How do Uremic Toxins Affect the Endothelium?

 , , and
, , and 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
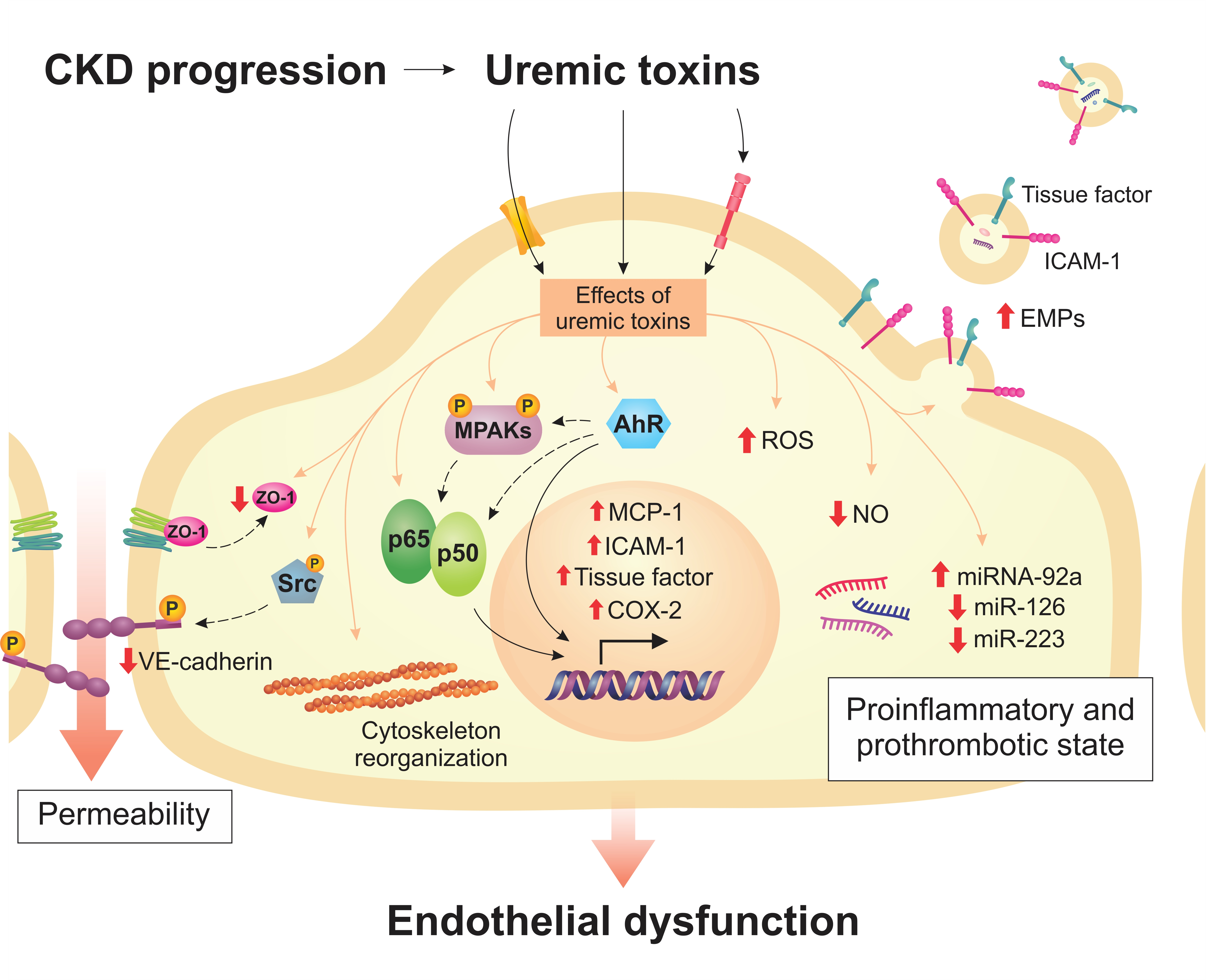
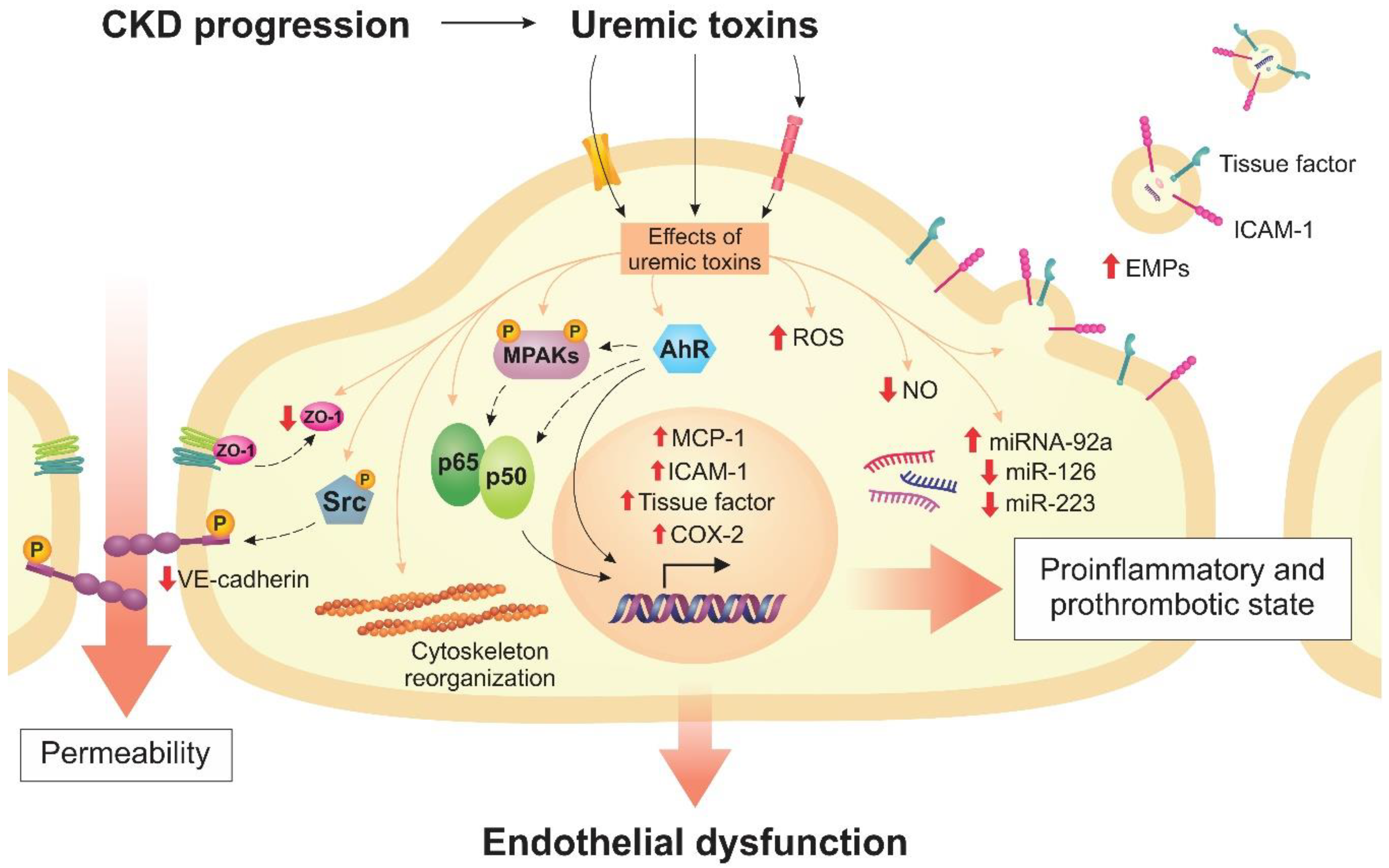
2. Endothelial Dysfunction in CKD
2.1. Uremic Toxins Induce Inflammation and Oxidative Stress in Endothelial Cells
2.2. Uremic Toxins Contribute to the Prothrombotic State of the Endothelium
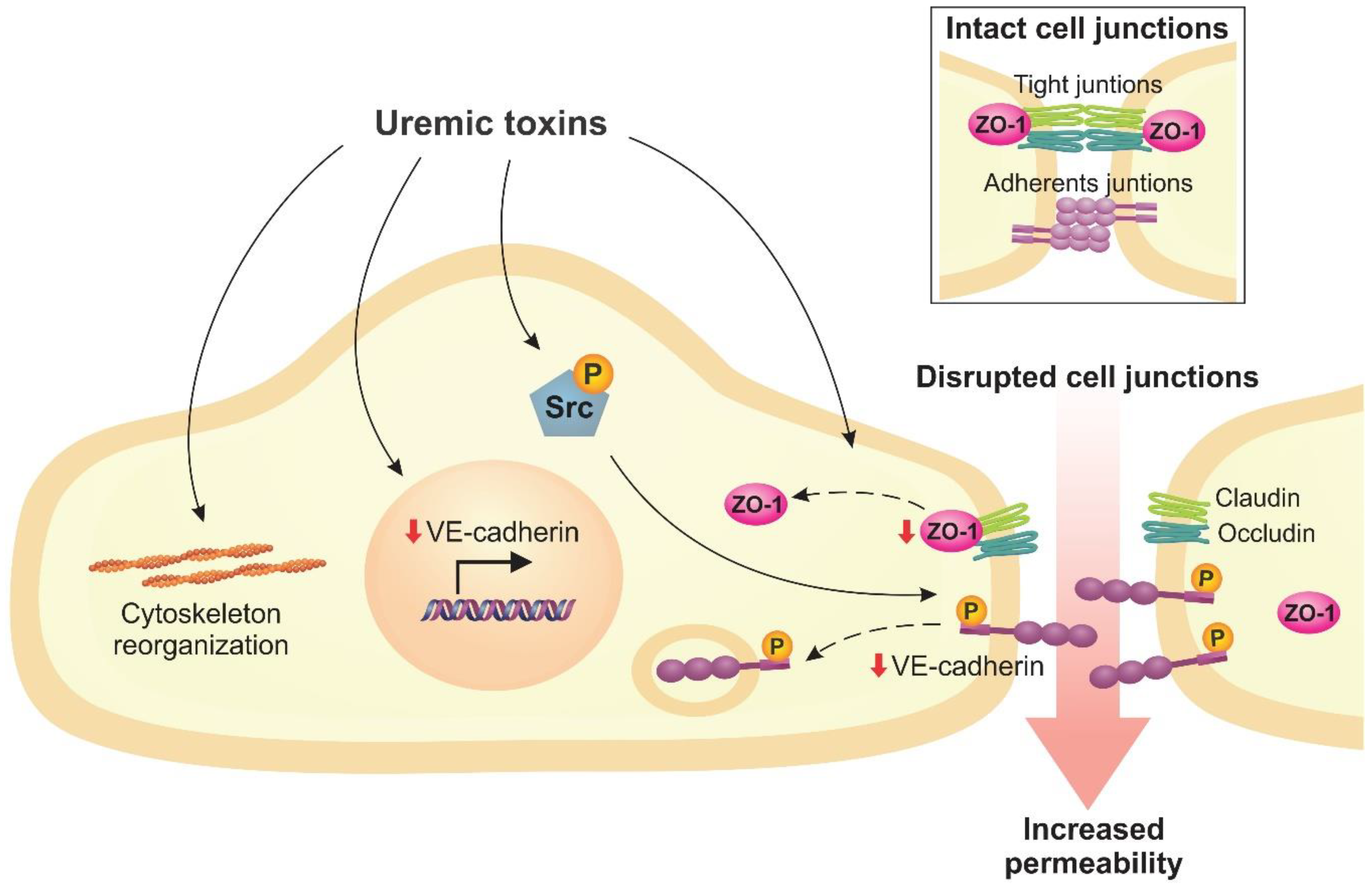
2.3. Uremic Toxins Increase Endothelial Permeability
2.4. Uremic Toxins Impairs Endothelial Protective Mechanisms
3. Transporters and Receptors of Uremic Toxins in the Endothelium
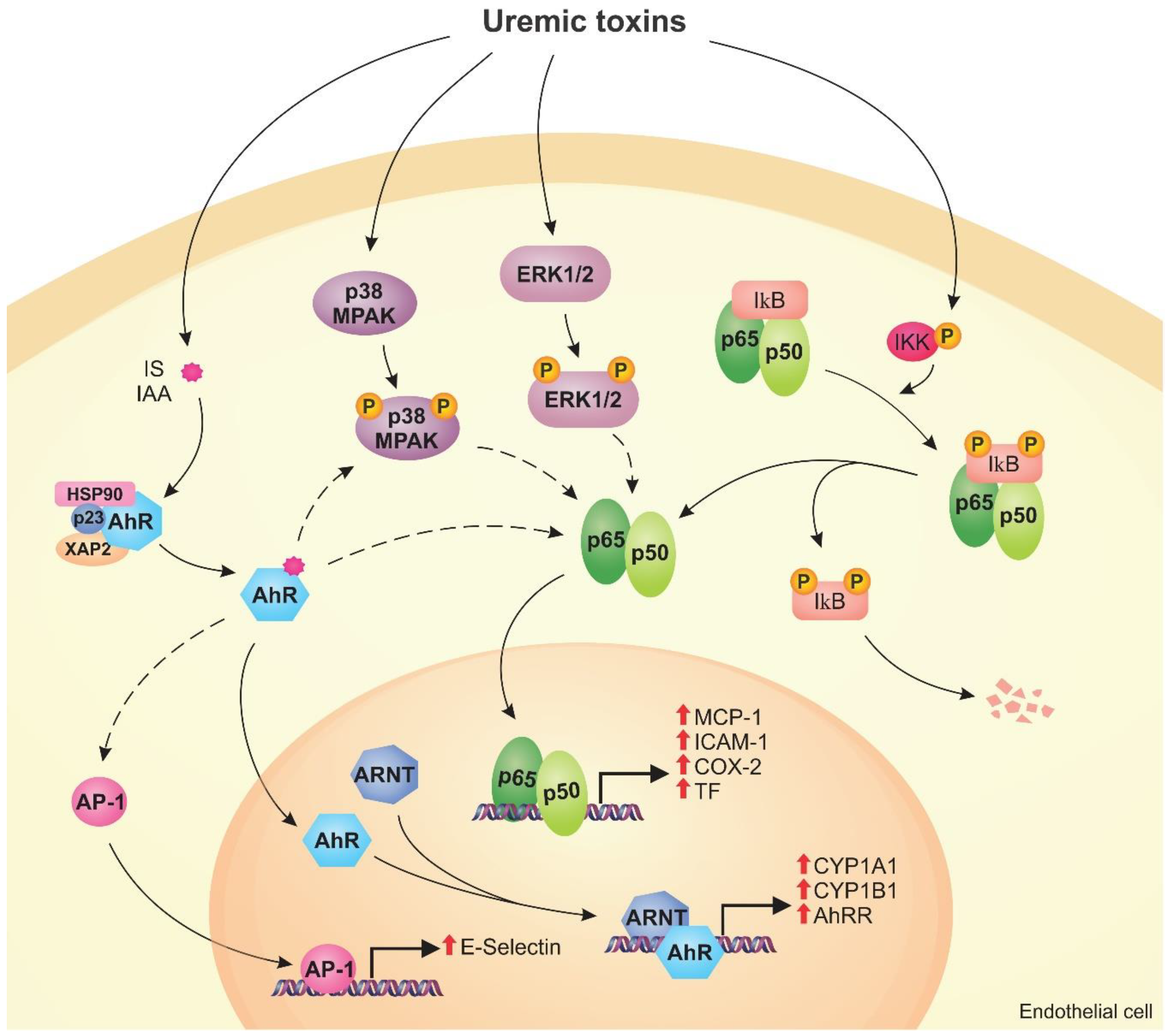
4. Cell Signaling Pathways Altered by Uremic Toxins
4.1. AhR Pathway
4.2. NF-κB Pathway
4.3. MAPK Pathway
5. Modulation of MicroRNAs by Uremic Toxins in Endothelial Cells
6. Uremic Toxins Induce the Formation of Endothelial Microparticles
7. Therapeutic Strategies for Uremic Toxins
8. Final Considerations
Author Contributions
Funding
Conflicts of Interest
References
- Jing, Y.J.; Ni, J.W.; Ding, F.H.; Fang, Y.H.; Wang, X.Q.; Wang, H.B.; Chen, X.N.; Chen, N.; Zhan, W.W.; Lu, L.; et al. p-Cresyl sulfate is associated with carotid arteriosclerosis in hemodialysis patients and promotes atherogenesis in apoE−/− mice. Kidney Int. 2016, 89, 439–449. [Google Scholar] [CrossRef]
- Wang, C.H.; Lai, Y.H.; Kuo, C.H.; Lin, Y.L.; Tsai, J.P.; Hsu, B.G. Association between serum indoxyl sulfate levels and endothelial function in non-dialysis chronic kidney disease. Toxins 2019, 11, 589. [Google Scholar] [CrossRef]
- Barreto, F.C.; Barreto, D.V.; Liabeuf, S.; Meert, N.; Glorieux, G.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A. Serum Indoxyl Sulfate Is Associated with Vascular Disease and Mortality in Chronic Kidney Disease Patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 1551–1558. [Google Scholar] [CrossRef]
- Wu, I.W.; Hsu, K.H.; Hsu, H.J.; Lee, C.C.; Sun, C.Y.; Tsai, C.J.; Wu, M.S. Serum free p-cresyl sulfate levels predict cardiovascular and all-cause mortality in elderly hemodialysis patients—A prospective cohort study. Nephrol. Dial. Transplant. 2012, 27, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Liabeuf, S.; Barreto, D.V.; Barreto, F.C.; Meert, N.; Glorieux, G.; Schepers, E.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z. A free p-cresylsulphate is a predictor of mortality in patients at different stages of chronic kidney disease. Nephrol. Dial. Transplant. 2010, 25, 1183–1191. [Google Scholar] [CrossRef] [PubMed]
