Quantitative Measurement of Melittin in Asian Honeybee Venom Using a New Method Including UPLC-QqTOF-MS

Abstract
1. Introduction
2. Results
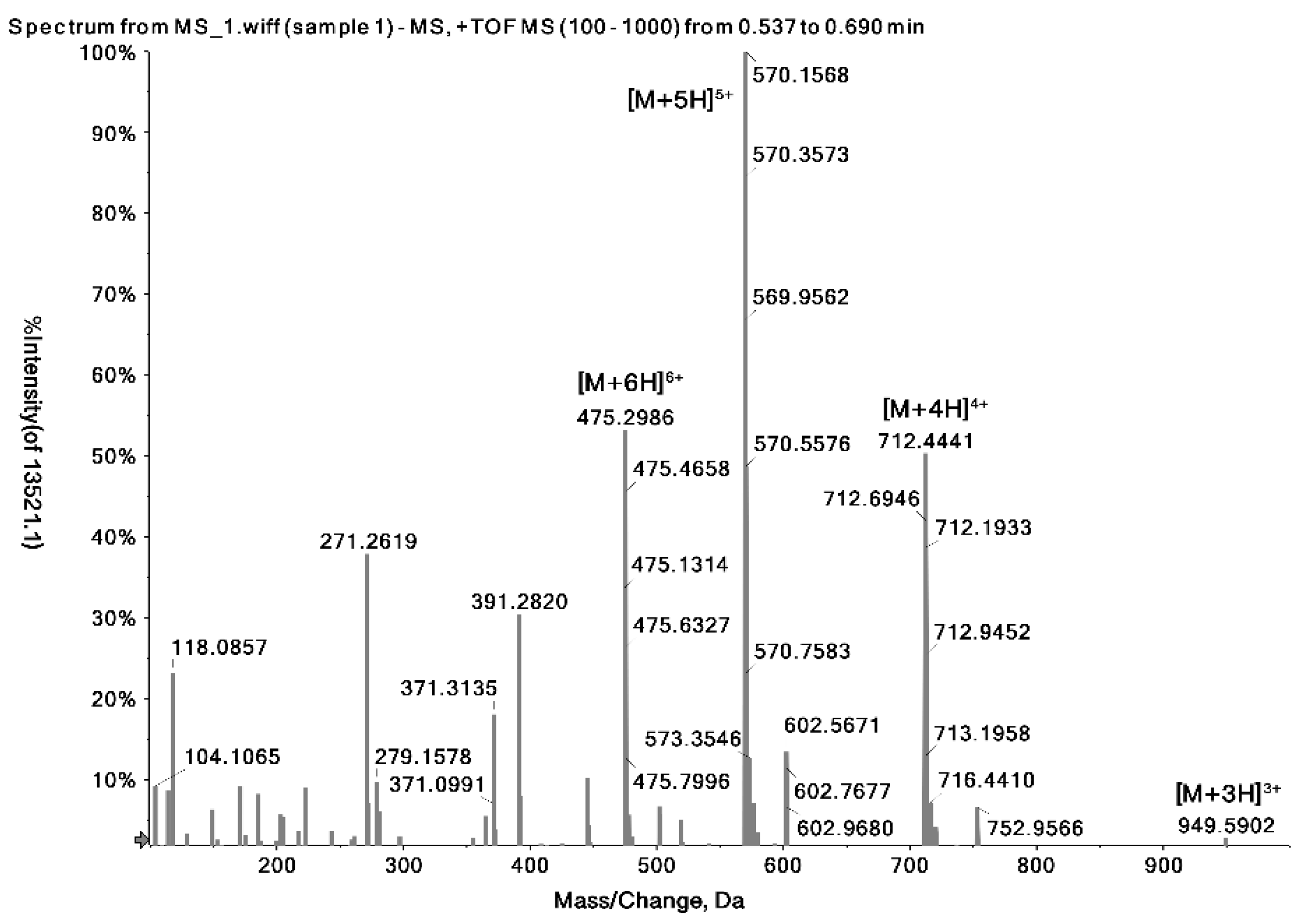
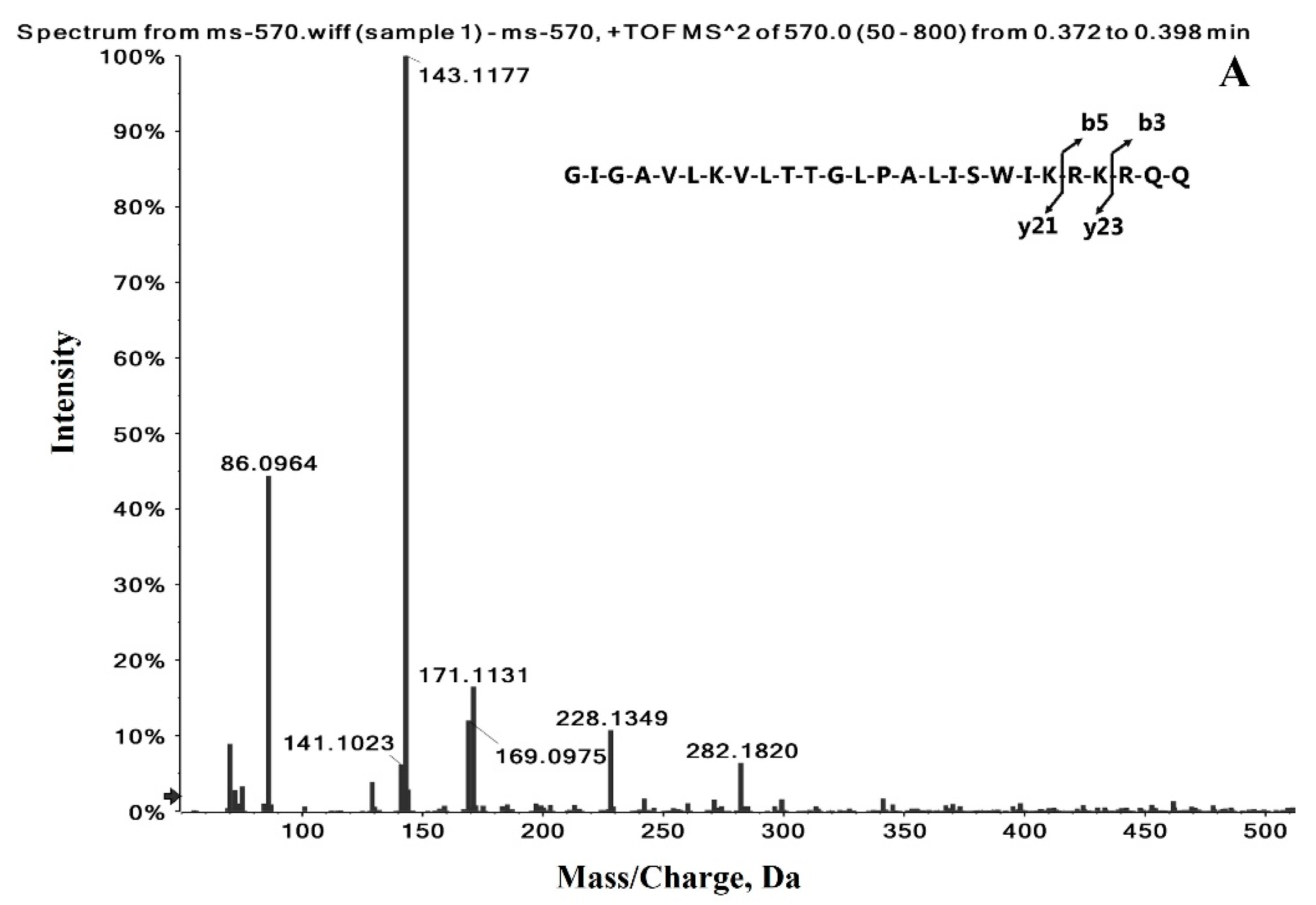
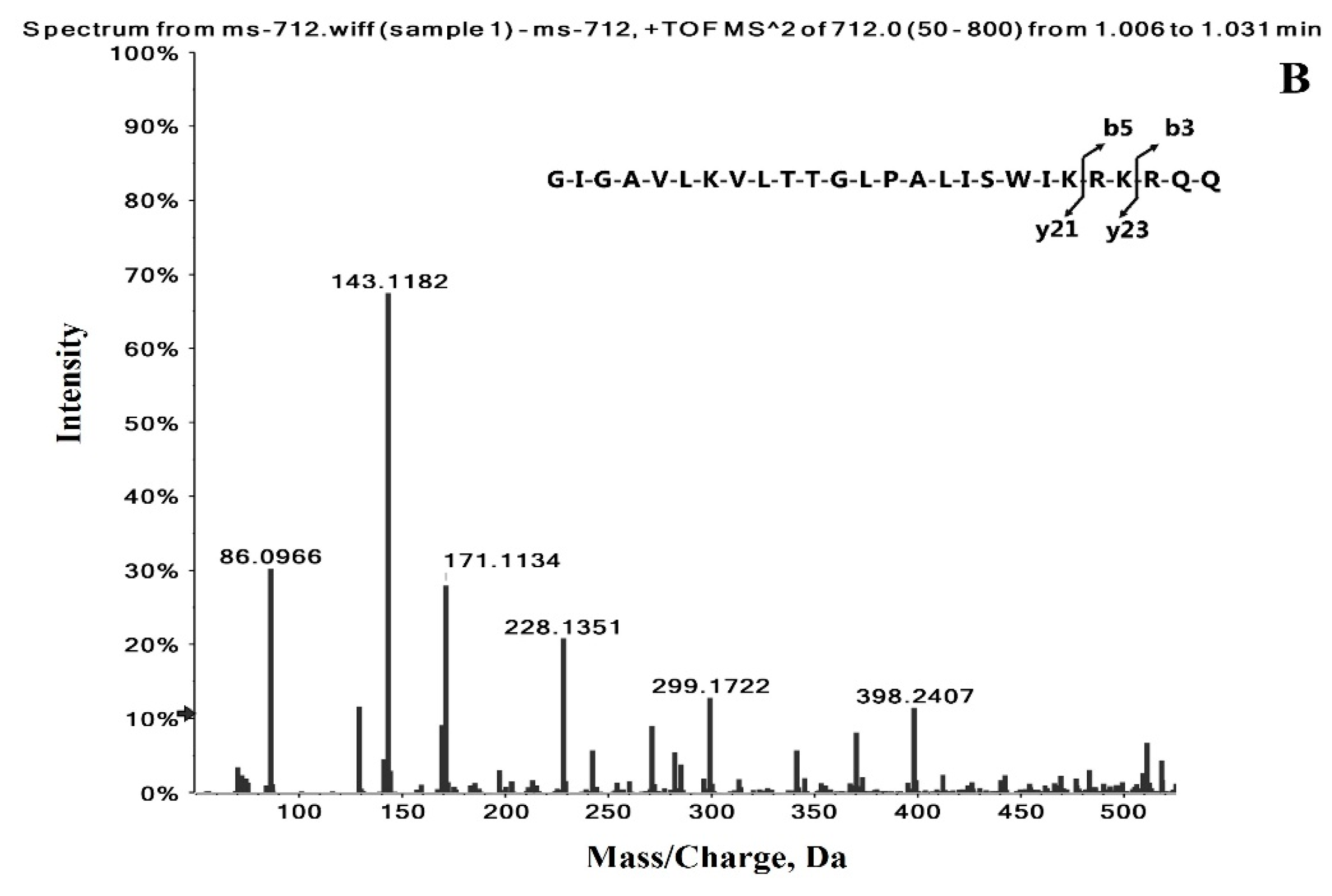
2.1. Identification of the Precursor and Fragment Ions of Melittin
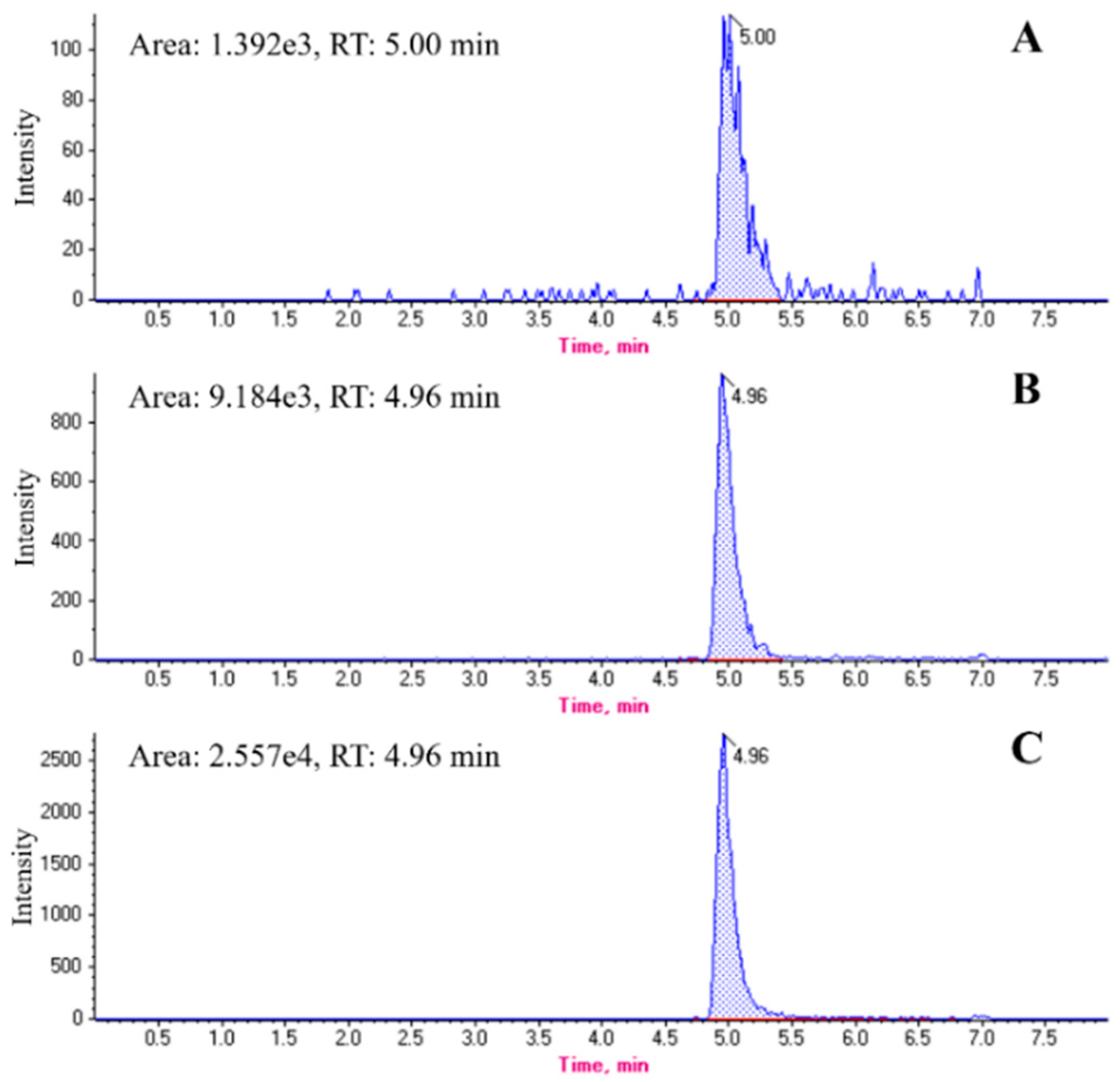
2.2. Chromatographic Profiles of Melittin
2.3. Method Validation
2.3.1. Linearity and the Matrix Effect
2.3.2. Limit of Detection (LOD) and LOQ
2.3.3. Accuracy and Precision
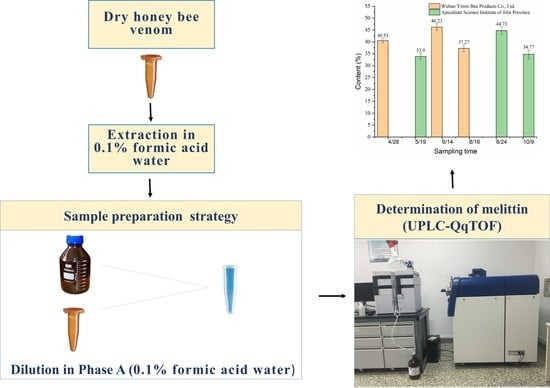
2.3.4. Melittin Content in Asian Honeybee Venom
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Chemicals and Reagents
5.2. Preparation of the Working and Calibration Standard Solutions
5.3. Preparation of the Quality Control (QC) Samples
5.4. Collection and Preparation of the Bee Venom Samples
5.5. Instrumentation and Analytical Conditions
5.6. Method Validation
5.6.1. Linearity and the Matrix Effect
5.6.2. LOD and LOQ
5.6.3. Precision and Accuracy
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Seo, B.K.; Han, K.; Kwon, O.; Jo, D.J.; Lee, J.H. Efficacy of bee venom acupuncture for chronic low back pain: A randomized, double-blinded, sham-controlled trial. Toxins 2017, 9, 361. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Bae, H. Anti-Inflammatory Applications of Melittin, a Major Component of Bee Venom: Detailed Mechanism of Action and Adverse Effects. Molecules 2016, 21, 616. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.; Choi, S.; Yang, E.J. The Effects of Bee Venom Acupuncture on the Central Nervous System and Muscle in an Animal hSOD1G93A Mutant. Toxins 2015, 7, 846–858. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.F.D.; Mongefuentes, V.; Gomes, F.M.M.; Lopes, K.S.; Anjos, L.C.D.; Campos, G.A.A.; Arenas, C.; Biolchi, A.; Goncalves, J.C.; Galante, P. Pharmacological Alternatives for the Treatment of Neurodegenerative Disorders: Wasp and Bee Venoms and Their Components as New Neuroactive Tools. Toxins 2015, 7, 3179–3209. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Shim, S.; Rhee, H.Y.; Park, H.; Jung, W.; Moon, S.; Park, J.; Ko, C.; Cho, K.; Park, S. Effectiveness of acupuncture and bee venom acupuncture in idiopathic Parkinson’s disease. Parkinsonism Relat. Disord. 2012, 18, 948–952. [Google Scholar] [CrossRef]
