Development of an Improved Method of Sample Extraction and Quantitation of Multi-Mycotoxin in Feed by LC-MS/MS

Abstract
1. Introduction
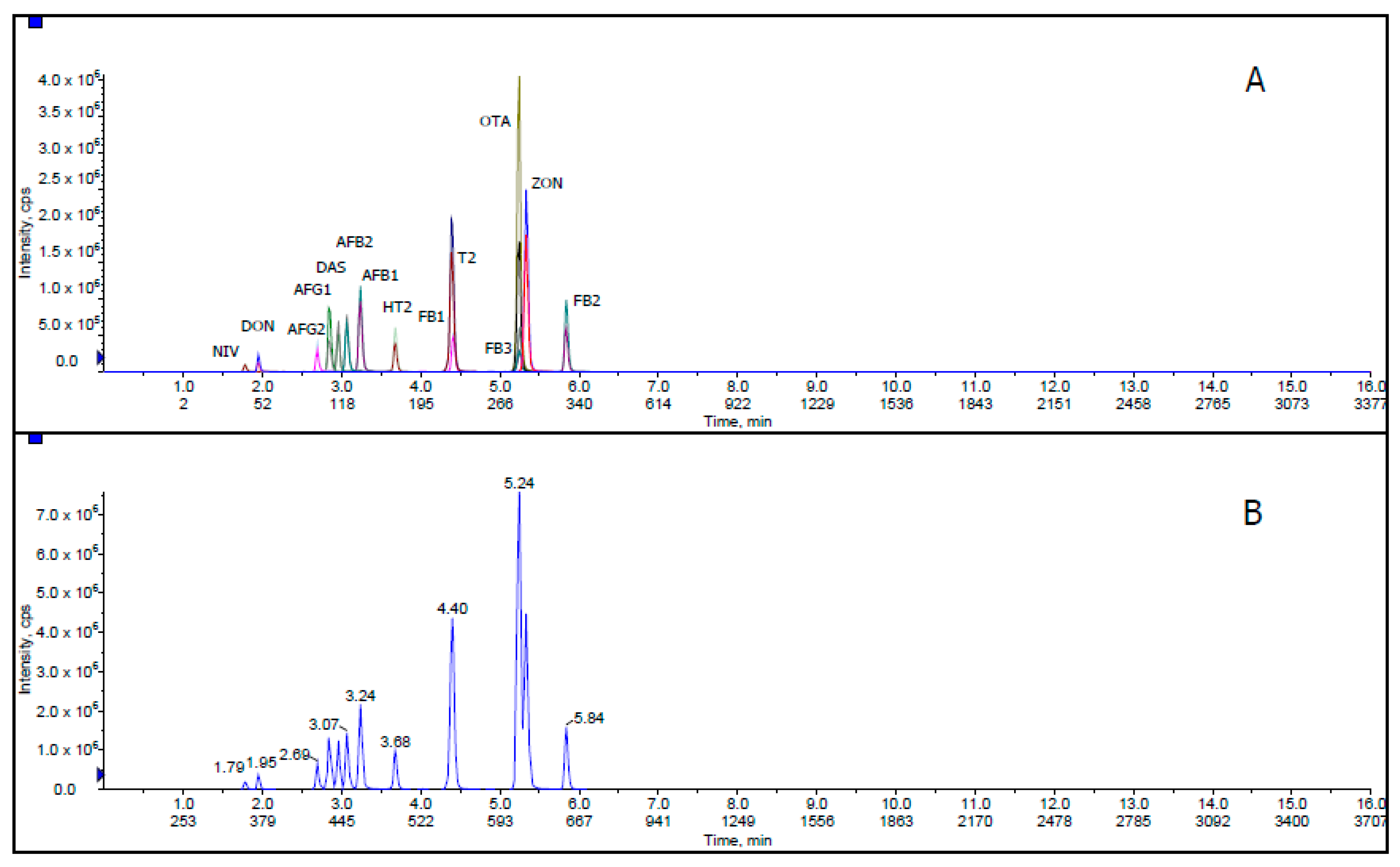
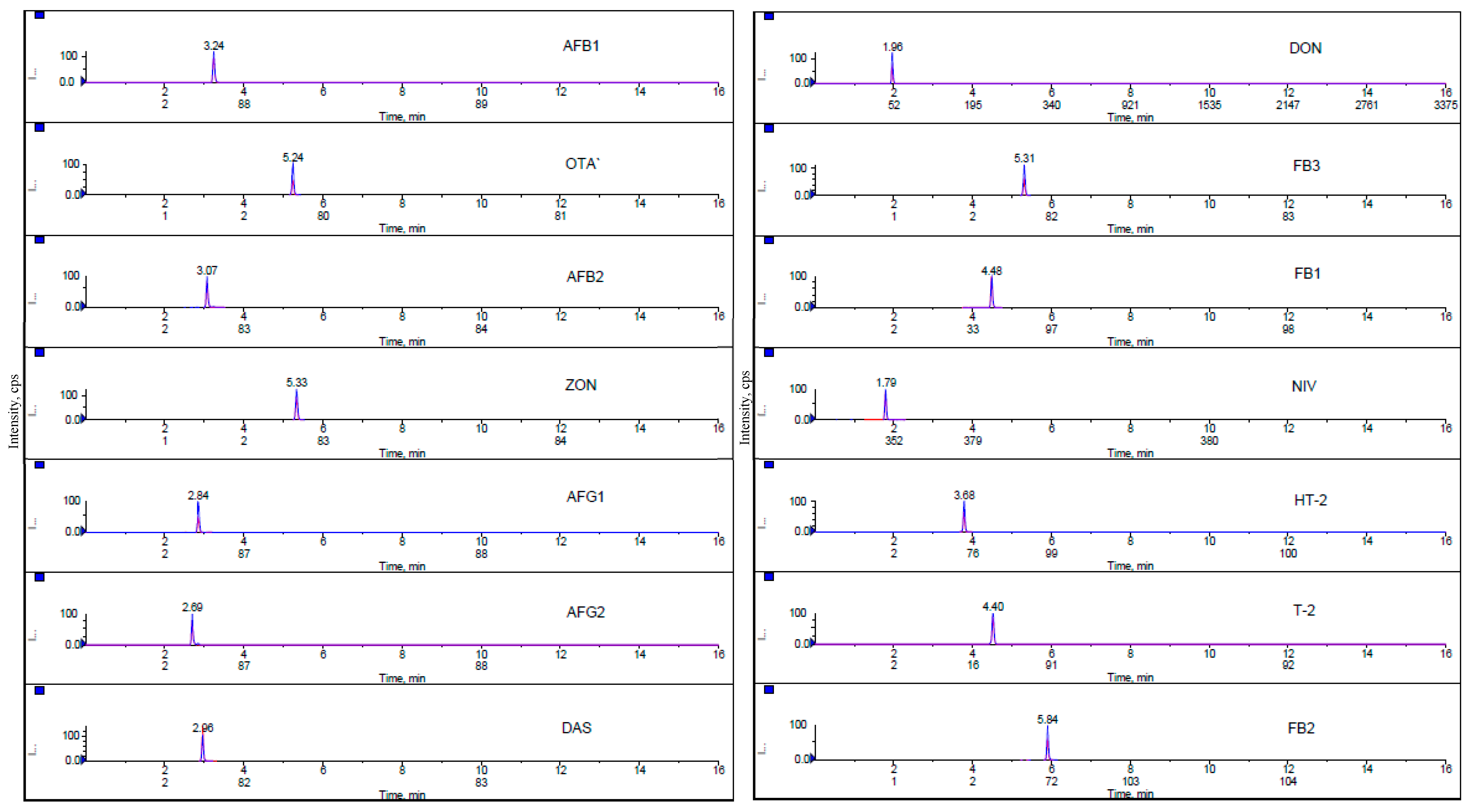
2. Results and Discussion
2.1. Optimization of Extraction Process
Solvent Mixture and Mobile Phase
2.2. Validation
3. Conclusions
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Sample Preservation and Storage
4.3. Sample Preparation
4.3.1. Immunoaffinity Column Method (Method A)
4.3.2. Solid-Phase Extraction (Method B)
4.3.3. QuEChERS Method (Method C)
4.3.4. Modified QuEChERS Method (method D)
4.4. Matrix-Matched Calibration Preparation
4.5. Equipment Conditions
4.5.1. Liquid Chromatography Separation Conditions
4.5.2. Mass Spectroscopy Conditions
4.6. Method Validation
4.6.1. Instrumental Linearity
4.6.2. Method Detection and Reporting Limit
4.6.3. Accuracy and Precision
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jelinek, C.F.; Pohland, A.E.; Wood, G.E.J. Worldwide occurrence of mycotoxins in foods and feeds-an update. Assoc. Off. Anal. Chem. 1989, 72, 223–230. [Google Scholar] [CrossRef]
- Bennett, J.W. Mycotoxins, mycotoxicoses, mycotoxicology and mycopathologia. Mycopathlogia 1987, 100, 3–5. [Google Scholar] [CrossRef]
- Abdulkadar, A.H.W.; Al-Ali, A.A.; Al-Kildi, A.M.; Al-Jedah, J.H. Mycotoxins in food products available in Qatar. Food Control 2004, 15, 543–548. [Google Scholar] [CrossRef]
- WHO Food Additives Series: 47 Safety Evaluation of Certain Mycotoxins in Food, Prepared by the Fifty-Sixth Meeting of the Joint FAO/WHO Expert Committee on Food Additives (JECFA). Available online: http://www.inchem.org/documents/jecfa/jecmono/v47je01.htm (accessed on 14 March 2014).
