BMAA, Methylmercury, and Mechanisms of Neurodegeneration in Dolphins: A Natural Model of Toxin Exposure

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Stranded Dolphins
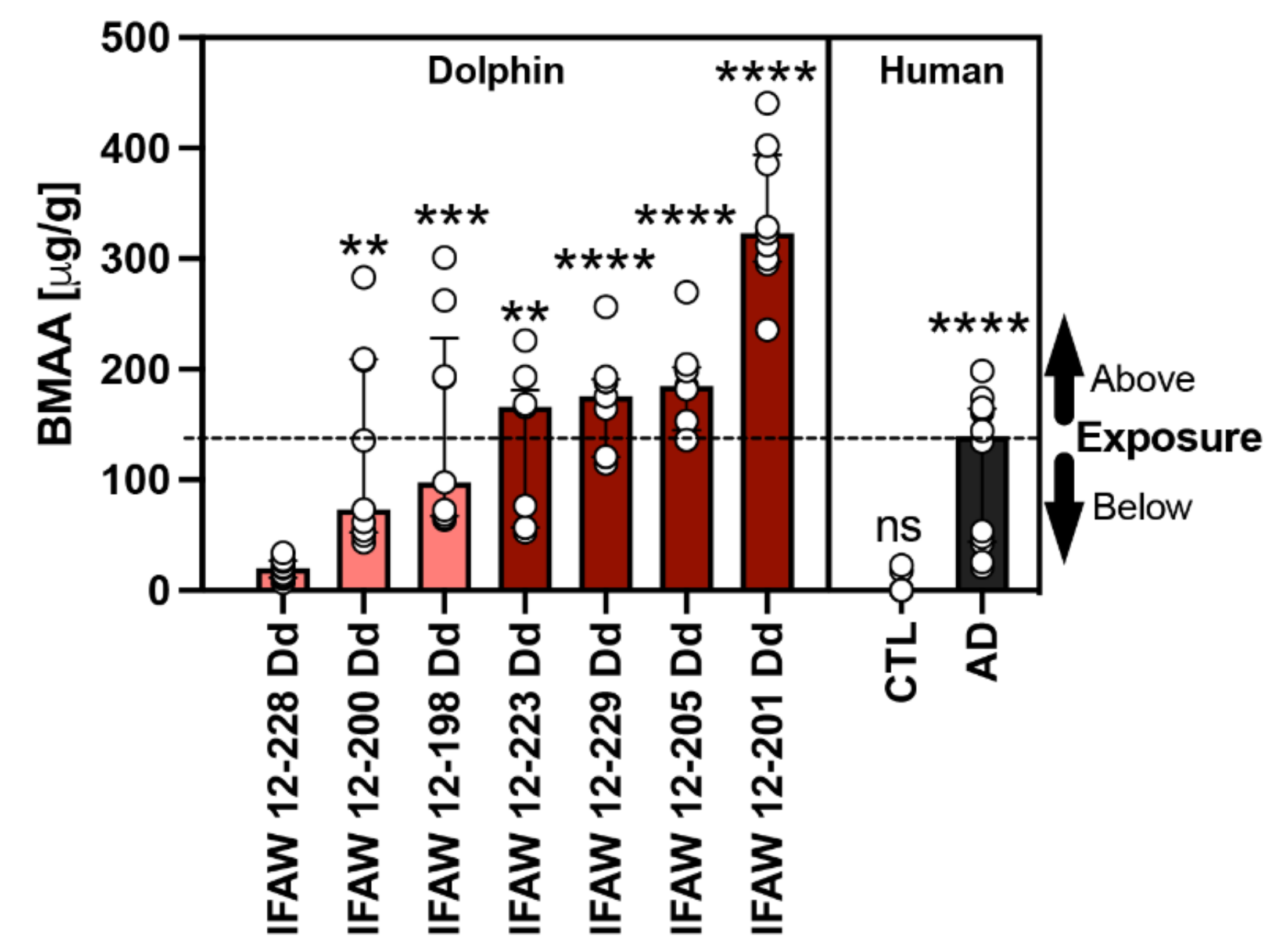
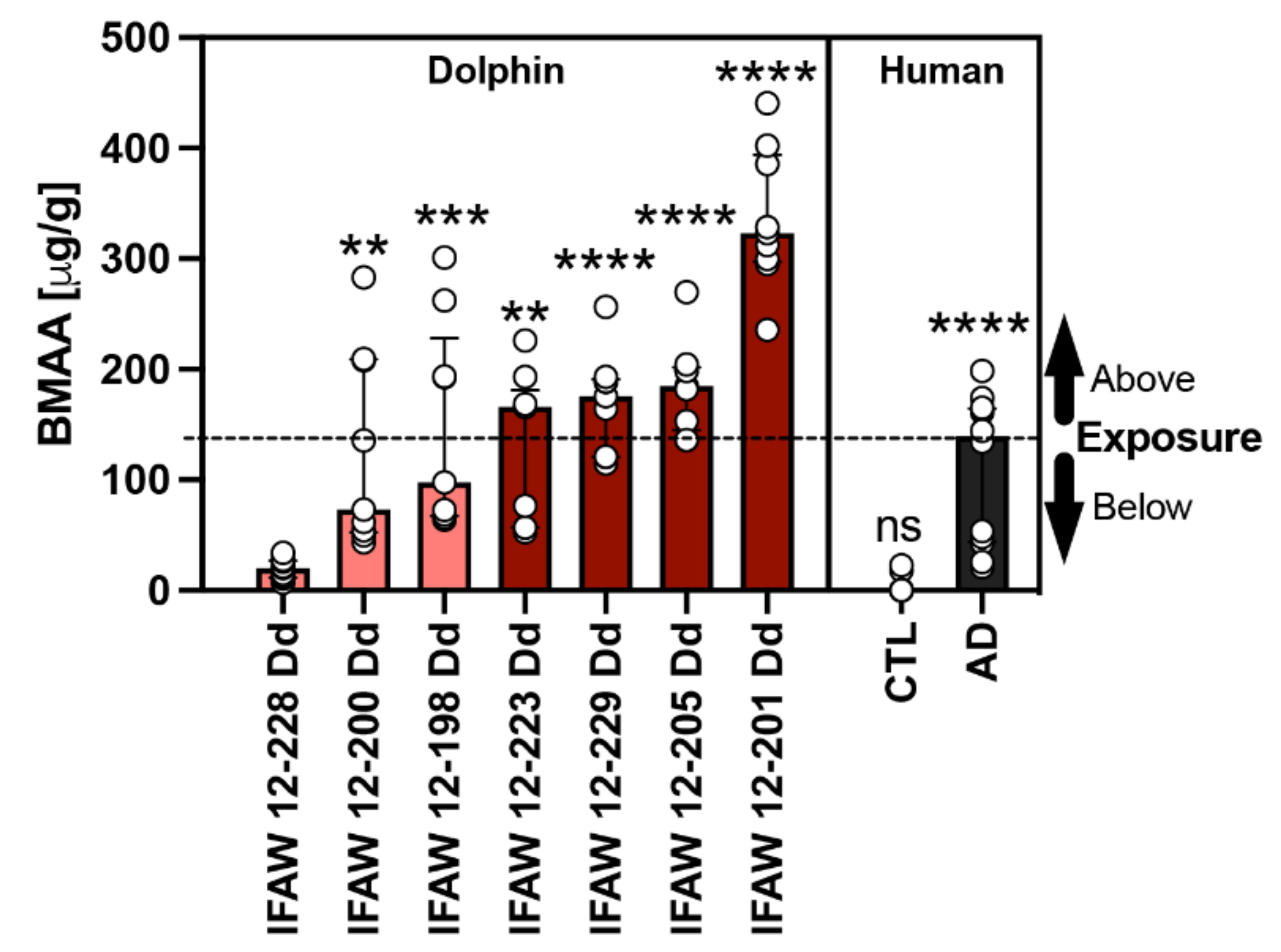
2.2. BMAA Exposure
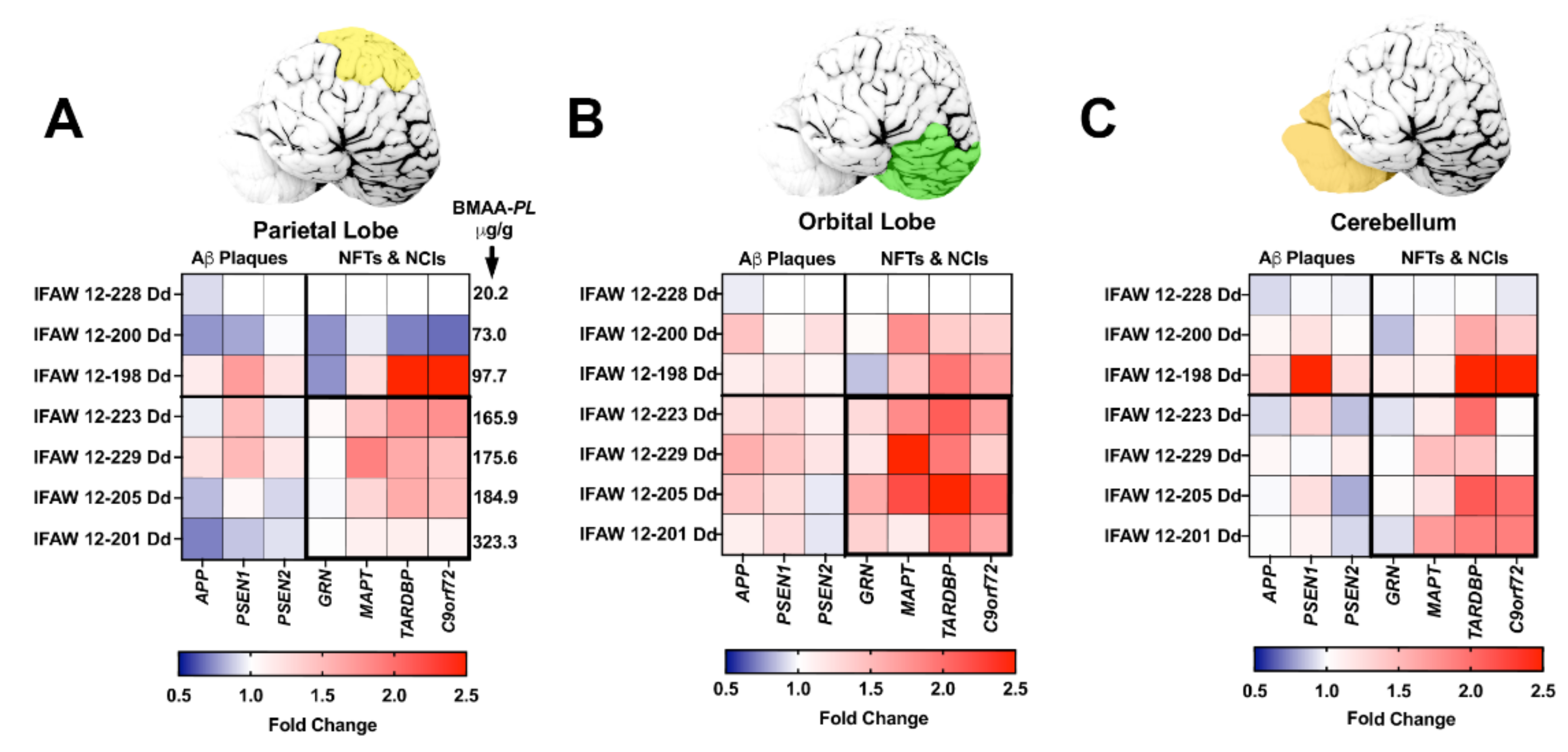
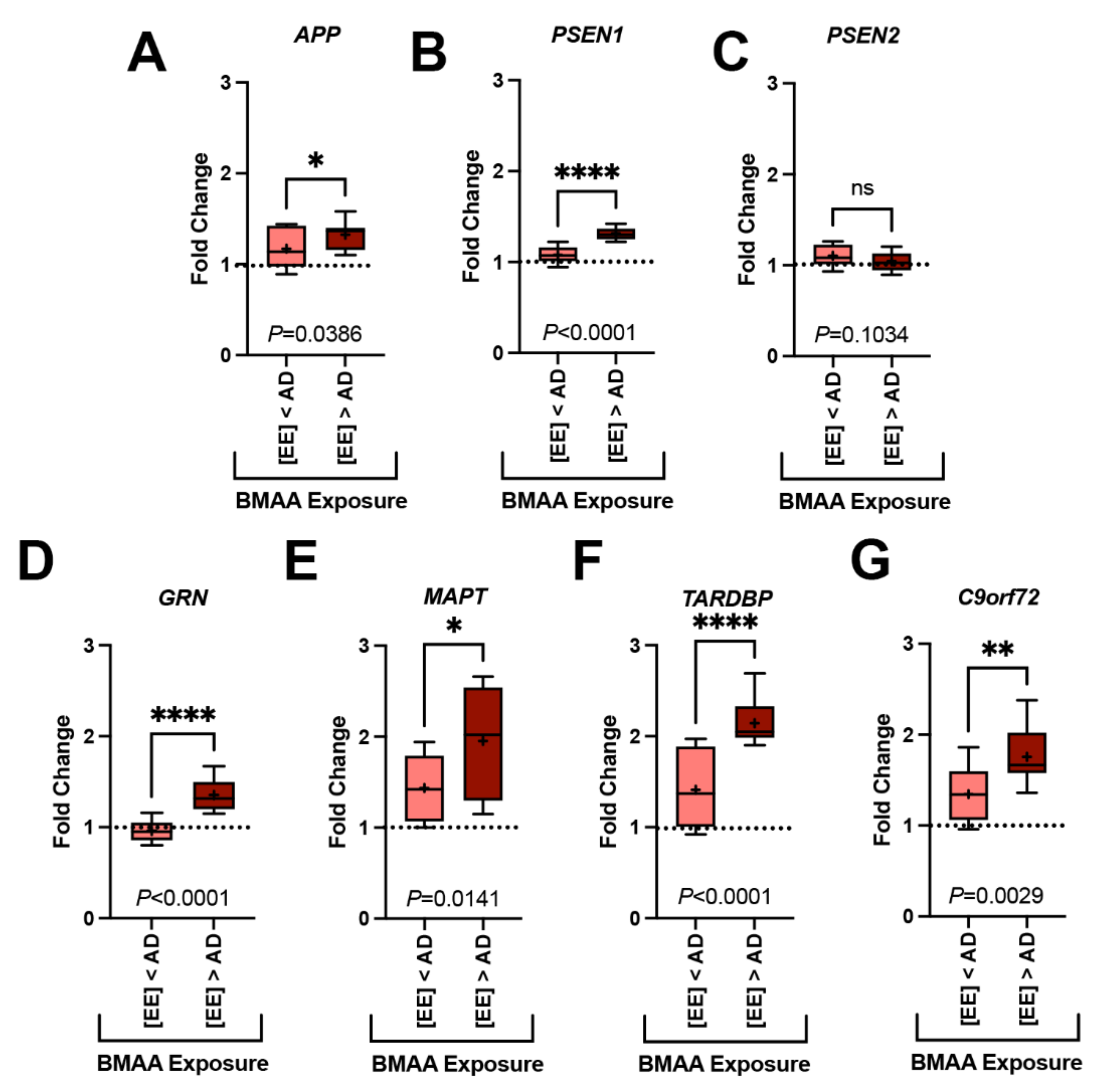
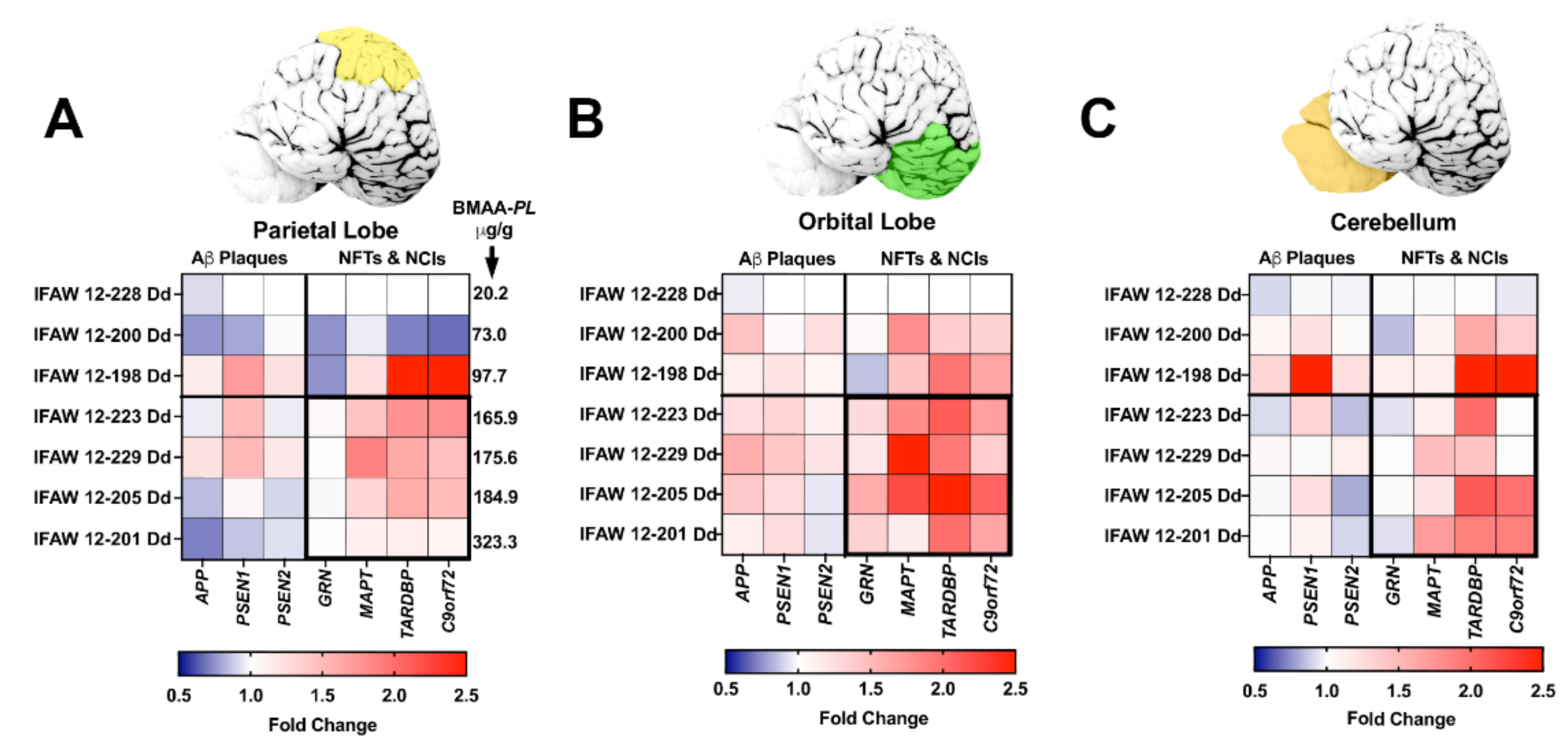
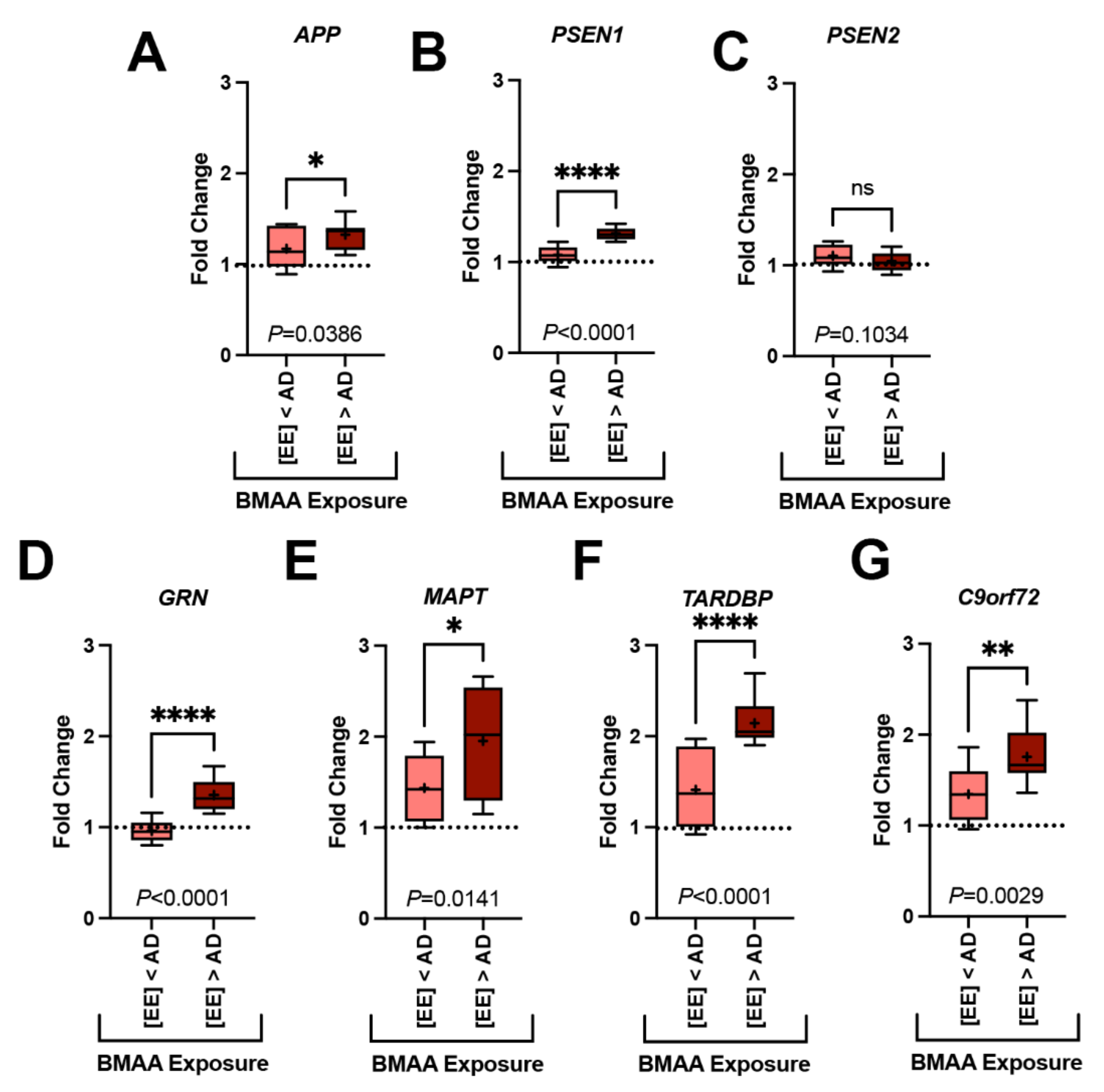
2.3. Gene Expression Markers
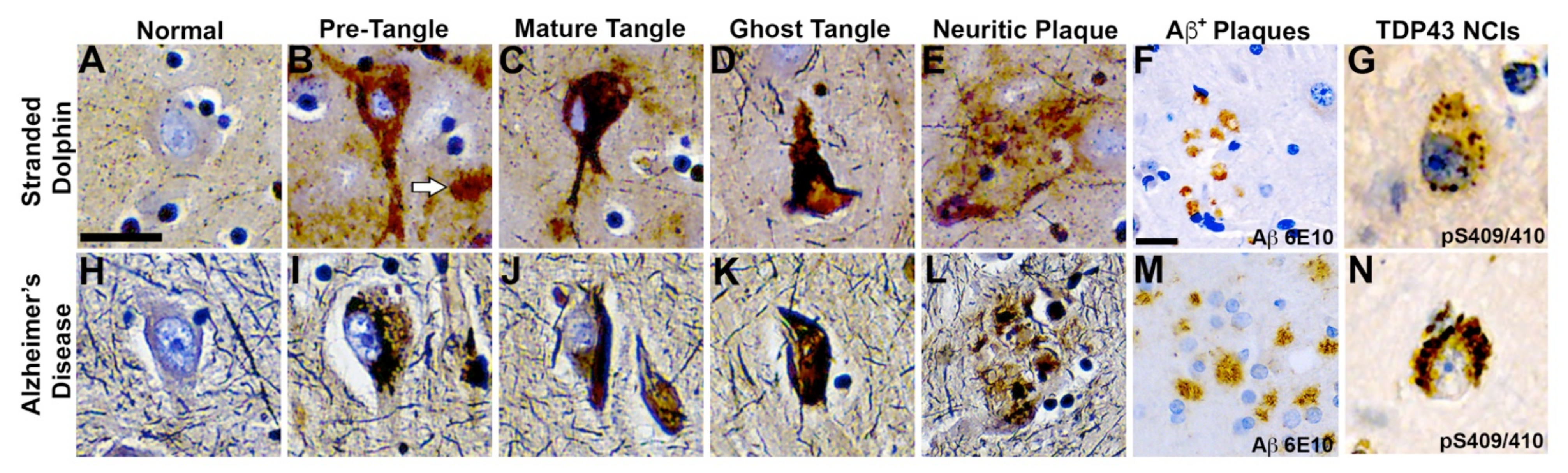
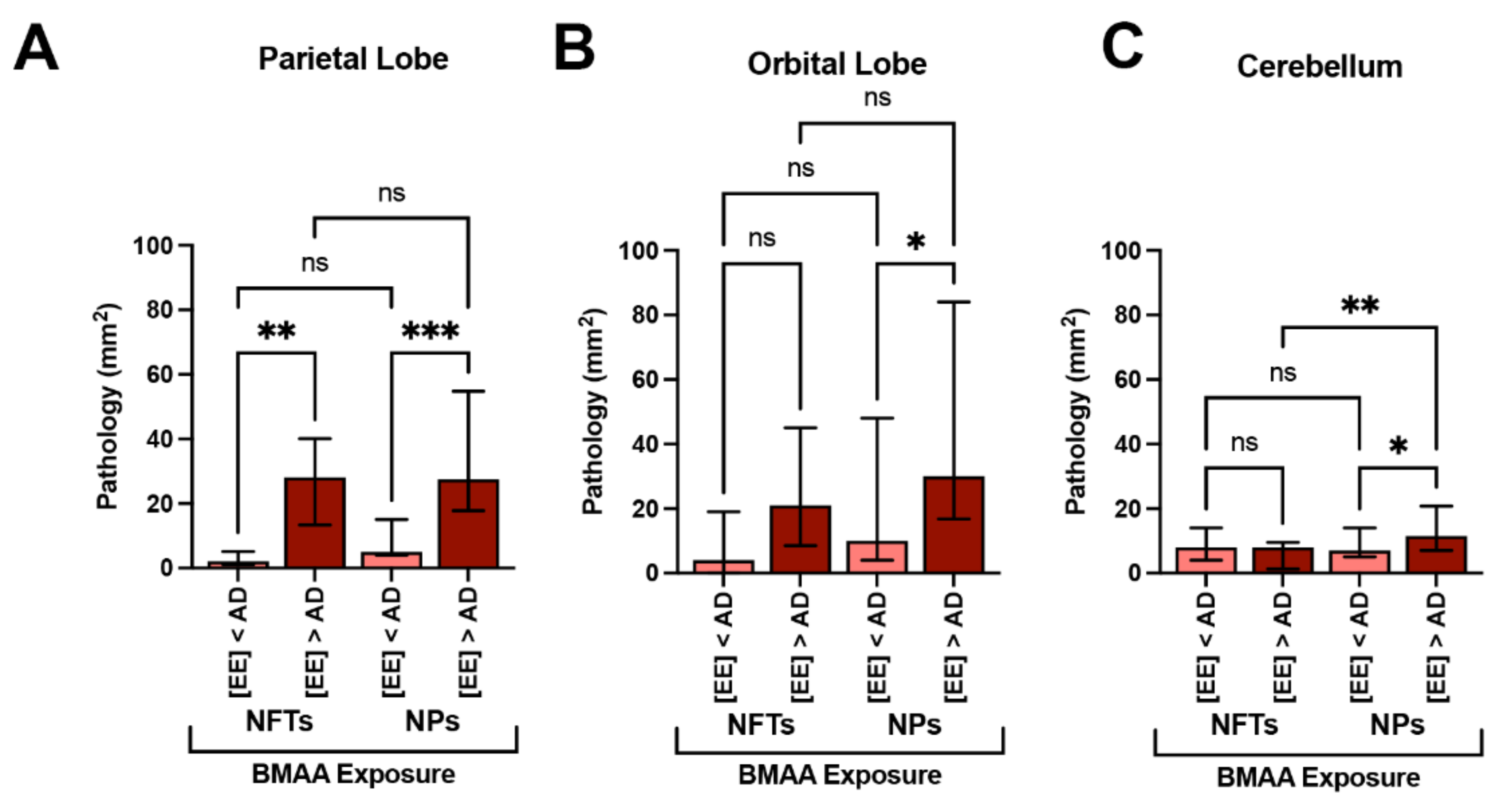
2.4. Neurofibrillary Tangles
2.5. Neuritic Plaques
2.6. TDP-43 Neuronal Intracytoplasmic Inclusions
2.7. Methylmercury Exposure
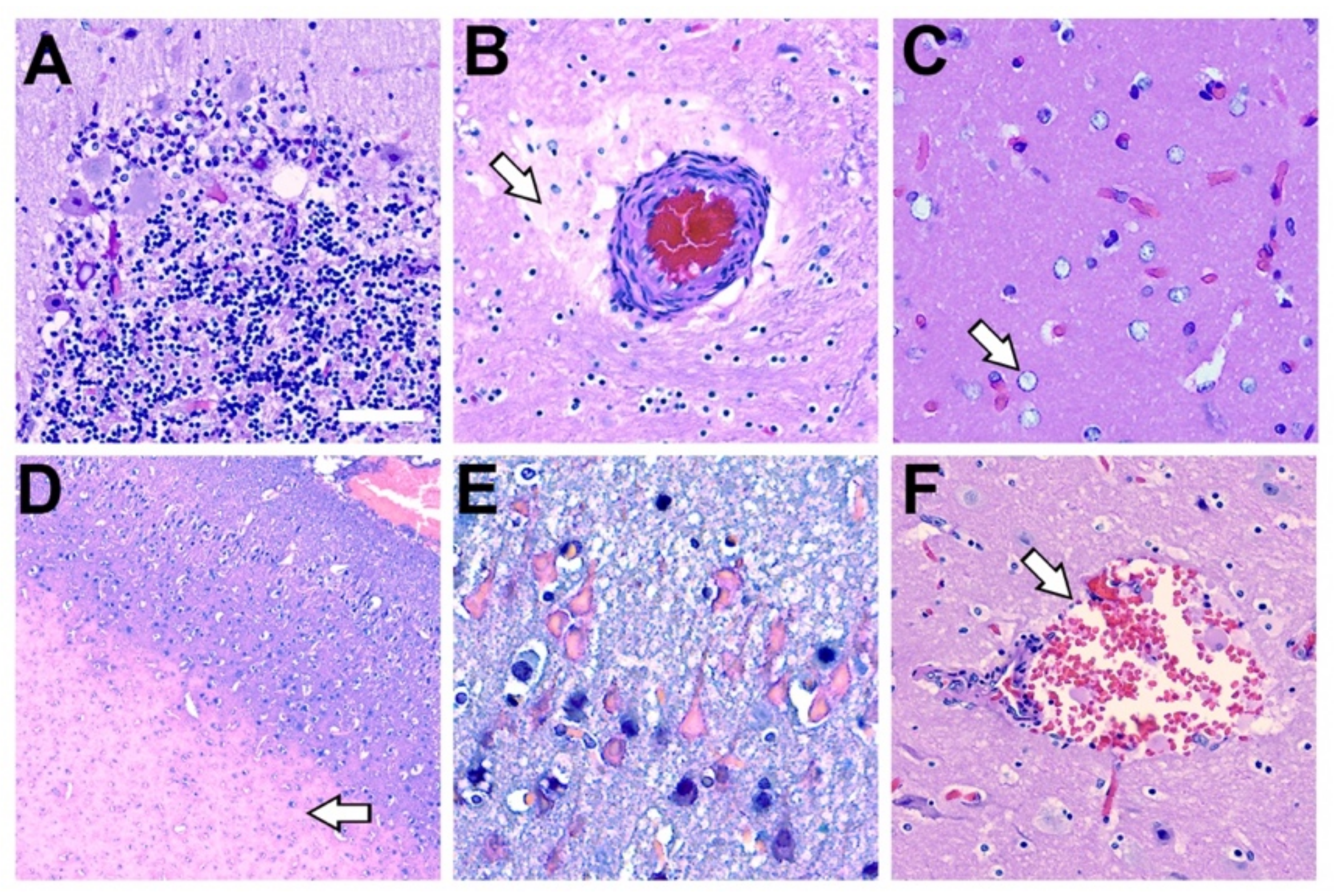
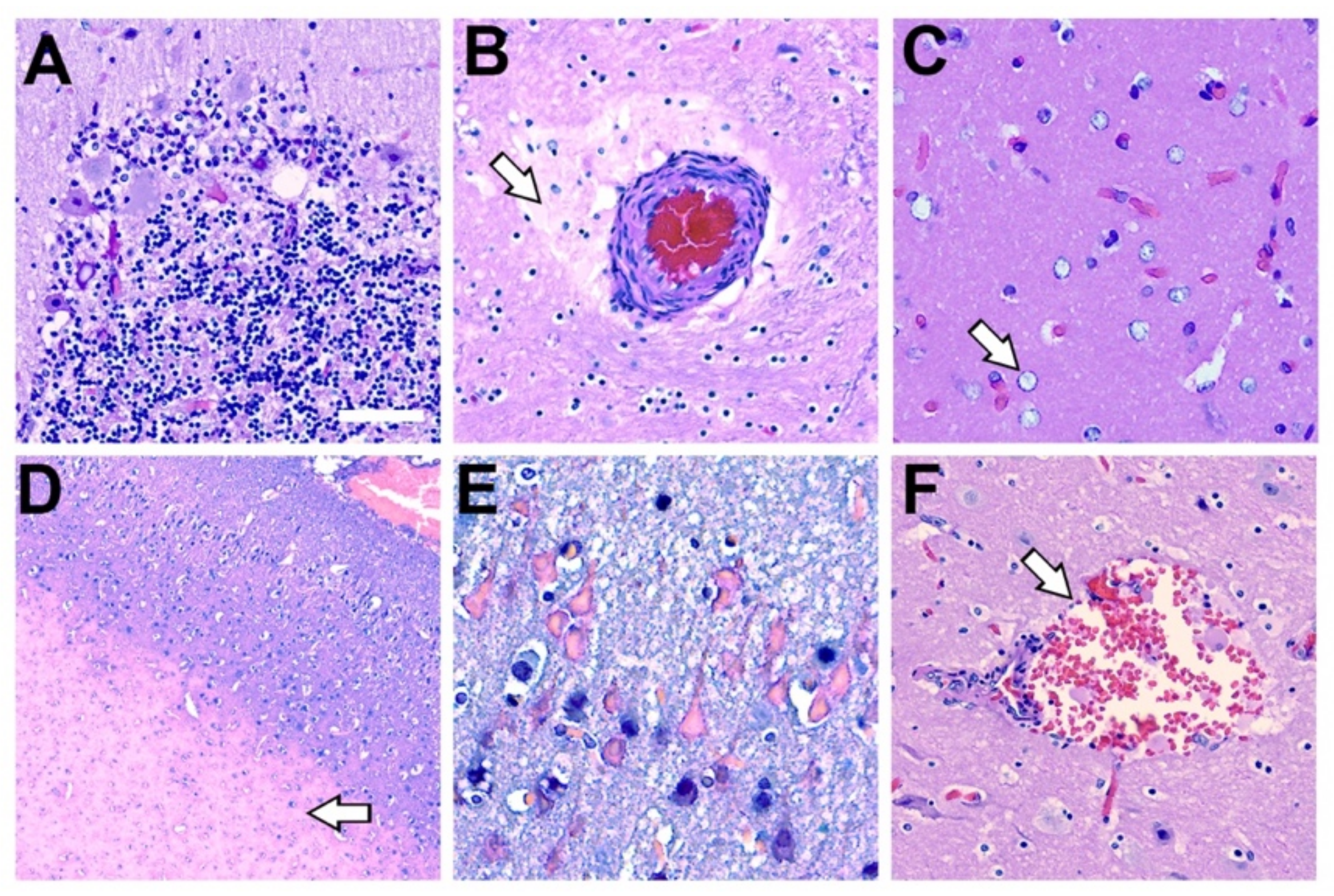
2.8. Additional Histopathological Findings
3. Discussion
4. Conclusions
5. Methods and Materials
5.1. Dolphins
5.2. Extraction of Dolphin RNAs
5.3. qPCR Analysis
5.4. HPLC-FD for BMAA Detection
5.5. PT-GC-AFS for MeHg Detection
5.6. Immunohistochemistry
5.7. Neuropathological Analysis
5.8. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Venn-Watson, S.K.; Jensen, E.D.; Smith, C.R.; Xitco, M.; Ridgway, S.H. Evaluation of annual survival and mortality rates and longevity of bottlenose dolphins (Tursiops truncatus) at the United States Navy Marine Mammal Program from 2004 through 2013. J. Am. Vet. Med Assoc. 2015, 246, 893–898. [Google Scholar] [CrossRef] [Green Version]
- Bogomolni, A.L.; Pugliares, K.R.; Sharp, S.M.; Patchett, K.; Harry, C.T.; LaRocque, J.M.; Touhey, K.M.; Moore, M. Mortality trends of stranded marine mammals on Cape Cod and southeastern Massachusetts, USA, 2000 to 2006. Dis. Aquat. Org. 2010, 88, 143–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jepson, P.D.; Deaville, R.; Acevedo-Whitehouse, K.; Barnett, J.; Brownlow, A.; Brownell, R.L., Jr.; Clare, F.C.; Davison, N.; Law, R.J.; Loveridge, J.; et al. What caused the UK’s largest common dolphin (Delphinus delphis) mass stranding event? PLoS ONE 2013, 8, e60953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fire, S.E.; Bogomolni, A.; DiGiovanni, R.A., Jr.; Early, G.; Leighfield, T.A.; Matassa, K.; Miller, G.A.; Moore, K.M.T.; Moore, M.; Niemeyer, M.; et al. An assessment of temporal, spatial and taxonomic trends in harmful algal toxin exposure in stranded marine mammals from the U.S. New England coast. PLoS ONE 2021, 16, e0243570. [Google Scholar] [CrossRef] [PubMed]