- Choi, S.; Chae, H.K.; Heo, H.; Hahm, D.; Kim, W.; Kim, S.K. Analgesic Effect of Melittin on Oxaliplatin-Induced Peripheral Neuropathy in Rats. Toxins 2019, 11, 396. [Google Scholar] [CrossRef]
- Li, D.; Lee, Y.; Kim, W.; Lee, K.; Bae, H.; Kim, S.K. Analgesic Effects of Bee Venom Derived Phospholipase A2 in a Mouse Model of Oxaliplatin-Induced Neuropathic Pain. Toxins 2015, 7, 2422–2434. [Google Scholar] [CrossRef]
- Kim, W.; Kim, M.J.; Go, D.; Min, B.I.; Na, H.S.; Kim, S.K. Combined Effects of Bee Venom Acupuncture and Morphine on Oxaliplatin-Induced Neuropathic Pain in Mice. Toxins 2016, 8, 33. [Google Scholar] [CrossRef]
- Orsolic, N. Bee venom in cancer therapy. Cancer Metastasis Rev. 2012, 31, 173–194. [Google Scholar] [CrossRef]
- Liu, C.; Hao, D.; Zhang, Q.; An, J.; Zhao, J.; Chen, B.; Zhang, L.; Yang, H. Application of bee venom and its main constituent melittin for cancer treatment. Cancer Chemother. Pharmacol. 2016, 78, 1113–1130. [Google Scholar] [CrossRef]
- Moga, M.; Dimienescu, O.G.; Arvătescu, C.A.; Ifteni, P.; Ples, L. Anticancer Activity of Toxins from Bee and Snake Venom—An Overview on Ovarian Cancer. Molecules 2018, 23, 692. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Pak, S.C.; Park, K. The Protective Effect of Bee Venom on Fibrosis Causing Inflammatory Diseases. Toxins 2015, 7, 4758–4772. [Google Scholar] [CrossRef] [PubMed]
- Banks, B.E.C.; Shipolini, R.A. Chemistry and pharmacology of honeybee venom. Venoms Hymenopt. Biochem. Pharmacol. Behav. Asp. 1986, 1986, 330–416. [Google Scholar]
- Choi, J.H.; Jang, A.Y.; Lin, S.; Lim, S.; Kim, D.; Park, K.; Han, S.M.; Yeo, J.H.; Seo, H.S. Melittin, a honeybee venom-derived antimicrobial peptide, may target methicillin-resistant Staphylococcus aureus. Mol. Med. Rep. 2015, 12, 6483–6490. [Google Scholar] [CrossRef] [PubMed]