- Miraglia, M.; Marvin, H.J.P.; Kleter, G.A.; Battilani, P.; Brera, C.; Coni, E.; Cubadda, F.; Croci, L.; De Santis, B.; Dekkers, S.; et al. Climate change and food safety: An emerging issue with special focus on Europe. Food Chem. Toxicol. 2009, 47, 1009–1021. [Google Scholar] [CrossRef] [PubMed]
- Becker-Algeri, T.A.; Castagnaro, D.; De Bortoli, K.; de Souza, C.; Drunkler, D.A.; Badiale-Furlong, E. Mycotoxins in bovine milk and dairy products: A review. J. Food Sci. 2016, 81, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Pereira, C.S.; Cunha, S.C.; Fernandez, J.O. Prevalent mycotoxins in animal feed: Occurrence and analytical method. Toxins 2019, 11, 290. [Google Scholar] [CrossRef] [PubMed]
- Milićević, D.R.; Škrinjar, M.; Baltić, T. Real and Perceived Risks for Mycotoxin Contamination in Foods and Feeds: Challenges for Food Safety Control. Toxins 2010, 2, 572–592. [Google Scholar] [CrossRef] [PubMed]
- Juan, C.; Gonzalez, L.; Soriano, J.M.; Molto, J.C.; Manes, J. Accelerated solvent extraction of ochratoxin A from rice samples. J. Agric. Food Chem. 2005, 53, 9348–9351. [Google Scholar] [CrossRef]
- Li, C.; Wu, Y.L.; Yang, T.; Huang-Fu, W.G. Rapid determination of fumonisins B1 and B2 in corn by liquid chromatography-tandem mass spectroscopy with ultrasonic extraction. J. Chromatogr. Sci. 2012, 50, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Bauer, J.; Gareis, M. Ochratoxin A in the food chain. J. Vet. Med. Ser. B 1987, 34, 613–627. [Google Scholar] [CrossRef]
- Tang, Y.Y.; Lin, H.Y.; Chen, Y.C.; Su, W.T.; Wang, S.C.; Chiueh, L.C.; Shin, Y.C. Development of a quantitative multi-mycotoxin method in rice, maze, wheat and peanut using UPLC-MS/MS. Food Anal. Methods 2013, 6, 727–736. [Google Scholar] [CrossRef]
- Malysheva, S.V.; Di Mavungu, J.D.; Boonen, J.; De Spiegeleer, B.; Goryacheva, I.Y.; Vanhaecke, L.; De Saeger, S. Improved positive electrospray ionization of patulin by adduct formation: Usefulness in liquid chromatography-tandem mass spectroscopy multi-mycotoxin analysis. J. Chromatogr. A 2012, 1270, 334–339. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Trufelli, H.; Palma, P.; Famiglini, G.; Cappiello, A. An overview of matrix effects in liquid chromatography-mass spectrometry. Mass Spectrom. Rev. 2011, 30, 491–509. [Google Scholar] [CrossRef] [PubMed]