- Danil, K.; Berman, M.; Frame, E.; Preti, A.; Fire, S.E.; Leighfield, T.; Carretta, J.; Carter, M.L.; Lefebvre, K. Marine algal toxins and their vectors in southern California cetaceans. Harmful Algae 2021, 103, 102000. [Google Scholar] [CrossRef] [PubMed]
- Murch, S.J.; Cox, P.A.; Banack, S.A.; Steele, J.C.; Sacks, O.W. Occurrence of beta-methylamino-l-alanine (BMAA) in ALS/PDC patients from Guam. Acta Neurol. Scand. 2004, 110, 267–269. [Google Scholar] [CrossRef] [PubMed]
- Banack, S.A.; Johnson, H.E.; Cheng, R.; Cox, P.A. Production of the neurotoxin BMAA by a marine cyanobacterium. Mar. Drugs 2007, 5, 180–196. [Google Scholar] [CrossRef]
- Pablo, J.; Banack, S.A.; Cox, P.A.; Johnson, T.E.; Papapetropoulos, S.; Bradley, W.G.; Buck, A.; Mash, D.C. Cyanobacterial neurotoxin BMAA in ALS and Alzheimer’s disease. Acta Neurol. Scand. 2009, 120, 216–225. [Google Scholar] [CrossRef]
- Berntzon, L.; Ronnevi, L.O.; Bergman, B.; Eriksson, J. Detection of BMAA in the human central nervous system. Neuroscience 2015, 292, 137–147. [Google Scholar] [CrossRef] [Green Version]
- Brand, L.E.; Pablo, J.; Compton, A.; Hammerschlag, N.; Mash, D.C. Cyanobacterial blooms and the occurrence of the neurotoxin beta-N-methylamino-L-alanine (BMAA) in South Florida aquatic food webs. Harmful Algae 2010, 9, 620–635. [Google Scholar] [CrossRef] [Green Version]
- Murch, S.J.; Cox, P.A.; Banack, S.A. A mechanism for slow release of biomagnified cyanobacterial neurotoxins and neurodegenerative disease in Guam. Proc. Natl. Acad. Sci. USA 2004, 101, 12228–12231. [Google Scholar] [CrossRef] [Green Version]
- Hammerschlag, N.; Davis, D.A.; Mondo, K.; Seely, M.S.; Murch, S.J.; Glover, W.B.; Divoll, T.; Evers, D.C.; Mash, D.C. Cyanobacterial Neurotoxin BMAA and Mercury in Sharks. Toxins 2016, 8, 238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, H.Z.; Yu, S.; Hsu, C.I.; Liu, J.; Acab, A.; Wu, R.; Tao, A.; Chiang, B.J.; Weiss, J.H. Intrathecal infusion of BMAA induces selective motor neuron damage and astrogliosis in the ventral horn of the spinal cord. Exp. Neurol. 