- Rattazzi, M.; Villalta, S.; De Lucchi, L.; Sponchiado, A.; Galliazzo, S.; Faggin, E.; Pagliara, V.; Zilli, C.; Callegari, E.; Caberlotto, L.; et al. Chronic kidney disease is associated with increased risk of venous thromboembolism recurrence. Thromb. Res. 2017, 160, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Betriu, A.; Martinez-Alonso, M.; Arcidiacono, M.V.; Cannata-Andia, J.; Pascual, J.; Valdivielso, J.M.; Fernandez, E. Prevalence of subclinical atheromatosis and associated risk factors in chronic kidney disease: The NEFRONA study. Nephrol. Dial. Transplant. 2014, 29, 1415–1422. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; Pletinck, A.; Schepers, E.; Glorieux, G. Biochemical and Clinical Impact of Organic Uremic Retention Solutes: A Comprehensive Update. Toxins 2018, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Boelaert, J.; Lynen, F.; Glorieux, G.; Schepers, E.; Neirynck, N.; Vanholder, R. Metabolic profiling of human plasma and urine in chronic kidney disease by hydrophilic interaction liquid chromatography coupled with time-of-flight mass spectrometry: A pilot study. Anal. Bioanal. Chem. 2017, 409, 2201–2211. [Google Scholar] [CrossRef] [PubMed]
- Duranton, F.; Cohen, G.; De Smet, R.; Rodriguez, M.; Jankowski, J.; Vanholder, R.; Argiles, A. Normal and pathologic concentrations of uremic toxins. J. Am. Soc. Nephrol. 2012, 23, 1258–1270. [Google Scholar] [CrossRef]
- Okuno, S.; Ishimura, E.; Kohno, K.; Fujino-Katoh, Y.; Maeno, Y.; Yamakawa, T.; Inaba, M.; Nishizawa, Y. Serum β2-microglobulin level is a significant predictor of mortality in maintenance haemodialysis patients. Nephrol. Dial. Transplant. 2009, 24, 571–577. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, R.B.; Liabeuf, S.; Okazaki, H.; Lenglet, A.; Desjardins, L.; Lemke, H.-D.; Vanholder, R.; Choukroun, G.; Massy, Z.A. The clinical impact of plasma leptin levels in a cohort of chronic kidney disease patients. Clin. Kidney J. 2013, 6, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Maciel, R.; Cunha, R.; Busato, V.; Franco, C.; Gregório, P.; Dolenga, C.; Nakao, L.; Massy, Z.; Boullier, A.; Pecoits-Filho, R.; et al. Uremia Impacts VE-Cadherin and ZO-1 Expression in Human Endothelial Cell-to-Cell Junctions. Toxins 2018, 10, 404. [Google Scholar] [CrossRef]
- Favretto, G.; Souza, L.M.; Gregório, P.C.; Cunha, R.S.; Maciel, R.A.P.; Sassaki, G.L.; Toledo, M.G.; Pecoits-Filho, R.; Souza, W.M.; Stinghen, A.E.M. Role of Organic Anion Transporters in the Uptake of Protein-Bound Uremic Toxins by Human Endothelial Cells and Monocyte Chemoattractant Protein-1 Expression. J. Vasc. Res. 2017, 54, 170–179. [Google Scholar] [CrossRef]
- Chen, Z.; Peng, I.-C.; Sun, W.; Su, M.-I.; Hsu, P.-H.; Fu, Y.; Zhu, Y.; DeFea, K.; Pan, S.; Tsai, M.-D.; et al. AMP-Activated Protein Kinase Functionally Phosphorylates Endothelial Nitric Oxide Synthase Ser633. Circ. Res. 2009, 104, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Miao, H.; Hu, Y.-L.; Shiu, Y.