- Matuszewski, B.K.; Constanzer, M.L.; Chavez-Eng, C.M. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS. Anal. Chem. 2003, 75, 3019–3030. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Steimling, J.A.; Konschnik, J.D.; Grossman, S.L.; Kahler, T.W. Quantitation of mycotoxins in four food matrices comparing stable isotope dilution assay (SIDA) with matrix-matched calibration methods by LC-MS/MS. J. AOAC. Int. 2019, 102, 1673–1680. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.W.; Subrahmanyam, S.; Piletsky, S.A. Analytical methods for determination of mycotoxins: A review. Anal. Chimica Acta 2009, 632, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.Z.; Richard, J.L.; Binder, J. A review of rapid methods for the analysis of mycotoxins. Mycopathologia 2006, 161, 261–273. [Google Scholar] [CrossRef]
- Soleimany, F.; Jinap, S.; Rahmani, A.; Khatib, A. Simultaneous detection of 12 mycotoxins in cereals using RP-HPLC-PDA-FLD with PHRED and a post-column derivatization system. Food Addit. Contam. 2011, 28, 494–501. [Google Scholar] [CrossRef]
- Ofitserova, M.; Nerkar, S.; Pickering, M.; Torma, L.; Thiex, N. Multiresidue mycotoxin analysis in corn grain by column high-performance liquid chromatography with postcolumn photochemical and chemical derivatization: Single-laboratory validation. J. AOAC Int. 2009, 92, 15–25. [Google Scholar] [CrossRef]
- Monbaliu, S.; Van Poucke, C.; Detavernier, C.; Dumoulin, F.; Van De Velde, M.; Schoeters, E.; Van Dyck, S.; Averkieva, O.; Van Peteghem, C.; De Seger, S. Occurrence of mycotoxins in feed as analyzed by a multi-mycotoxin LC-MS/Ms method. J. Agric. Food Chem. 2010, 58, 66–71. [Google Scholar] [CrossRef]
- Ediage, E.N.; Di Mavungu, J.D.; Monbaliu, S.; Van Peteghem, C.; De Saeger, S. A validated multianalyte LC–MS/MS method for quantification of 25 mycotoxins in cassava flour, peanut cake and maize samples. J. Agric. Food Chem. 2011, 59, 5173–5180. [Google Scholar] [CrossRef] [PubMed]
- Lattanzio, V.M.; Solfrizzo, M.; Powers, S.; Viscounti, A. Simultaneous determination of aflatoxins, ochratoxin A and Fusarium toxins in maize by liquid chromatography tandem mass spectrometry after multitoxin immunoaffinity clean-up. Rapid Commun. Mass Spectrom. 2007, 21, 3253–3261. [Google Scholar] [CrossRef]
- Solfrizzo, M.; Gambacorta, L.; Bibi, R.; Ciriaci, M.; Paoloni, A.; Pecorelli, I. Multimycotoxin analysis by LC-MS/MS in cereal food and feed: Comparison of different approaches for extraction, purification, and calibration. J. AOAC Int. 2018, 101, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Agilent Application Note 5991-8962EN. Analysis of mycotoxins in Food matrices using the Agilent Ultivo triple Quadrupole LC/MS. 2019. Available online: https://www.agilent.com/cs/library/applications/5991-8962EN_Ultivo_AppNote.pdf (accessed on 2 July 2019).