2014, 261, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Cox, P.A.; Davis, D.A.; Mash, D.C.; Metcalf, J.S.; Banack, S.A. Dietary exposure to an environmental toxin triggers neurofibrillary tangles and amyloid deposits in the brain. Proc. Biol. Sci. 2016, 283, 20152397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, D.A.; Mondo, K.; Stern, E.; Annor, A.K.; Murch, S.J.; Coyne, T.M.; Brand, L.E.; Niemeyer, M.E.; Sharp, S.; Bradley, W.G.; et al. Cyanobacterial neurotoxin BMAA and brain pathology in stranded dolphins. PLoS ONE 2019, 14, e0213346. [Google Scholar] [CrossRef] [PubMed]
- Gunn-Moore, D.; Kaidanovich-Beilin, O.; Gallego Iradi, M.C.; Gunn-Moore, F.; Lovestone, S. Alzheimer’s disease in humans and other animals: A consequence of postreproductive life span and longevity rather than aging. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2017, 14, 195–204. [Google Scholar] [CrossRef] [Green Version]
- Sarasa, M.; Pesini, P. Natural non-trasgenic animal models for research in Alzheimer’s disease. Curr. Alzheimer Res. 2009, 6, 171–178. [Google Scholar] [CrossRef] [Green Version]
- Page-Karjian, A.; Lo, C.F.; Ritchie, B.; Harms, C.; Rotstein, D.S.; Han, S.; Hassan, S.M.; Lehner, A.F.; Buchweitz, J.P.; Thayer, V.G.; et al. Anthropogenic Contaminants and Histopathological Findings in Stranded Cetaceans in the Southeastern United States, 2012–2018. Front. Mar. Sci. 2020, 7, 630. [Google Scholar] [CrossRef]
- Reif, J.S.; Schaefer, A.M.; Bossart, G.D. Atlantic Bottlenose Dolphins (Tursiops truncatus) as A Sentinel for Exposure to Mercury in Humans: Closing the Loop. Vet. Sci. 2015, 2, 407–422. [Google Scholar] [CrossRef] [Green Version]
- Metcalf, J.S.; Codd, G.A. Co-Occurrence of Cyanobacteria and Cyanotoxins with Other Environmental Health Hazards: Impacts and Implications. Toxins 2020, 12, 629. [Google Scholar] [CrossRef]
- Rush, T.; Liu, X.; Lobner, D. Synergistic toxicity of the environmental neurotoxins methylmercury and beta-N-methylamino-L-alanine. Neuroreport 2012, 23, 216–219. [Google Scholar] [CrossRef]
- Bossart, G.D. Marine mammals as sentinel species for oceans and human health. Vet. Pathol. 2011, 48, 676–690. [Google Scholar] [CrossRef] [Green Version]
- Mirra, S.S.; Heyman, A.; McKeel, D.; Sumi, S.M.; Crain, B.J.; Brownlee, L.M.; Vogel, F.S.; Hughes, J.P.; van Belle, G.; Berg, L. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 1991, 41, 479–486. [Google Scholar] [CrossRef]