-T.; Yuan, S.; Zhao, Y.; Kaunas, R.; Wang, Y.; Jin, G.; Usami, S.; Chien, S. Effects of Flow Patterns on the Localization and Expression of VE-Cadherin at Vascular Endothelial Cell Junctions: In vivo and in vitro Investigations. J. Vasc. Res. 2005, 42, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Amabile, N.; Guerin, A.P.; Leroyer, A.P.; Mallat, Z.; Nguyen, C.; Boddaert, J.; London, G.M.; Tedgui, A.; Boulanger, C.M. Circulating Endothelial Microparticles Are Associated with Vascular Dysfunction in Patients with End-Stage Renal Failure. J. Am. Soc. Nephrol. 2005, 16, 3381–3388. [Google Scholar] [CrossRef]
- Boulanger, C.M.; Amabile, N.; Guérin, A.P.; Pannier, B.; Leroyer, A.S.; Nguyen, C.; Mallat, Z.; Tedgui, A.; London, G.M. In vivo shear stress determines circulating levels of endothelial microparticles in end-stage renal disease. Hypertension 2007, 49, 902–908. [Google Scholar] [CrossRef]
- Verbeke, F.H.; Agharazii, M.; Boutouyrie, P.; Pannier, B.; Guérin, A.P.; London, G.M. Local Shear Stress and Brachial Artery Functions in End-Stage Renal Disease. J. Am. Soc. Nephrol. 2007, 18, 621–628. [Google Scholar] [CrossRef]
- Park, D.W.; Kruger, G.H.; Rubin, J.M.; Hamilton, J.; Gottschalk, P.; Dodde, R.E.; Shih, A.J.; Weitzel, W.F. In Vivo Vascular Wall Shear Rate and Circumferential Strain of Renal Disease Patients. Ultrasound Med. Biol. 2013, 39, 241–252. [Google Scholar] [CrossRef]
- Jourde-Chiche, N.; Dou, L.; Cerini, C.; Dignat-George, F.; Brunet, P. Vascular Incompetence in Dialysis Patients-Protein-Bound Uremic Toxins and Endothelial Dysfunction. Semin. Dial. 2011, 24, 327–337. [Google Scholar] [CrossRef]
- Eloueyk, A.; Osta, B.; Alameldinne, R.; Awad, D. Uremic Serum Induces Inflammation in Cultured Human Endothelial Cells and Triggers Vascular Repair Mechanisms. Inflammation 2019, 42, 2003–2010. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Yoo, T.H.; Hwang, Y.; Lee, G.H.; Kim, B.; Jang, J.; Yu, H.T.; Kim, M.C.; Cho, J.Y.; Lee, C.J.; et al. Indoxyl sulfate (IS)-mediated immune dysfunction provokes endothelial damage in patients with end-stage renal disease (ESRD). Sci. Rep. 2017, 7, 3057. [Google Scholar] [CrossRef] [PubMed]
- Jourde-Chiche, N.; Fakhouri, F.; Dou, L.; Bellien, J.; Burtey, S.; Frimat, M.; Jarrot, P.A.; Kaplanski, G.; Le Quintrec, M.; Pernin, V.; et al. Endothelium structure and function in kidney health and disease. Nat. Rev. Nephrol. 2019, 15, 87–108. [Google Scholar] [CrossRef]
- Vanholder, R.; De Smet, R. Pathophysiologic effects of uremic retention solutes. J. Am. Soc. Nephrol. 1999, 10, 1815–1823. [Google Scholar] [PubMed]
- Ito, S.; Osaka, M.; Edamatsu, T.; Itoh, Y.; Yoshida, M. Crucial Role of the Aryl Hydrocarbon Receptor (AhR) in Indoxyl Sulfate-Induced Vascular Inflammation. J. Atheroscler. Thromb. 2016, 23, 960–975. [Google Scholar] [CrossRef]