- Picoli, T.; Peter, C.M.; Zani, J.L.; Waller, S.B.; Lopes, M.G.; Boesche, K.N.; de Oliveira Hübner, S.; Fischer, G. Melittin and its potential in the destruction and inhibition of the biofilm formation by Staphylococcus aureus, Escherichia coli and Pseudomonas aeruginosa isolated from bovine milk. Microb. Pathog. 2017, 112, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Socarras, K.M.; Theophilus, P.A.; Torres, J.P.; Gupta, K.; Sapi, E. Antimicrobial activity of bee venom and melittin against Borrelia burgdorferi. Antibiotics 2017, 6, 31. [Google Scholar] [CrossRef]
- Falco, A.; Barrajón-Catalán, E.; Menéndez-Gutiérrez, M.P.; Coll, J.; Micol, V.; Estepa, A. Melittin-loaded immunoliposomes against viral surface proteins, a new approach to antiviral therapy. Antivir. Res. 2013, 97, 218–221. [Google Scholar] [CrossRef]
- Hood, J.L.; Jallouk, A.P.; Campbell, N.; Ratner, L.; Wickline, S.A. Cytolytic nanoparticles attenuate HIV-1 infectivity. Antivir. Ther. 2013, 18, 95–103. [Google Scholar] [CrossRef]
- Cheng, B.; Thapa, B.; Remant, K.; Xu, P. Dual secured nano-melittin for the safe and effective eradication of cancer cells. J. Mater. Chem. 2015, 3, 25–29. [Google Scholar] [CrossRef]
- Plasay, M.; Wahid, S.; Natzir, R.; Miskad, U.A. Selective cytotoxicity Assay in anticancer drug of Melittin Isolated from Bee Venom (Apis cerana indica) to several human cell lines: HeLa, WiDr and Vero. J. Chem. Pharm. Sci. 2016, 9, 2674–2676. [Google Scholar]
- Rady, I.; Siddiqui, I.A.; Rady, M.; Mukhtar, H. Melittin, a major peptide component of bee venom, and its conjugates in cancer therapy. Cancer Lett. 2017, 402, 16–31. [Google Scholar] [CrossRef] [PubMed]
- Baracchi, D.; Turillazzi, S. Differences in venom and cuticular peptides in individuals of Apis mellifera (Hymenoptera: Apidae) determined by MALDI-TOF MS. J. Insect Physiol. 2010, 56, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Baracchi, D.; Francese, S.; Turillazzi, S. Beyond the antipredatory defence: Honeybee venom function as a component of social immunity. Toxicon Off. J. Int. Soc. Toxinol. 2011, 58, 550–557. [Google Scholar] [CrossRef]
