ochratoxin A and citrinin. Toxins 2013, 5, 1574–1586. [Google Scholar] [CrossRef] [PubMed]
- Abrunhosa, L.; Paterson, R.R.M.; Venâncio, A. Biodegradation of ochratoxin a for food and feed decontamination. Toxins 2010, 2, 1078–1099. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Dohnal, V.; Huang, L.; Kuca, K.; Wang, X.; Chen, G.; Yuan, Z. Metabolic pathways of Ochratoxin A. Curr. Drug Metab. 2011, 12, 1–10. [Google Scholar] [CrossRef]
- Muñoz, K.; Blaszkewicz, M.; Degen, G.H. Simultaneous analysis of ochratoxin A and its major metabolite ochratoxin alpha in plasma and urine for an advanced biomonitoring of the mycotoxin. J. Chromatogr. B 2010, 878, 2623–2629. [Google Scholar] [CrossRef] [PubMed]
- Cramer, B.; Osteresch, B.; Muñoz, K.A.; Hillmann, H.; Sibrowski, W.; Humpf, H. Biomonitoring using dried blood spots: Detection of ochratoxin A and its degradation product 2′R-ochratoxin A in blood from coffee drinkers. Mol. Nutr. Food Res. 2015, 59, 1837–1843. [Google Scholar] [CrossRef]
- Krogh, P. Role of ochratoxin in disease causation. Food Chem. Toxicol. 1992, 30, 213–224. [Google Scholar] [CrossRef]
- Long, D.T.; Voice, T.C. Role of exposure analysis in solving the mystery of balkan endemic nephropathy. Croat. Med. J. 2007, 48, 300–311. [Google Scholar]
- Belmadani, A.; Tramu, G.; Betbeder, A.M.; Creppy, E.E. Subchronic effects of ochratoxin A on young adult rat brain and partial prevention by aspartame, a sweetener. Hum. Exp. Toxicol. 1998, 17, 380–386. [Google Scholar] [CrossRef]
- Belmadani, A.; Steyn, P.S.; Tramu, G.; Betbeder, A.-M.; Baudrimont, I.; Creppy, E.E. Selective toxicity of ochratoxin A in primary cultures from different brain regions. Arch. Toxicol. 1999, 73, 108–114. [Google Scholar] [CrossRef]
- Sava, V.; Reunova, O.; Velasquez, A.; Harbison, R.; Sánchez-Ramos, J. Acute neurotoxic effects of the fungal metabolite ochratoxin A. NeuroToxicology 2006, 27, 82–92. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Boesch-Saadatmandi, C.; Lou, Y.; Wolffram, S.; Huebbe, P.; Rimbach, G. Ochratoxin A induces apoptosis in neuronal cells. Genes Nutr. 2009, 4, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Sava, V.; Reunova, O.; Velasquez, A.; Sanchez-Ramos, J. Can low level exposure to ochratoxin-A cause parkinsonism. J. Neurol. Sci. 2006, 249, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Lim, W.; You, S.; Song, G. Ochratoxin A exerts neurotoxicity in human astrocytes through mitochondria-dependent apoptosis and intracellular calcium overload. Toxicol. Lett. 2019, 313, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Razafimanjato, H.; Garmy, N.; Guo, X.-J.; Varini, K.; Di Scala, C.; Di Pasquale, E.; Taïeb, N.; Maresca, M. The food-associated fungal neurotoxin ochratoxin A inhibits the absorption of glutamate by astrocytes through a decrease in cell surface expression of the excitatory amino-acid transporters GLAST and GLT-1. NeuroToxicology 2010, 31, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Scudamore, K.A.; Clarke, J.H.; Hetmanski, M.T. Isolation of Penicillium strains producing ochratoxin A, citrinin, xanthomegnin, viomellein and vioxanthin from stored cereal grains. Lett. Appl. Microbiol. 1993, 17, 82–87. [Google Scholar] [CrossRef]