- Claro, L.; Moreno-Amaral, A.; Gadotti, A.; Dolenga, C.; Nakao, L.; Azevedo, M.; de Noronha, L.; Olandoski, M.; de Moraes, T.; Stinghen, A.; et al. The Impact of Uremic Toxicity Induced Inflammatory Response on the Cardiovascular Burden in Chronic Kidney Disease. Toxins 2018, 10, 384. [Google Scholar] [CrossRef]
- Maciel, R.A.P.; Rempel, L.C.T.; Bosquetti, B.; Finco, A.B.; Pecoits-Filho, R.; Souza, W.M.d.; Stinghen, A.E.M. p-cresol but not p-cresyl sulfate stimulate MCP-1 production via NF-κB p65 in human vascular smooth muscle cells. J. Bras. Nefrol. 2016, 38, 153–160. [Google Scholar] [CrossRef]
- Stinghen, A.E.M.; Gonçalves, S.M.; Martines, E.G.; Nakao, L.S.; Riella, M.C.; Aita, C.A.; Pecoits-Filho, R. Increased plasma and endothelial cell expression of chemokines and adhesion molecules in chronic kidney disease. Nephron Clin. Pract. 2009, 111, c117–c126. [Google Scholar] [CrossRef]
- Ribeiro, V.; Bosquetti, B.; Gonçalves, S.M.; Bucharles, S.G.E.; Rempel, L.; Maciel, R.A.P.; Oliveira, R.B.D.; Pecoits-Filho, R.; Stinghen, A.E.M. Uremic serum inhibits in vitro expression of chemokine SDF-1: Impact of uremic toxicity on endothelial injury. J. Bras. Nefrol. 2014, 36, 123–131. [Google Scholar] [CrossRef]
- Becherucci, F.; Mazzinghi, B.; Ronconi, E.; Peired, A.; Lazzeri, E.; Sagrinati, C.; Romagnani, P.; Lasagni, L. The role of endothelial progenitor cells in acute kidney injury. Blood Purif. 2009, 27, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Yuen, D.A.; Kuliszewski, M.A.; Liao, C.; Rudenko, D.; Leong-Poi, H.; Chan, C.T. Nocturnal hemodialysis is associated with restoration of early-outgrowth endothelial progenitor-like cell function. Clin. J. Am. Soc. Nephrol. 2011, 6, 1345–1353. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.-C.; Liang, C.-J.; Huang, T.-M.; Liu, C.-W.; Wang, S.-H.; Young, G.-H.; Tsai, J.-S.; Tseng, Y.-C.; Peng, Y.-S.; Wu, V.-C.; et al. Indoxyl sulfate enhances IL-1β-induced E-selectin expression in endothelial cells in acute kidney injury by the ROS/MAPKs/NFκB/AP-1 pathway. Arch. Toxicol. 2016, 90, 2779–2792. [Google Scholar] [CrossRef]
- Ito, S.; Osaka, M.; Higuchi, Y.; Nishijima, F.; Ishii, H.; Yoshida, M. Indoxyl sulfate induces leukocyte-endothelial interactions through up-regulation of E-selectin. J. Biol. Chem. 2010, 285, 38869–38875. [Google Scholar] [CrossRef]
- Six, I.; Gross, P.; Rémond, M.C.; Chillon, J.M.; Poirot, S.; Drueke, T.B.; Massy, Z.A. Deleterious vascular effects of indoxyl sulfate and reversal by oral adsorbent AST-120. Atherosclerosis 2015, 243, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Six, I.; Maizel, J.; Barreto, F.C.; Rangrez, A.Y.; Dupont, S.; Slama, M.; Tribouilloy, C.; Choukroun, G.; Maziere, J.C.; Bode-Boeger, S.; et al. Effects of phosphate on vascular function under normal conditions and influence of the uraemic state. Cardiovasc. Res. 2012, 96, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Wei, R.; Wang, Y.; Su, T.; Li, P.; Chen, X. The uremic toxin hippurate promotes endothelial dysfunction via the activation of Drp1-mediated mitochondrial fission. Redox Biol. 2018, 16, 303–313. [Google Scholar] [CrossRef]