- Rybak-Chmielewska, H.; Szczêsna, T. HPLC study of chemical composition of honeybee (Apis mellifera L.) venom. J. Apic. Sci. 2004, 48, 103–109. [Google Scholar]
- Kokot, Z.J.; Matysiak, J. Simultaneous determination of major constituents of honeybee venom by LC-DAD. Chromatographia 2009, 69, 1401–1405. [Google Scholar] [CrossRef]
- Kokot, Z.J.; Matysiak, J.; Urbaniak, B.; Derezinski, P. New CZE-DAD method for honeybee venom analysis and standardization of the product. Anal. Bioanal. Chem. 2011, 399, 2487–2494. [Google Scholar] [CrossRef] [PubMed]
- Matysiak, J.; Schmelzer, C.E.; Neubert, R.H.; Kokot, Z.J. Characterization of honeybee venom by MALDI-TOF and nanoESI-QqTOF mass spectrometry. J. Pharm. Biomed. Anal. 2011, 54, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Haghi, G.; Hatami, A.; Mehran, M. Qualitative and quantitative evaluation of melittin in honeybee venom and drug products containing honeybee venom. J. Apic. Sci. 2013, 57, 37–44. [Google Scholar] [CrossRef][Green Version]
- Zhou, J.; Zhao, J.; Zhang, S.; Shen, J.; Qi, Y.; Xue, X.; Li, Y.; Wu, L.; Zhang, J.; Chen, F. Quantification of melittin and apamin in bee venom lyophilized powder from Apis mellifera by liquid chromatography–diode array detector–tandem mass spectrometry. Anal. Biochem. 2010, 404, 171–178. [Google Scholar] [CrossRef]
- Akhmerova, D.; Stavrianidi, A.; Rodin, I.; Shpigun, O. Application of high performance liquid chromatography-tandem mass spectrometry for detection of melittin, a characteristic peptide of bee venom. Inorg. Mater. 2015, 51, 1431–1437. [Google Scholar] [CrossRef]
- Morin, L.; Mess, J.; Ramagiri, S.; Garofolo, F. Elimination of selectivity and quantification issues during ursodiol regulated bioanalysis by using a high resolution quadrupole time-of-flight mass spectrometer. In Proceedings of the 59th American Society for Mass Spectrometry Conference, Denver, CO, USA, 5–9 June 2011. [Google Scholar]
- Morin, L.P.; Mess, J.N.; Garofolo, F. Large-molecule quantification: Sensitivity and selectivity head-to-head comparison of triple quadrupole with Q-TOF. Bioanalysis 2013, 5, 1181–1193. [Google Scholar] [CrossRef] [PubMed]