- Patterson, M.F.; Damoglou, A.P. Conversion of the mycotoxin citrinin into dihydrocitrinone and ochratoxin A by Penicillium viridicatum. Appl. Microbiol. Biotechnol. 1987, 26, 574–578. [Google Scholar] [CrossRef]
- Dunn, B.B.; Stack, M.E.; Park, D.L.; Joshi, A.; Friedman, L.; King, R.L. Isolation and identification of dihydrocitrinone, a urinary metabolite of citrinin in rats. J. Toxicol. Environ. Health. Part A 1983, 12, 283–289. [Google Scholar] [CrossRef]
- Blaszkewicz, M.; Muñoz, K.; Degen, G.H. Methods for analysis of citrinin in human blood and urine. Arch. Toxicol. 2013, 87, 1087–1094. [Google Scholar] [CrossRef]
- Gerding, J.; Cramer, B.; Humpf, H.-U. Determination of mycotoxin exposure in Germany using an LC-MS/MS multibiomarker approach. Mol. Nutr. Food Res. 2014, 58, 2358–2368. [Google Scholar] [CrossRef]
- Ali, N.; Blaszkewicz, M.; Degen, G.H. Occurrence of the mycotoxin citrinin and its metabolite dihydrocitrinone in urines of German adults. Arch. Toxicol. 2014, 89, 573–578. [Google Scholar] [CrossRef]
- Gerding, J.; Ali, N.; Schwartzbord, J.; Cramer, B.; Brown, D.L.; Degen, G.H.; Humpf, H.-U. A comparative study of the human urinary mycotoxin excretion patterns in Bangladesh, Germany, and Haiti using a rapid and sensitive LC-MS/MS approach. Mycotoxin Res. 2015, 31, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Ali, N.; Blaszkewicz, M.; Mohanto, N.C.; Rahman, M.; Alim, A.; Hossain, K.; Degen, G.H. First results on citrinin biomarkers in urines from rural and urban cohorts in Bangladesh. Mycotoxin Res. 2014, 31, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Flajs, D.; Peraica, M. Toxicological properties of citrinin. Arch. Ind. Hyg. Toxicol. 2009, 60, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.; Sasmal, D.; Bandyopadhyay, S.; Bagchi, G.; Chatterjee, T.; Dey, S. Hematological changes produced in mice by ochratoxin A and citrinin. Toxicology 1983, 26, 55–62. [Google Scholar] [CrossRef]
- Phillips, R.D.; Berndt, W.O.; Hayes, A. Distribution and excretion of [14C]citrinin in rats. Toxicology 1979, 12, 285–298. [Google Scholar] [CrossRef]
- Berndt, W.; Hayes, A. The effect of probenecid on citrinin-induced nephrotoxicity. Toxicol. Appl. Pharmacol. 1982, 64, 118–124. [Google Scholar] [CrossRef]
- Chen, C.-C.; Chan, W.-H. Inhibition of citrinin-induced apoptotic biochemical signaling in human hepatoma G2 cells by resveratrol. Int. J. Mol. Sci. 2009, 10, 3338–3357. [Google Scholar] [CrossRef] [PubMed]
- Föllmann, W.; Behm, C.; Degen, G.H. Toxicity of the mycotoxin citrinin and its metabolite dihydrocitrinone and of mixtures of citrinin and ochratoxin A in vitro. Arch. Toxicol. 2014, 88, 1097–1107. [Google Scholar] [CrossRef] [PubMed]
- Speijers, G.; Speijers, M. Combined toxic effects of mycotoxins. Toxicol. Lett. 2004, 153, 91–98. [Google Scholar] [CrossRef]