- Masai, N.; Tatebe, J.; Yoshino, G.; Morita, T. Indoxyl sulfate stimulates monocyte chemoattractant protein-1 expression in human umbilical vein endothelial cells by inducing oxidative stress through activation of the NADPH oxidase-nuclear factor-kappaB pathway. Circ. J. 2010, 74, 2216–2224. [Google Scholar] [CrossRef]
- Koizumi, M.; Tatebe, J.; Watanabe, I.; Yamazaki, J.; Ikeda, T.; Morita, T. Aryl Hydrocarbon Receptor Mediates Indoxyl Sulfate-Induced Cellular Senescence in Human Umbilical Vein Endothelial Cells. J. Atheroscler. Thromb. 2014, 21, 904–916. [Google Scholar] [CrossRef]
- Pei, J.; Juni, R.; Harakalova, M.; Duncker, D.J.; Asselbergs, F.W.; Koolwijk, P.; van Hinsbergh, V.; Verhaar, M.C.; Mokry, M.; Cheng, C. Indoxyl Sulfate Stimulates Angiogenesis by Regulating Reactive Oxygen Species Production via CYP1B1. Toxins 2019, 11, 454. [Google Scholar] [CrossRef]
- Tbahriti, H.F.; Kaddous, A.; Bouchenak, M.; Mekki, K. Effect of Different Stages of Chronic Kidney Disease and Renal Replacement Therapies on Oxidant-Antioxidant Balance in Uremic Patients. Biochem. Res. Int. 2013, 2013, 358985. [Google Scholar] [CrossRef] [PubMed]
- Pieniazek, A.; Gwozdzinski, L.; Hikisz, P.; Gwozdzinski, K. Indoxyl Sulfate Generates Free Radicals, Decreases Antioxidant Defense, and Leads to Damage to Mononuclear Blood Cells. Chem. Res. Toxicol. 2018, 31, 869–875. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Gua, C.; Wu, B.; Chen, Y. Increased circulating trimethylamine N-oxide contributes to endothelial dysfunction in a rat model of chronic kidney disease. Biochem. Biophys. Res. Commun. 2018, 495, 2071–2077. [Google Scholar] [CrossRef]
- Dou, L.; Jourde-Chiche, N.; Faure, V.; Cerini, C.; Berland, Y.; Dignat-George, F.; Brunet, P. The uremic solute indoxyl sulfate induces oxidative stress in endothelial cells. J. Thromb. Haemost. 2007, 5, 1302–1308. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.-C.; Li, L.-C.; Chen, J.-B.; Chang, H.-W. Indoxyl Sulfate-Induced Oxidative Stress, Mitochondrial Dysfunction, and Impaired Biogenesis Are Partly Protected by Vitamin C and N-Acetylcysteine. Sci. World J. 2015, 2015, 620826. [Google Scholar] [CrossRef] [PubMed]
- Wassmann, S.; Wassmann, K.; Nickenig, G. Modulation of Oxidant and Antioxidant Enzyme Expression and Function in Vascular Cells. Hypertension 2004, 44, 381–386. [Google Scholar] [CrossRef]
- Incalza, M.A.; D’Oria, R.; Natalicchio, A.; Perrini, S.; Laviola, L.; Giorgino, F. Oxidative stress and reactive oxygen species in endothelial dysfunction associated with cardiovascular and metabolic diseases. Vascul. Pharmacol. 2018, 100, 1–19. [Google Scholar] [CrossRef]