- Kiefer, P.; Portais, J.C.; Vorholt, J.A. Quantitative metabolome analysis using liquid chromatography–high-resolution mass spectrometry. Anal. Biochem. 2008, 382, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Chung, I.M.; Kim, H.; Jung, M.Y. High resolution LC–ESI-TOF-mass spectrometry method for fast separation, identification, and quantification of 12 isoflavones in soybeans and soybean products. Food Chem. 2015, 176, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Collins, B.C.; Gillet, L.C.; Rosenberger, G.; Rost, H.L.; Vichalkovski, A.; Gstaiger, M.; Aebersold, R. Quantifying protein interaction dynamics by SWATH mass spectrometry: Application to the 14-3-3 system. Nat. Methods 2013, 10, 1246–1253. [Google Scholar] [CrossRef]
- Collins, B.C.; Hunter, C.L.; Liu, Y.; Schilling, B.; Rosenberger, G.; Bader, S.L.; Chan, D.W.; Gibson, B.W.; Gingras, A.C.; Held, J.M. Multi-laboratory assessment of reproducibility, qualitative and quantitative performance of SWATH-mass spectrometry. Nat. Commun. 2017, 8, 291. [Google Scholar] [CrossRef]
- Raghuraman, H.; Chattopadhyay, A. Effect of ionic strength on folding and aggregation of the hemolytic peptide melittin in solution. Biopolymers 2006, 83, 111–121. [Google Scholar] [CrossRef]
- Xu, W.; Jiang, H.; Titsch, C.; Gadkari, S.; Batog, A.; Wang, B.; Hippeli, L.; Yamamoto, B.; Chadwick, K.D.; Wheeler, J. Concerted application of LC–MS and ligand binding assays to better understand exposure of a large molecule drug. Bioanalysis 2018, 10, 1261–1272. [Google Scholar] [CrossRef]
- Schilling, B.; Meyer, J.G.; Wei, L.; Ott, M.; Verdin, E. High-Resolution Mass Spectrometry to Identify and Quantify Acetylation Protein Targets. Methods Mol. Biol. 2019, 1983, 3–16. [Google Scholar]
- Chen, Z.; Alelyunas, Y.; Wrona, M.; Kehler, J.R.; Szapacs, M.; Evans, C.A. Microflow UPLC and high-resolution MS as a sensitive and robust platform for quantitation of intact peptide hormones. Bioanalysis 2019, 11, 1275–1289. [Google Scholar] [CrossRef]