- Benson, K.; Cramer, S.; Galla, H.-J. Impedance-based cell monitoring: Barrier properties and beyond. Fluids Barriers CNS 2013, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- Gollapudi, S.; Kim, C.H.; Tran, B.-N.; Sangha, S.; Gupta, S. Probenecid reverses multidrug resistance in multidrug resistance-associated protein-overexpressing HL60/AR and H69/AR cells but not in P-glycoprotein-overexpressing HL60/Tax and P388/ADR cells. Cancer Chemother Pharm. 1997, 40, 150–158. [Google Scholar] [CrossRef]
- Lohmann, C.; Hüwel, S.; Galla, H.-J. Predicting blood-brain barrier permeability of drugs: Evaluation of different in vitro assays. J. Drug Target. 2002, 10, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Sokol, P.P.; Ripich, G.; Holohan, P.D.; Ross, C.R. Mechanism of ochratoxin A transport in kidney. J. Pharmacol. Exp. Ther. 1988, 246, 460–465. [Google Scholar]
- Dai, J.; Park, G.; Perry, J.L.; Il’Ichev, Y.V.; Bow, D.A.J.; Pritchard, J.B.; Faucet, V.; Pfohl-Leszkowicz, A.; Manderville, R.A.; Simon, J.D. Molecular aspects of the transport and toxicity of ochratoxin A. Acc. Chem. Res. 2004, 37, 874–881. [Google Scholar] [CrossRef]
- Anzai, N.; Jutabha, P.; Endou, H. Molecular Mechanism of ochratoxin A transport in the kidney. Toxins 2010, 2, 1381–1398. [Google Scholar] [CrossRef]
- Tsuda, M.; Sekine, T.; Takeda, M.; Cha, S.H.; Kanai, Y.; Kimura, M.; Endou, H. Transport of ochratoxin A by renal multispecific organic anion transporter 1. J. Pharmacol. Exp. Ther. 1999, 289, 1301–1305. [Google Scholar] [PubMed]
- Jung, K.Y.; Takeda, M.; Kim, D.K.; Tojo, A.; Narikawa, S.; Yoo, B.S.; Hosoyamada, M.; Cha, S.H.; Sekine, T.; Endou, H. Characterization of ochratoxin A transport by human organic anion transporters. Life Sci. 2001, 69, 2123–2135. [Google Scholar] [CrossRef]
- Babu, E.; Takeda, M.; Narikawa, S.; Kobayashi, Y.; Enomoto, A.; Tojo, A.; Cha, S.H.; Sekine, T.; Sakthisekaran, D.; Endou, H. Role of human organic anion transporter 4 in the transport of ochratoxin A. Biochim. Biophys. Acta (BBA) Bioenerg. 2002, 1590, 64–75. [Google Scholar] [CrossRef]
- Jutabha, P.; Anzai, N.; Hayashi, K.; Domae, M.; Uchida, K.; Endou, H.; Sakurai, H. A novel human organic anion transporter NPT4 mediates the transport of ochratoxin A. J. Pharmacol. Sci. 2011, 116, 392–396. [Google Scholar] [CrossRef]
- Ose, A.; Kusuhara, H.; Endo, C.; Tohyama, K.; Miyajima, M.; Kitamura, S.; Sugiyama, Y. Functional characterization of mouse organic anion transporting peptide 1a4 in the uptake and efflux of drugs across the blood-brain barrier. Drug Metab. Dispos. 2009, 38, 168–176. [Google Scholar] [CrossRef]
- Schwerdt, G.; Freudinger, R.; Silbernagl, S.; Gekle, M. Apical uptake of radiolabelled ochratoxin A into Madin–Darby canine kidney cells. Toxicology 1998, 131, 193–202. [Google Scholar] [CrossRef]
- Groves, C.E.; Morales, M.; Wright, S.H. Peritubular transport of ochratoxin A in rabbit renal proximal tubules. J. Pharmacol. Exp. Ther. 1998, 284, 943–948. [Google Scholar] [PubMed]