- Field, N.C.; Metcalf, J.S.; Caller, T.A.; Banack, S.A.; Cox, P.A.; Stommel, E.W. Linking beta-methylamino-L-alanine exposure to sporadic amyotrophic lateral sclerosis in Annapolis, MD. Toxicon Off. J. Int. Soc. Toxinol. 2013, 70, 179–183. [Google Scholar] [CrossRef]
- Banack, S.A.; Metcalf, J.S.; Bradley, W.G.; Cox, P.A. Detection of cyanobacterial neurotoxin beta-N-methylamino-l-alanine within shellfish in the diet of an ALS patient in Florida. Toxicon Off. J. Int. Soc. Toxinol. 2014, 90, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Masseret, E.; Banack, S.; Boumediene, F.; Abadie, E.; Brient, L.; Pernet, F.; Juntas-Morales, R.; Pageot, N.; Metcalf, J.; Cox, P.; et al. Dietary BMAA exposure in an amyotrophic lateral sclerosis cluster from southern France. PLoS ONE 2013, 8, e83406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banack, S.A.; Murch, S.J.; Cox, P.A. Neurotoxic flying foxes as dietary items for the Chamorro people, Marianas Islands. J. Ethnopharmacol. 2006, 106, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Monson, C.S.; Banack, S.A.; Cox, P.A. Conservation implications of Chamorro consumption of flying foxes as a possible cause of amyotrophic lateral sclerosis–parkinsonism dementia complex in Guam. Conserv. Biol. 2003, 17, 678–686. [Google Scholar] [CrossRef]
- Finch, C.E.; Kulminski, A.M. The Alzheimer’s Disease Exposome. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2019, 15, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Eto, K.; Marumoto, M.; Takeya, M. The pathology of methylmercury poisoning (Minamata disease): The 50th Anniversary of Japanese Society of Neuropathology. Neuropathol. Off. J. Jpn. Soc. Neuropathol. 2010, 30, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Cox, P.A.; Sacks, O.W. Cycad neurotoxins, consumption of flying foxes, and ALS-PDC disease in Guam. Neurology 2002, 58, 956–959. [Google Scholar] [CrossRef]
- Bell, E.A. The discovery of BMAA, and examples of biomagnification and protein incorporation involving other non-protein amino acids. Amyotroph. Lateral Scler. Off. Publ. World Fed. Neurol. Res. Group Motor Neuron Dis. 2009, 10 (Suppl. 2), 21–25. [Google Scholar] [CrossRef]
- Foley, M.M.; Seidel, I.; Sevier, J.; Wendt, J.; Kogan, M. One man’s swordfish story: The link between Alzheimer’s disease and mercury exposure. Complement. Ther. Med. 2020, 52, 102499. [Google Scholar] [CrossRef]