- Barzenhanson, K.A.; Wilkes, R.A.; Aristilde, L. Quantitation of carbohydrate monomers and dimers by liquid chromatography coupled with high-resolution mass spectrometry. Carbohydr. Res. 2018, 468, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, J.M.; Smith, J.; Rzewuski, S.; Legidoquigley, C.; Lauber, M. High sensitivity LC-MS profiling of antibody-drug conjugates with difluoroacetic acid ion pairing. mAbs 2019, 11, 1358–1366. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Berger, S.J.; Martinovic, S.; Pasatolic, L.; Anderson, G.A.; Shen, Y.; Zhao, R.; Smith, R.D. Direct mass spectrometric analysis of intact proteins of the yeast large ribosomal subunit using capillary LC/FTICR. Proc. Natl. Acad. Sci. USA 2002, 99, 5942–5947. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Ou, J.; Liu, Z.; Liu, J.; Lin, H.; Wang, F.; Zou, H. Separation of intact proteins by using polyhedral oligomeric silsesquioxane based hybrid monolithic capillary columns. J. Chromatogr. A 2013, 1317, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.C.; Hogenboom, A.C.; Zappey, H.; Irth, H. Effect of the mobile phase composition on the separation and detection of intact proteins by reversed-phase liquid chromatography-electrospray mass spectrometry. J. Chromatogr. 2002, 957, 187–199. [Google Scholar] [CrossRef]
- Apffel, A.; Fischer, S.M.; Goldberg, G.; Goodley, P.C.; Kuhlmann, F.E. Enhanced sensitivity for peptide mapping with electrospray liquid chromatography-mass spectrometry in the presence of signal suppression due to trifluoroacetic acid-containing mobile phases. J. Chromatogr. 1995, 712, 177–190. [Google Scholar] [CrossRef]
- Mirza, U.A.; Chait, B.T. Effects of anions on the positive ion electrospray ionization mass spectra of peptides and proteins. Anal. Chem. 1994, 66, 2898–2904. [Google Scholar] [CrossRef]
- Nshanian, M.; Lakshmanan, R.; Chen, H.; Loo, R.R.O.; Loo, J.A. Enhancing sensitivity of liquid chromatography–mass spectrometry of peptides and proteins using supercharging agents. Int. J. Mass Spectrom. 2017, 427, 157–164. [Google Scholar] [CrossRef]
- Salm, P.; Taylor, P.J.; Lynch, S.V.; Warnholtz, C.; Pillans, P.I. A rapid HPLC-mass spectrometry cyclosporin method suitable for current monitoring practices. Clin. Biochem. 2005, 38, 667–673. [Google Scholar] [CrossRef]