- Jin, L.; Jagatheesan, G.; Lynch, J.; Guo, L.; Conklin, D.J. Crotonaldehyde-induced vascular relaxation and toxicity: Role of endothelium and transient receptor potential ankyrin-1 (TRPA1). Toxicol. Appl. Pharmacol. 2020, 398, 115012. [Google Scholar] [CrossRef]
- Benítez-Angeles, M.; Morales-Lázaro, S.L.; Juárez-González, E.; Rosenbaum, T. TRPV1: Structure, Endogenous Agonists, and Mechanisms. Int. J. Mol. Sci. 2020, 21, 3421. [Google Scholar] [CrossRef]
- Chen, Y.-L.; Sonkusare, S.K. Endothelial TRPV4 channels and vasodilator reactivity. In Current Topics in Membranes; Elsevier Inc.: East Lansing, MI, USA, 2020; pp. 89–117. [Google Scholar] [CrossRef]
- Wang, Y.; Cui, L.; Xu, H.; Liu, S.; Zhu, F.; Yan, F.; Shen, S.; Zhu, M. TRPV1 agonism inhibits endothelial cell inflammation via activation of eNOS/NO pathway. Atherosclerosis 2017, 260, 13–19. [Google Scholar] [CrossRef]
- Rodrigues, S.D.; Santos, S.S.; Meireles, T.; Romero, N.; Glorieux, G.; Pecoits-Filho, R.; Zhang, D.D.; Nakao, L.S. Uremic toxins promote accumulation of oxidized protein and increased sensitivity to hydrogen peroxide in endothelial cells by impairing the autophagic flux. Biochem. Biophys. Res. Commun. 2020, 523, 123–129. [Google Scholar] [CrossRef] [PubMed]
- LaRocca, T.J.; Henson, G.D.; Thorburn, A.; Sindler, A.L.; Pierce, G.L.; Seals, D.R. Translational evidence that impaired autophagy contributes to arterial ageing. J. Physiol. 2012, 590, 3305–3316. [Google Scholar] [CrossRef] [PubMed]
- Grootaert, M.O.J.; Roth, L.; Schrijvers, D.M.; De Meyer, G.R.Y.; Martinet, W. Defective Autophagy in Atherosclerosis: To Die or to Senesce? Oxid. Med. Cell. Longev. 2018, 2018, 7687083. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.-J.; Wei, R.; Wang, Y.; Su, T.; Di, P.; Li, Q.; Yang, X.; Li, P.; Chen, X. Blood coagulation system in patients with chronic kidney disease: A prospective observational study. BMJ Open 2017, 7, e014294. [Google Scholar] [CrossRef]
- Wattanakit, K.; Cushman, M.; Stehman-Breen, C.; Heckbert, S.R.; Folsom, A.R. Chronic kidney disease increases risk for venous thromboembolism. J. Am. Soc. Nephrol. 2008, 19, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Folsom, A.R.; Lutsey, P.L.; Astor, B.C.; Wattanakit, K.; Heckbert, S.R.; Cushman, M. Chronic kidney disease and venous thromboembolism: A prospective study. Nephrol. Dial. Transplant. 2010, 25, 3296–3301. [Google Scholar] [CrossRef]
- Kamiński, T.W.; Pawlak, K.; Karbowska, M.; Myśliwiec, M.; Pawlak, D. Indoxyl sulfate—The uremic toxin linking hemostatic system disturbances with the prevalence of cardiovascular disease in patients with chronic kidney disease. BMC Nephrol. 2017, 18, 35. [Google Scholar] [CrossRef]