- Wu, J.; Hilborn, E.D.; Schaeffer, B.A.; Urquhart, E.; Coffer, M.M.; Lin, C.J.; Egorov, A.I. Acute health effects associated with satellite-determined cyanobacterial blooms in a drinking water source in Massachusetts. Environ. Health Glob. Access Sci. Source 2021, 20, 83. [Google Scholar] [CrossRef]
- Taylor, D.L.; Calabrese, N.M. Mercury content of blue crabs (Callinectes sapidus) from southern New England coastal habitats: Contamination in an emergent fishery and risks to human consumers. Mar. Pollut. Bull. 2018, 126, 166–178. [Google Scholar] [CrossRef] [Green Version]
- Brand, L.E. Human exposure to cyanobacteria and BMAA. Amyotroph. Lateral Scler. Off. Publ. World Fed. Neurol. Res. Group Motor Neuron Dis. 2009, 10 (Suppl. 2), 85–95. [Google Scholar] [CrossRef]
- Hong, Y.S.; Kim, Y.M.; Lee, K.E. Methylmercury exposure and health effects. J. Prev. Med. Public Health 2012, 45, 353–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lance, E.; Arnich, N.; Maignien, T.; Bire, R. Occurrence of beta-N-methylamino-l-alanine (BMAA) and Isomers in Aquatic Environments and Aquatic Food Sources for Humans. Toxins 2018, 10, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckman, K.L.; Mason, R.P.; Seelen, E.; Taylor, V.F.; Balcom, P.H.; Chipman, J.; Chen, C.Y. Patterns in forage fish mercury concentrations across Northeast US estuaries. Environ. Res. 2021, 194, 110629. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Basile, M.; Mash, D.C. Cerebral uptake and protein incorporation of cyanobacterial toxin beta-N-methylamino-L-alanine. Neuroreport 2013, 24, 779–784. [Google Scholar] [CrossRef] [PubMed]
- Waidyanatha, S.; Ryan, K.; Sanders, J.M.; McDonald, J.D.; Wegerski, C.J.; Doyle-Eisle, M.; Garner, C.E. Disposition of beta-N-methylamino-l-alanine (L-BMAA), a neurotoxin, in rodents following a single or repeated oral exposure. Toxicol. Appl. Pharmacol. 2018, 339, 151–160. [Google Scholar] [CrossRef]
- Duncan, M.W.; Villacreses, N.E.; Pearson, P.G.; Wyatt, L.; Rapoport, S.I.; Kopin, I.J.; Markey, S.P.; Smith, Q.R. 2-amino-3-(methylamino)-propanoic acid (BMAA) pharmacokinetics and blood-brain barrier permeability in the rat. J. Pharmacol. Exp. Ther. 1991, 258, 27–35. [Google Scholar]