- Gao, S.; Zhang, Z.; Karnes, H.T. Sensitivity enhancement in liquid chromatography/atmospheric pressure ionization mass spectrometry using derivatization and mobile phase additives. J. Chromatogr. 2005, 825, 98–110. [Google Scholar] [CrossRef]
- Wang, S.; Cyronak, M.J.; Yang, E. Does a stable isotopically labeled internal standard always correct analyte response? A matrix effect study on a LC/MS/MS method for the determination of carvedilol enantiomers in human plasma. J. Pharm. Biomed. Anal. 2007, 43, 701–707. [Google Scholar] [CrossRef]
- Ismaiel, O.A.; Halquist, M.S.; Elmamly, M.Y.; Shalaby, A.; Karnes, H.T. Monitoring phospholipids for assessment of matrix effects in a liquid chromatography-tandem mass spectrometry method for hydrocodone and pseudoephedrine in human plasma. J. Chromatogr. 2007, 859, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Bruins, C.H.P.; Jeronimusstratingh, C.; Ensing, K.; Van Dongen, W.D.; De Jong, G.J. On-line coupling of solid-phase extraction with mass spectrometry for the analysis of biological samples I. Determination of clenbuterol in urine. J. Chromatogr. A 1999, 863, 115–122. [Google Scholar] [CrossRef]
- Gros, M.; Petrovic, M.; Barcelo, D. Development of a multi-residue analytical methodology based on liquid chromatography–tandem mass spectrometry (LC–MS/MS) for screening and trace level determination of pharmaceuticals in surface and wastewaters. Talanta 2006, 70, 678–690. [Google Scholar] [CrossRef] [PubMed]
- John, H.; Huynh, K.D.; Hedtmann, C.; Walden, M.; Schulz, A.; Anspach, F.B.; Forssmann, W. In vitro degradation of the antimicrobial human peptide HEM-γ 130–146 in plasma analyzed by a validated quantitative LC–MS/MS procedure. Anal. Biochem. 2005, 341, 173–186. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide Name | Molecular Weight (Da) | Sequence | Formula | Observed Charge States | Most Abundant Isotopes Mass (Da) |
|---|---|---|---|---|---|
| Melittin | 2846.46 | GIGAVLKVLTTGLPALISWIKRKRQQ | C131H229N39O31 | +3 | 949.5902 |
| +4 | 712.4441 | ||||
| +5 | 570.1568 | ||||
| +6 | 475.2986 |