- Rasmussen, R.R.; Storm, I.M.L.D.; Rasmussen, P.H.; Smedsgaard, J.; Nielsen, K.F. Multi-mycotoxin analysis of maize silage by LC-MS/MS. Anal. Bioanal. Chem. 2010, 397, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Malachova, A.; Stranska, M.; Vaclavikova, M.; Elliott, C.T.; Black, C.; Meneely, J.; Hajslova, J.; Ezekiel, C.N.; Schuhmacher, R.; Krska, R. Advanced LC-MS-based methods to study the co-occurrence and metabolization of multiple mycotoxins in cereals and cereal-based food. Anal. Bioanal. Chem. 2018, 410, 801–825. [Google Scholar] [CrossRef]
- Kah, M.; Brown, C.D. Log D: Lipophilicity for ionisable compounds. Chemosphere 2008, 72, 1401–1408. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Analyte | Spike Level µg/kg | Method A Rec. (%) and RSD (%) | Method B Rec. (%) and RSD (%) | Method C Rec. (%) and RSD (%) | Method D Rec. (%) and RSD (%) |
|---|---|---|---|---|---|
| DON | 40 | 94.4 and 7.1 | 100.2 and 8.4 | 83.6 and 1.9 | 85.0 and 4.2 |
| DAS | 20 | 10.6 and 9.9 | 12.1 and 18.5 | 82.6 and 5.5 | 96.1 and 3.7 |
| FB1 | 40 | 31.7 and 19.6 | 38.9 and 19.9 | 62.8 and 6.3 | 75.7 and 4.2 |
| FB2 | 40 | 55.5 and 9.5 | 58.5 and 9.0 | 54.7 and 3.0 | 78.9 and 5.3 |
| FB3 | 40 | 58.7 and 7.7 | 60.5 and 4.5 | 67.6 and 7.1 | 76.9 and 4.7 |
| HT-2 | 40 | 98.4 and 6.5 | 121.6 and 4.0 | 93.5 and 0.9 | 95.9 and 4.0 |
| T-2 | 40 | 94.9 and 7.1 | 113.4 and 6.5 | 87.3 and 4.1 | 99.0 and 2.4 |
| OTA | 20 | 7.9 and 8.5 | 9.3 and 7.2 | 58.9 and 3.2 | 86.0 and 1.3 |
| ZON | 40 | 62.5 and 6.6 | 103.8 and 2.9 | 96.7 and 2.9 | 81.8 and 6.0 |
| AFG1 | 2.0 | 108.4 and 3.5 | 109.5 and 4.2 | 87.5 and 4.2 | 82.9 and 2.8 |
| AFG2 | 2.0 | 111.3 and 4.4 | 112.0 and 1.9 | 91.6 and 3.4 | 88.8 and 5.4 |
| AFB1 | 2.0 | 81.8 and 4.3 | 86.7 and 6.6 | 78.5 and 3.4 | 79.2 and 0.94 |
| AFB2 | 2.0 | 83.6 and 5.7 | 94.3 and 5.9 | 84.4 and 2.9 | 84.5 and 5.2 |
| NIV | 80 | ND | ND | ND | 58.8 and 3.7 |
| Sample Type | Sheep Food | Dried Distillers Grain | Dairy Food | Fish Food | Goat Starter | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Toxin | Assessed Value, µg/kg | Lab Value, µg/kg | z-Score | Assessed Value, µg/kg | Lab Value, µg/kg | z-Score | Assessed Value, µg/kg | Lab Value, µg/kg | z-Score | Assessed Value, µg/kg | Lab Value, µg/kg | z-Score | Assessed Value, µg/kg | Lab Value, µg/kg | z-Score |
| Don | 2181 | 993 | −1.8 | 9673 | 4559 | −1.8 | 5354 | 4036 | −0.8 | 2366 | 988 | −1.9 | 2221 | 1615 | −0.91 |
| FB1 | 4432 | 1770 | −2.0 | 1306 | 560 | −1.8 | 2905 | 2016 | −1.0 | 2470 | 991 | −2.0 | 3190 | 3321 | 0.14 |