- Rand, M.D.; Caito, S.W. Variation in the biological half-life of methylmercury in humans: Methods, measurements and meaning. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 129301. [Google Scholar] [CrossRef]
- Dunlop, R.A.; Cox, P.A.; Banack, S.A.; Rodgers, K.J. The non-protein amino acid BMAA is misincorporated into human proteins in place of l-serine causing protein misfolding and aggregation. PLoS ONE 2013, 8, e75376. [Google Scholar] [CrossRef] [Green Version]
- Davis, D.A.; Cox, P.A.; Banack, S.A.; Lecusay, P.D.; Garamszegi, S.P.; Hagan, M.J.; Powell, J.T.; Metcalf, J.S.; Palmour, R.M.; Beierschmitt, A.; et al. l-Serine Reduces Spinal Cord Pathology in a Vervet Model of Preclinical ALS/MND. J. Neuropathol. Exp. Neurol. 2020, 79, 393–406. [Google Scholar] [CrossRef]
- Spencer, P.S.; Hugon, J.; Ludolph, A.; Nunn, P.B.; Ross, S.M.; Roy, D.N.; Schaumburg, H.H. Discovery and partial characterization of primate motor-system toxins. Ciba Found. Symp. 1987, 126, 221–238. [Google Scholar]
- Spencer, P.S.; Nunn, P.B.; Hugon, J.; Ludolph, A.C.; Ross, S.M.; Roy, D.N.; Robertson, R.C. Guam amyotrophic lateral sclerosis-parkinsonism-dementia linked to a plant excitant neurotoxin. Science 1987, 237, 517–522. [Google Scholar] [CrossRef]
- Cox, P.A.; Banack, S.A.; Murch, S.J. Biomagnification of cyanobacterial neurotoxins and neurodegenerative disease among the Chamorro people of Guam. Proc. Natl. Acad. Sci. USA 2003, 100, 13380–13383. [Google Scholar] [CrossRef] [Green Version]
- Oyanagi, K.; Yamazaki, M.; Hashimoto, T.; Asakawa, M.; Wakabayashi, K.; Takahashi, H. Hippocampal sclerosis in the parkinsonism-dementia complex of Guam: Quantitative examination of neurons, neurofibrillary tangles, and TDP-43 immunoreactivity in CA1. Neuropathol. Off. J. Jpn. Soc. Neuropathol. 2015, 35, 224–235. [Google Scholar] [CrossRef]
- Nelson, P.T.; Dickson, D.W.; Trojanowski, J.Q.; Jack, C.R.; Boyle, P.A.; Arfanakis, K.; Rademakers, R.; Alafuzoff, I.; Attems, J.; Brayne, C.; et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): Consensus working group report. Brain 2019, 142, 1503–1527. [Google Scholar] [CrossRef] [Green Version]
- Mackenzie, I.R.; Rademakers, R.; Neumann, M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 2010, 9, 995–1007. [Google Scholar] [CrossRef]