- Chen, J.; Hamm, L.L.; Mohler, E.R.; Hudaihed, A.; Arora, R.; Chen, C.-S.; Liu, Y.; Browne, G.; Mills, K.T.; Kleinpeter, M.A.; et al. Interrelationship of Multiple Endothelial Dysfunction Biomarkers with Chronic Kidney Disease. PLoS ONE 2015, 10, e0132047. [Google Scholar]
- Grover, S.P.; Mackman, N. Tissue Factor: An Essential Mediator of Hemostasis and Trigger of Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 709–725. [Google Scholar] [CrossRef]
- Gondouin, B.; Cerini, C.; Dou, L.; Sallée, M.; Duval-Sabatier, A.; Pletinck, A.; Calaf, R.; Lacroix, R.; Jourde-Chiche, N.; Poitevin, S.; et al. Indolic uremic solutes increase tissue factor production in endothelial cells by the aryl hydrocarbon receptor pathway. Kidney Int. 2013, 84, 733–744. [Google Scholar] [CrossRef]
- Addi, T.; Poitevin, S.; McKay, N.; El Mecherfi, K.E.; Kheroua, O.; Jourde-Chiche, N.; de Macedo, A.; Gondouin, B.; Cerini, C.; Brunet, P.; et al. Mechanisms of tissue factor induction by the uremic toxin indole-3 acetic acid through aryl hydrocarbon receptor/nuclear factor-kappa B signaling pathway in human endothelial cells. Arch. Toxicol. 2019, 93, 121–136. [Google Scholar] [CrossRef] [PubMed]
- Roberts, W.; Michno, A.; Aburima, A.; Nassem, K.M. Nitric oxide inhibits von Willebrand factor-mediated platelet adhesion and spreading through regulation of integrin Î ± IIb Î2 3 and myosin light chain. J. Thromb. Haemost. 2009, 7, 2106–2115. [Google Scholar] [CrossRef] [PubMed]
- Stinghen, A.E.M.; Chillon, J.M.; Massy, Z.A.; Boullier, A. Differential effects of indoxyl sulfate and inorganic phosphate in a murine cerebral endothelial cell line (bEnd.3). Toxins 2014, 6, 1742–1760. [Google Scholar] [CrossRef] [PubMed]
- Tumur, Z.; Niwa, T. Indoxyl sulfate inhibits nitric oxide production and cell viability by inducing oxidative stress in vascular endothelial cells. Am. J. Nephrol. 2009, 29, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Stevens, K.K.; Denby, L.; Patel, R.K.; Mark, P.B.; Kettlewell, S.; Smith, G.L.; Clancy, M.J.; Delles, C.; Jardine, A.G. Deleterious effects of phosphate on vascular and endothelial function via disruption to the nitric oxide pathway. Nephrol. Dial. Transplant. 2016, 32, gfw252. [Google Scholar] [CrossRef]
- Shafi, T.; Hostetter, T.H.; Meyer, T.W.; Hwang, S.; Hai, X.; Melamed, M.L.; Banerjee, T.; Coresh, J.; Powe, N.R. Serum Asymmetric and Symmetric Dimethylarginine and Morbidity and Mortality in Hemodialysis Patients. Am. J. Kidney Dis. 2017, 70, 48–58. [Google Scholar] [CrossRef]
- Kajimoto, H.; Kai, H.; Aoki, H.; Yasuoka, S.; Anegawa, T.; Aoki, Y.; Ueda, S.; Okuda, S.; Imaizumi, T. Inhibition of eNOS phosphorylation mediates endothelial dysfunction in renal failure: New effect of asymmetric dimethylarginine. Kidney Int. 2012, 81, 762–768. [Google Scholar] [CrossRef]