| Analyte | Calibration Curve Type | Slope (a ± Sa) | Intercept (b ± Sb) | Regression Coefficient (r) |
|---|---|---|---|---|
| Melittin | 1 | 2880.54 ± 2.3 | 16,066.18 ± 4.7 | 0.99977 |
| 2 | 2845.86 ± 1.8 | 22,401.19 ± 5.7 | 0.99731 |
| Spiked Level (μg/mL) | Mean Recovery in Intra-Day SD (n = 5, %) | Intra-Day RSD (n = 5, %) | Mean Recovery in Inter-Day (n = 15, %) | Inter-Day RSD (n = 15, %) |
|---|---|---|---|---|
| 1 | 87.35 ± 8.3 | 9.5 | 84.88 ± 8.9 | 10.6 |
| 5 | 91.65 ± 4.0 | 5.4 | 92.32 ± 5.7 | 6.2 |
| 10 | 90.14 ± 4.2 | 4.6 | 91.07 ± 4.3 | 4.7 |
| Sampling Location | Sampling Date | Batch Number | Content (%) | Daily Mean Content (%) | SD |
|---|---|---|---|---|---|
| Wuhan | 28 April 2015 | W-1 | 41.8 | 40.53 | 0.95 |
| W-2 | 39.5 | ||||
| W-3 | 40.29 | ||||
| 14 June 2015 | W-4 | 44.3 | 46.23 | 1.40 | |
| W-5 | 47.6 | ||||
| W-6 | 46.79 | ||||
| 14 August 2015 | W-7 | 37.91 | 37.27 | 1.42 | |
| W-8 | 38.6 | ||||
| W-9 | 35.3 | ||||
| Jilin | 19 May 2015 | J-1 | 35.6 | 33.9 | 1.35 |
| J-2 | 32.3 | ||||
| J-3 | 33.8 | ||||
| 24 August 2015 | J-4 | 43.2 | 44.73 | 1.58 | |
| J-5 | 46.9 | ||||
| J-6 | 44.29 | ||||
| 9 October 2015 | J-7 | 35.51 | 34.77 | 1.87 | |
| J-8 | 36.6 | ||||
| J-9 | 32.2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, S.; Wang, J.; Guo, Z.; Wang, Y.; Liu, C. Quantitative Measurement of Melittin in Asian Honeybee Venom Using a New Method Including UPLC-QqTOF-MS. Toxins 2020, 12, 437. https://doi.org/10.3390/toxins12070437
Huang S, Wang J, Guo Z, Wang Y, Liu C. Quantitative Measurement of Melittin in Asian Honeybee Venom Using a New Method Including UPLC-QqTOF-MS. Toxins. 2020; 12(7):437. https://doi.org/10.3390/toxins12070437
Chicago/Turabian StyleHuang, Sheng, Jianhua Wang, Zeqin Guo, Yan Wang, and Chundong Liu. 2020. "Quantitative Measurement of Melittin in Asian Honeybee Venom Using a New Method Including UPLC-QqTOF-MS" Toxins 12, no. 7: 437. https://doi.org/10.3390/toxins12070437
APA StyleHuang, S., Wang, J., Guo, Z., Wang, Y., & Liu, C. (2020). Quantitative Measurement of Melittin in Asian Honeybee Venom Using a New Method Including UPLC-QqTOF-MS. Toxins, 12(7), 437. https://doi.org/10.3390/toxins12070437