- Josephs, K.A.; Whitwell, J.L.; Knopman, D.S.; Hu, W.T.; Stroh, D.A.; Baker, M.; Rademakers, R.; Boeve, B.F.; Parisi, J.E.; Smith, G.E.; et al. Abnormal TDP-43 immunoreactivity in AD modifies clinicopathologic and radiologic phenotype. Neurology 2008, 70, 1850–1857. [Google Scholar] [CrossRef] [Green Version]
- Geser, F.; Winton, M.J.; Kwong, L.K.; Xu, Y.; Xie, S.X.; Igaz, L.M.; Garruto, R.M.; Perl, D.P.; Galasko, D.; Lee, V.M.; et al. Pathological TDP-43 in parkinsonism-dementia complex and amyotrophic lateral sclerosis of Guam. Acta Neuropathol. 2008, 115, 133–145. [Google Scholar] [CrossRef]
- Crary, J.F.; Trojanowski, J.Q.; Schneider, J.A.; Abisambra, J.F.; Abner, E.L.; Alafuzoff, I.; Arnold, S.E.; Attems, J.; Beach, T.G.; Bigio, E.H.; et al. Primary age-related tauopathy (PART): A common pathology associated with human aging. Acta Neuropathol. 2014, 128, 755–766. [Google Scholar] [CrossRef] [Green Version]
- Ash, P.E.A.; Dhawan, U.; Boudeau, S.; Lei, S.; Carlomagno, Y.; Knobel, M.; Al Mohanna, L.F.A.; Boomhower, S.R.; Newland, M.C.; Sherr, D.H.; et al. Heavy Metal Neurotoxicants Induce ALS-Linked TDP-43 Pathology. Toxicol. Sci. 2019, 167, 105–115. [Google Scholar] [CrossRef]
- Munoz-Saez, E.; de Munck, E.; Arahuetes, R.M.; Solas, M.T.; Martinez, A.M.; Miguel, B.G. beta-N-methylamino-L-alanine induces changes in both GSK3 and TDP-43 in human neuroblastoma. J. Toxicol. Sci. 2013, 38, 425–430. [Google Scholar] [CrossRef] [Green Version]
- Scott, L.L.; Downing, T.G. A Single Neonatal Exposure to BMAA in a Rat Model Produces Neuropathology Consistent with Neurodegenerative Diseases. Toxins 2017, 10, 22. [Google Scholar] [CrossRef] [Green Version]
- Rice, K.M.; Walker, E.M., Jr.; Wu, M.; Gillette, C.; Blough, E.R. Environmental mercury and its toxic effects. J. Prev. Med. Public Health 2014, 47, 74–83. [Google Scholar] [CrossRef]
- Li, X.; Pan, J.; Wei, Y.; Ni, L.; Xu, B.; Deng, Y.; Yang, T.; Liu, W. Mechanisms of oxidative stress in methylmercury-induced neurodevelopmental toxicity. Neurotoxicology 2021, 85, 33–46. [Google Scholar] [CrossRef]
- Yorifuji, T. Lessons From an Early-stage Epidemiological Study of Minamata Disease. J. Epidemiol. 2020, 30, 12–14. [Google Scholar] [CrossRef] [Green Version]
- U.S. Food and Drug Administration. Mercury Concentrations in Fish from the FDA Monitoring Program (1990–2010); U.S. Food and Drug Administration: Sliver Spring, MD, USA, 2017.