- Schrickx, J.; Lektarau, Y.; Fink-Gremmels, J. Ochratoxin A secretion by ATP-dependent membrane transporters in Caco-2 cells. Arch. Toxicol. 2005, 80, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Potschka, H.; Baltes, S.; Löscher, W. Inhibition of multidrug transporters by verapamil or probenecid does not alter blood-brain barrier penetration of levetiracetam in rats. Epilepsy Res. 2004, 58, 85–91. [Google Scholar] [CrossRef]
- Smeets, P.H.; Van Aubel, R.A.; Wouterse, A.C.; Heuvel, J.J.V.D.; Russel, F.G. Contribution of multidrug resistance protein 2 (mrp2/abcc2) to the renal excretion of p-aminohippurate (pah) and identification of mrp4 (abcc4) as a novel pah transporter. J. Am. Soc. Nephrol. 2004, 15, 2828–2835. [Google Scholar] [CrossRef]
- Hagelberg, S.; Hult, K.; Fuchs, R. Toxicokinetics of ochratoxin A in several species and its plasma-binding properties. J. Appl. Toxicol. 1989, 9, 91–96. [Google Scholar] [CrossRef]
- Sueck, F.; Poór, M.; Faisal, Z.; Gertzen, C.G.W.; Cramer, B.; Lemli, B.; Kunsági-Máté, S.; Gohlke, H.; Humpf, H.-U. Interaction of ochratoxin A and its thermal degradation product 2′R-ochratoxin a with human serum albumin. Toxins 2018, 10, 256. [Google Scholar] [CrossRef] [PubMed]
- Poór, M.; Lemli, B.; Bálint, M.; Hetenyi, C.; Sali, N.; Kőszegi, T.; Kunsagi-Mate, S. Interaction of citrinin with human serum albumin. Toxins 2015, 7, 5155–5166. [Google Scholar] [CrossRef] [PubMed]
- Cramer, B.; Königs, M.; Humpf, H.-U. Identification and in vitro cytotoxicity of ochratoxin A degradation products formed during coffee roasting. J. Agric. Food Chem. 2008, 56, 5673–5681. [Google Scholar] [CrossRef] [PubMed]
- Beyer, M.; Ferse, I.; Humpf, H.-U. Large-scale production of selected type A trichothecenes: The use of HT-2 toxin and T-2 triol as precursors for the synthesis of d3-T-2 and d3-HT-2 toxin. Mycotoxin Res. 2009, 25, 41–52. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | pc [10−6 cm/s] | t [h] |
|---|---|---|
| 14C sucrose | 0.43 ± 0.20 | 0.17–1.33 |
| 1 µM OTA | 0.38 ± 0.08 | 18 |
| 1 µM OTα | 0.68 ± 0.15 | 18 |
| 1 µM CIT | 1.79 ± 0.40 | 18 |
| 1 µM DHCIT | 0.51 ± 0.14 | 18 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Behrens, M.; Hüwel, S.; Galla, H.-J.; Humpf, H.-U. Efflux at the Blood-Brain Barrier Reduces the Cerebral Exposure to Ochratoxin A, Ochratoxin α, Citrinin and Dihydrocitrinone. Toxins 2021, 13, 327. https://doi.org/10.3390/toxins13050327
Behrens M, Hüwel S, Galla H-J, Humpf H-U. Efflux at the Blood-Brain Barrier Reduces the Cerebral Exposure to Ochratoxin A, Ochratoxin α, Citrinin and Dihydrocitrinone. Toxins. 2021; 13(5):327. https://doi.org/10.3390/toxins13050327
Chicago/Turabian StyleBehrens, Matthias, Sabine Hüwel, Hans-Joachim Galla, and Hans-Ulrich Humpf. 2021. "Efflux at the Blood-Brain Barrier Reduces the Cerebral Exposure to Ochratoxin A, Ochratoxin α, Citrinin and Dihydrocitrinone" Toxins 13, no. 5: 327. https://doi.org/10.3390/toxins13050327
APA StyleBehrens, M., Hüwel, S., Galla, H.-J., & Humpf, H.-U. (2021). Efflux at the Blood-Brain Barrier Reduces the Cerebral Exposure to Ochratoxin A, Ochratoxin α, Citrinin and Dihydrocitrinone. Toxins, 13(5), 327. https://doi.org/10.3390/toxins13050327