| FB2 | 1332 | 460 | −2.1 | 373 | 155 | −1.8 | 844 | 698 | −0.6 | 581 | 233 | −1.9 | 933 | 875 | −0.20 |
| FB3 | 595 | 234 | −1.9 | 189 | 100 | −1.4 | 357 | 380 | 0.2 | 337 | 164 | −1.6 | ND | ND | ND |
| HT-2 | 96.1 | 57.7 | −1.2 | 76.0 | 58.4 | −0.7 | 74 | 97 | −1.0 | 37.2 | 27 | −0.8 | 234 | 288 | 0.72 |
| T-2 | 91.5 | 30.0 | −2.0 | 55.0 | 25.5 | −1.6 | 97 | 60.5 | −1.2 | 69.5 | 28 | −1.8 | 262 | 214 | -0.57 |
| OTA | 107 | 63.8 | 1.2 | 7.6 | 6.3 | −0.5 | 151 | 200 | 1.0 | 186.7 | 103 | −1.4 | 316 | 364 | 0.48 |
| ZON | 335 | 126 | −1.9 | 574 | 231.1 | −1.9 | 885 | 730 | −0.6 | 368.3 | 144 | −1.9 | 377 | 342 | -0.30 |
| AFG1 | 0.59 | 0.62 | 0.14 | ND | ND | ND | ND | ND | ND | ND | ND | ND | 1.37 | 1.6 | 0.46 |
| AFG2 | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND | ND |
| AFB1 | 24.3 | 14.1 | −1.2 | 15.1 | 4.4 | −2.1 | 38.5 | 28.4 | −0.8 | 53.7 | 17.6 | −2.0 | 85.8 | 89.5 | 0.13 |
| AFB2 | 1.5 | 0.6 | −1.7 | 1.3 | 1.0 | −0.6 | 7.8 | 6.2 | −0.6 | 3.5 | 1.1 | −1.9 | 16 | 23 | 1.3 |
| Analyte | Type | Q1 (m/z) | Q3 (m/z) | Retention Time (min) | DP (Volts) | CE (Volts) | CXP (Volts) |
|---|---|---|---|---|---|---|---|
| DON | [M+H]+ | 297.2 | 249.2 231.1 | 1.93 | 37 | 16 18 | 16 16 |
| DAS | [M+H]+ | 384.0 | 307.1 105.1 | 3.00 | 54 | 9 40 | 27 20 |
| FB1 | [M+H]+ | 722.2 | 334.3 352.4 | 4.23 | 75 | 53 50 | 16 16 |
| FB2 | [M+H]+ | 706.1 | 336.3 318.4 | 5.67 | 73 | 48 50 | 14 14 |
| FB3 | [M+H]+ | 706.1 | 336.3 318.4 | 5.04 | 73 | 48 50 | 14 14 |
| HT-2 | [M+H]+ | 442.2 | 263.2 215.1 | 3.71 | 39 | 25 28 | 14 11 |
| T-2 | [M+H]+ | 484.2 | 215.2 185.1 | 4.42 | 38 | 29 38 | 15 16 |
| OTA | [M+H]+ | 404.0 | 239.0 358.1 | 5.14 | 42 | 31 19 | 13 15 |
| ZON | [M+H]+ | 319.2 | 283.2 187.1 | 5.26 | 60 | 17 26 | 15 15 |
| AFG1 | [M+H]+ | 329.0 | 243.1 283.1 | 2.78 | 73 | 37 35 | 16 16 |
| AFG2 | [M+H]+ | 331.1 | 245.1 257.1 | 2.64 | 70 | 41 42 | 16 16 |
| AFB1 | [M+H]+ | 313.1 | 285.1 241.1 | 3.17 | 58 | 32 51 | 14 18 |
| AFB2 | [M+H]+ | 315.2 | 287.2 259.1 | 2.99 | 43 | 36 41 | 12 17 |
| NIV | [M+H]+ | 313.1 | 175.1 115.1 | 1.77 | 96 | 21 73 | 12 8 |
| Toxin | Spike (µg/mL) | SD | MDL | RL |
|---|---|---|---|---|
| DON | 0.08 | 0.00023 | 0.00713 | 0.01426 |
| DAS | 0.04 | 0.00115 | 0.00360 | 0.00720 |
| FB1 | 0.24 | 0.01588 | 0.04991 | 0.09983 |
| FB2 | 0.24 | 0.01357 | 0.04266 | 0.08533 |
| FB3 | 0.08 | 0.00348 | 0.01094 | 0.02189 |
| HT-2 | 0.08 | 0.00115 | 0.00362 | 0.00724 |
| T-2 | 0.08 | 0.00122 | 0.00383 | 0.00766 |
| OTA | 0.04 | 0.00159 | 0.00498 | 0.00997 |
| ZON | 0.08 | 0.00269 | 0.00845 | 0.01690 |
| AFG1 | 0.004 | 0.00010 | 0.00032 | 0.00064 |
| AFG2 | 0.004 | 0.00016 | 0.00049 | 0.00098 |