- Godfrey, M.E.; Wojcik, D.P.; Krone, C.A. Apolipoprotein E genotyping as a potential biomarker for mercury neurotoxicity. J. Alzheimer’s Dis. JAD 2003, 5, 189–195. [Google Scholar] [CrossRef] [Green Version]
- Siblerud, R.; Mutter, J.; Moore, E.; Naumann, J.; Walach, H. A Hypothesis and Evidence That Mercury May be an Etiological Factor in Alzheimer’s Disease. Int. J. Environ. Res. Public. Health 2019, 16, 5152. [Google Scholar] [CrossRef] [Green Version]
- Yokoo, E.M.; Valente, J.G.; Grattan, L.; Schmidt, S.L.; Platt, I.; Silbergeld, E.K. Low level methylmercury exposure affects neuropsychological function in adults. Environ. Health Glob. Access Sci. Source 2003, 2, 8. [Google Scholar] [CrossRef] [Green Version]
- Eto, K.; Takizawa, Y.; Akagi, H.; Haraguchi, K.; Asano, S.; Takahata, N.; Tokunaga, H. Differential diagnosis between organic and inorganic mercury poisoning in human cases—The pathologic point of view. Toxicol. Pathol. 1999, 27, 664–671. [Google Scholar] [CrossRef]
- Bjorkman, L.; Lundekvam, B.F.; Laegreid, T.; Bertelsen, B.I.; Morild, I.; Lilleng, P.; Lind, B.; Palm, B.; Vahter, M. Mercury in human brain, blood, muscle and toenails in relation to exposure: An autopsy study. Environ. Health A Glob. Access Sci. Source 2007, 6, 30. [Google Scholar] [CrossRef] [Green Version]
- Davis, L.E.; Kornfeld, M.; Mooney, H.S.; Fiedler, K.J.; Haaland, K.Y.; Orrison, W.W.; Cernichiari, E.; Clarkson, T.W. Methylmercury poisoning: Long-term clinical, radiological, toxicological, and pathological studies of an affected family. Ann. Neurol. 1994, 35, 680–688. [Google Scholar] [CrossRef]
- Rao, S.D.; Banack, S.A.; Cox, P.A.; Weiss, J.H. BMAA selectively injures motor neurons via AMPA/kainate receptor activation. Exp. Neurol. 2006, 201, 244–252. [Google Scholar] [CrossRef]
- Silva, D.F.; Candeias, E.; Esteves, A.R.; Magalhaes, J.D.; Ferreira, I.L.; Nunes-Costa, D.; Rego, A.C.; Empadinhas, N.; Cardoso, S.M. Microbial BMAA elicits mitochondrial dysfunction, innate immunity activation, and Alzheimer’s disease features in cortical neurons. J. Neuroinflamm. 2020, 17, 332. [Google Scholar] [CrossRef] [PubMed]
- Hiraoka, H.; Nakahara, K.; Kaneko, Y.; Akiyama, S.; Okuda, K.; Iwawaki, T.; Fujimura, M.; Kumagai, Y.; Takasugi, N.; Uehara, T. Modulation of Unfolded Protein Response by Methylmercury. Biol. Pharm. Bull. 2017, 40, 1595–1598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunlop, R.A.; Powell, J.T.; Metcalf, J.S.; Guillemin, G.J.; Cox, P.A. L-Serine-Mediated Neuroprotection Includes the Upregulation of the ER Stress Chaperone Protein Disulfide Isomerase (PDI). Neurotox. Res. 2018, 33, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, C.E.; Alexander, K.A. Unchartered waters: Climate change likely to intensify infectious disease outbreaks causing mass mortality events in marine mammals. Glob. Chang. Biol. 2020, 26, 4284–4301. [Google Scholar] [CrossRef] [PubMed]
- Di Guardo, G. Alzheimer’s disease, cellular prion protein, and dolphins. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2018, 14, 259–260. [Google Scholar] [CrossRef] [PubMed]
- Guzman-Verri, C.; Gonzalez-Barrientos, R.; Hernandez-Mora, G.; Morales, J.A.; Baquero-Calvo, E.; Chaves-Olarte, E.; Moreno, E. Brucella ceti and brucellosis in cetaceans. Front. Cell. Infect. Microbiol. 2012, 2, 3. [Google Scholar] [CrossRef] [Green Version]
- Davison, N.J.; Brownlow, A.; Doeschate, M.T.; Dale, E.J.; Foster, G.; Muchowski, J.; Perrett, L.L.; Rocchi, M.; Whatmore, A.M.; Dagleish, M.P. Neurobrucellosis due to Brucella ceti ST26 in Three Sowerby’s Beaked Whales (Mesoplodon bidens). J. Comp. Pathol. 2021, 182, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Mora, G.; Gonzalez-Barrientos, R.; Morales, J.A.; Chaves-Olarte, E.; Guzman-Verri, C.; Barquero-Calvo, E.; De-Miguel, M.J.; Marin, C.M.; Blasco, J.M.; Moreno, E. Neurobrucellosis in stranded dolphins, Costa Rica. Emerg. Infect. Dis. 2008, 14, 1430–1433. [Google Scholar] [CrossRef]
- Geraci, J.R.; Lounsbury, V.L.; Yates, N. Marine Mammals Ashore, A Field Guide for Strandings, 2nd ed.; National Aquarium in Baltimore, Inc.: Baltimore, MD, USA, 2005; p. 382. [Google Scholar]
- LeDuc, R.G.; Perrin, W.F.; Dizon, A.E. Phylogenetic Relationships Among the Delphinid Cetaceans Based on Full Cytochrome B Sequences. Mar. Mammal Sci. 1999, 15, 619–648. [Google Scholar] [CrossRef]
- Chen, I.H.; Chou, L.S.; Chou, S.J.; Wang, J.H.; Stott, J.; Blanchard, M.; Jen, I.F.; Yang, W.C. Selection of suitable reference genes for normalization of quantitative RT-PCR in peripheral blood samples of bottlenose dolphins (Tursiops truncatus). Sci. Rep. 2015, 5, 15425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]
- USEPA. Method 1630, Methyl Mercury in Water by Distillation, Aqueous Ethylation, Purge and Trap, and Cold Vapor Atomic Fluorescence Spectrometry; Office of Water, Ed.; USEPA: Washington, DC, USA, 2001.
- Oelschlager, H.H.; Haas-Rioth, M.; Fung, C.; Ridgway, S.H.; Knauth, M. Morphology and evolutionary biology of the dolphin (Delphinus sp.) brain—MR imaging and conventional histology. Brain Behav. Evol. 2008, 71, 68–86. [Google Scholar] [CrossRef] [PubMed]
- Mirra, S.S.; Hart, M.N.; Terry, R.D. Making the diagnosis of Alzheimer’s disease. A primer for practicing pathologists. Arch. Pathol. Lab. Med. 1993, 117, 132–144. [Google Scholar]
- Hof, P.R.; Chanis, R.; Marino, L. Cortical complexity in cetacean brains. Anat. Rec. Part A Discov. Mol. Cell. Evol. Biol. 2005, 287, 1142–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Agency ID | Exposure Category | BMAAϕ (μg/g) | MeHg (μg/g) | BMAA:MeHg |
|---|---|---|---|---|
| IFAW 12-228 Dd * | [EE] < AD | 20.2 | 0.411 | 49:1 |
| IFAW 12-200 Dd | 73.0 | 0.163 | 448:1 | |
| IFAW 12-198 Dd | 97.7 | 0.252 | 388:1 | |
| IFAW 12-223 Dd | [EE] > AD | 166.0 | 0.315 | 527:1 |
| IFAW 12-229 Dd | 175.6 | 0.278 | 632:1 | |
| IFAW 12-205 Dd | 185.0 | 0.165 | 1121:1 | |
| IFAW 12-201 Dd | 323.3 | 0.395 | 818:1 | |
| Median (IQR) | 166.0 (112.0) | 0.278 (0.230) | 572:1 (430.0) | |
| Min–Max | 20.2–323.3 | 0.163–0.411 | 49:1–1121:1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Davis, D.A.; Garamszegi, S.P.; Banack, S.A.; Dooley, P.D.; Coyne, T.M.; McLean, D.W.; Rotstein, D.S.; Mash, D.C.; Cox, P.A. BMAA, Methylmercury, and Mechanisms of Neurodegeneration in Dolphins: A Natural Model of Toxin Exposure. Toxins 2021, 13, 697. https://doi.org/10.3390/toxins13100697
Davis DA, Garamszegi SP, Banack SA, Dooley PD, Coyne TM, McLean DW, Rotstein DS, Mash DC, Cox PA. BMAA, Methylmercury, and Mechanisms of Neurodegeneration in Dolphins: A Natural Model of Toxin Exposure. Toxins. 2021; 13(10):697. https://doi.org/10.3390/toxins13100697
Chicago/Turabian StyleDavis, David A., Susanna P. Garamszegi, Sandra Anne Banack, Patrick D. Dooley, Thomas M. Coyne, Dylan W. McLean, David S. Rotstein, Deborah C. Mash, and Paul Alan Cox. 2021. "BMAA, Methylmercury, and Mechanisms of Neurodegeneration in Dolphins: A Natural Model of Toxin Exposure" Toxins 13, no. 10: 697. https://doi.org/10.3390/toxins13100697
APA StyleDavis, D. A., Garamszegi, S. P., Banack, S. A., Dooley, P. D., Coyne, T. M., McLean, D. W., Rotstein, D. S., Mash, D. C., & Cox, P. A. (2021). BMAA, Methylmercury, and Mechanisms of Neurodegeneration in Dolphins: A Natural Model of Toxin Exposure. Toxins, 13(10), 697. https://doi.org/10.3390/toxins13100697