| AFB1 | 0.004 | 0.00012 | 0.00037 | 0.00073 |
| AFB2 | 0.004 | 0.00006 | 0.00018 | 0.00035 |
| NIV | 0.16 | 0.00456 | 0.01433 | 0.02867 |
| Mycotoxins | Concentration (µg/g) | Mean Recovery (%) | SD (µg/g) | RSD (%) | Mycotoxins | Concentration (µg/g) | Mean Recovery (%) | SD (µg/g) | RSD (%) |
|---|---|---|---|---|---|---|---|---|---|
| DON | 0.06 | 87.3 | 2.9 | 3.3 | OTA | 0.03 | 86.9 | 6.1 | 7.1 |
| 0.08 | 86.1 | 2.1 | 2.4 | 0.04 | 89.0 | 89.0 | 4.3 | ||
| 0.2 | 86.0 | 1.9 | 2.2 | 0.1 | 94.7 | 94.7 | 7.3 | ||
| DAS | 0.03 | 93.8 | 5.8 | 6.2 | ZON | 0.06 | 78.7 | 2.2 | 2.9 |
| 0.04 | 96.1 | 7.6 | 7.9 | 0.08 | 81.9 | 5.7 | 7.0 | ||
| 0.1 | 91.7 | 2.3 | 2.5 | 0.02 | 87.2 | 5.5 | 6.3 | ||
| FB1 | 0.24 | 75.8 | 8.9 | 10.6 | AFG1 | 0.003 | 84.7 | 10.5 | 12.3 |
| 0.6 | 74.4 | 6.7 | 9.1 | 0.004 | 86.3 | 7.3 | 8.5 | ||
| 2.4 | 80.4 | 1.7 | 2.1 | 0.01 | 81.9 | 6.1 | 7.4 | ||
| FB2 | 0.24 | 71.3 | 5.3 | 8.7 | AFG2 | 0.003 | 99.1 | 8.7 | 8.8 |
| 0.6 | 74.0 | 2.9 | 4.0 | 0.004 | 98.0 | 4.9 | 5.0 | ||
| 2.4 | 74.6 | 3.5 | 5.1 | 0.01 | 92.3 | 2.6 | 2.9 | ||
| FB3 | 0.08 | 74.2 | 6.7 | 9.0 | AFB1 | 0.003 | 79.6 | 5.7 | 7.2 |
| 0.2 | 77.6 | 3.9 | 5.1 | 0.004 | 82.0 | 3.3 | 4.1 | ||
| 0.8 | 78.2 | 4.6 | 5.9 | 0.01 | 79.3 | 3.5 | 4.4 | ||
| HT-2 | 0.06 | 97.5 | 2.5 | 2.6 | AFB2 | 0.003 | 86.1 | 6.2 | 7.2 |
| 0.08 | 97.0 | 4.2 | 4.3 | 0.004 | 86.5 | 6.9 | 7.9 | ||
| 0.5 | 84.1 | 4.4 | 5.2 | 0.01 | 81.9 | 4.4 | 5.3 | ||
| T-2 | 0.06 | 96.1 | 3.5 | 3.6 | NIV | 0.12 | 55.0 | 3.7 | 6.7 |
| 0.08 | 98.4 | 3.4 | 3.5 | 0.16 | 56.7 | 3.6 | 6.3 | ||
| 0.2 | 98.4 | 4.3 | 4.4 | 0.4 | 55.0 | 4.1 | 7.5 | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakhjavan, B.; Ahmed, N.S.; Khosravifard, M. Development of an Improved Method of Sample Extraction and Quantitation of Multi-Mycotoxin in Feed by LC-MS/MS. Toxins 2020, 12, 462. https://doi.org/10.3390/toxins12070462
Nakhjavan B, Ahmed NS, Khosravifard M. Development of an Improved Method of Sample Extraction and Quantitation of Multi-Mycotoxin in Feed by LC-MS/MS. Toxins. 2020; 12(7):462. https://doi.org/10.3390/toxins12070462
Chicago/Turabian StyleNakhjavan, Bahar, Nighat Sami Ahmed, and Maryam Khosravifard. 2020. "Development of an Improved Method of Sample Extraction and Quantitation of Multi-Mycotoxin in Feed by LC-MS/MS" Toxins 12, no. 7: 462. https://doi.org/10.3390/toxins12070462
APA StyleNakhjavan, B., Ahmed, N. S., & Khosravifard, M. (2020). Development of an Improved Method of Sample Extraction and Quantitation of Multi-Mycotoxin in Feed by LC-MS/MS. Toxins, 12(7), 462. https://doi.org/10.3390/toxins12070462