- Gross, S.; Tilly, P.; Hentsch, D.; Vonesch, J.L.; Fabre, J.E. Vascular wall-produced prostaglandin E2 exacerbates arterial thrombosis and atherothrombosis through platelet EP3 receptors. J. Exp. Med. 2007, 204, 311–320. [Google Scholar] [CrossRef]
- Dou, L.; Sallée, M.; Cerini, C.; Poitevin, S.; Gondouin, B.; Jourde-Chiche, N.; Fallague, K.; Brunet, P.; Calaf, R.; Dussol, B.; et al. The cardiovascular effect of the uremic solute indole-3 acetic acid. J. Am. Soc. Nephrol. 2015, 26, 876–887. [Google Scholar] [CrossRef]
- Karbowska, M.; Kaminski, T.W.; Marcinczyk, N.; Misztal, T.; Rusak, T.; Smyk, L.; Pawlak, D. The uremic toxin indoxyl sulfate accelerates thrombotic response after vascular injury in animal models. Toxins 2017, 9, 229. [Google Scholar] [CrossRef]
- Karbowska, M.; Kaminski, T.W.; Znorko, B.; Domaniewski, T.; Misztal, T.; Rusak, T.; Pryczynicz, A.; Guzinska-Ustymowicz, K.; Pawlak, K.; Pawlak, D. Indoxyl Sulfate Promotes Arterial Thrombosis in Rat Model via Increased Levels of Complex TF/VII, PAI-1, Platelet Activation as Well as Decreased Contents of SIRT1 and SIRT3. Front. Physiol. 2018, 9, 1623. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, T.W.; Pawlak, K.; Karbowska, M.; Mysliwiec, M.; Grzegorzewski, W.; Kuna, J.; Pawlak, D. Association between uremic toxin-anthranilic acid and fibrinolytic system activity in predialysis patients at different stages of chronic kidney disease. Int. Urol. Nephrol. 2018, 50, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Lutz, J.; Menke, J.; Sollinger, D.; Schinzel, H.; Thürmel, K. Haemostasis in chronic kidney disease. Nephrol. Dial. Transplant. 2014, 29, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.C.; Huang, S.Y.; Wu, C.C.; Hsu, C.F. P-Cresylsulfate, the Protein-Bound Uremic Toxin, Increased Endothelial Permeability Partly Mediated by Src-Induced Phosphorylation of VE-Cadherin. Toxins 2020, 12, 62. [Google Scholar] [CrossRef] [PubMed]
- Cuenca, M.V.; Ferrantelli, E.; Meinster, E.; Pouw, S.M.; Kovačević, I.; de Menezes, R.X.; Niessen, H.W.; Beelen, R.H.J.; Hordijk, P.L.; Vervloet, M.G. Vitamin D attenuates endothelial dysfunction in uremic rats and maintains human endothelial stability. J. Am. Heart Assoc. 2018, 7, e008776. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.; Wang, C.P.; Yu, T.H.; Tai, P.Y.; Liang, S.S.; Hung, W.C.; Wu, C.C.; Huang, S.H.; Lee, Y.J.; Chen, S.C. Protein-bounded uremic toxin p-cresylsulfate induces vascular permeability alternations. Histochem. Cell Biol. 2018, 149, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Assefa, E.G.; Yan, Q.; Gezahegn, S.B.; Salissou, M.T.M.; He, S.; Wu, N.; Zuo, X.; Ying, C. Role of resveratrol on indoxyl sulfate-induced endothelial hyperpermeability via aryl hydrocarbon receptor (AHR)/Src-dependent pathway. Oxid. Med. Cell. Longev. 2019, 2019. [Google Scholar] [CrossRef]
- Cuenca, M.V.; van Bezu, J.; Beelen, R.H.J.; Vervloet, M.G.; Hordijk, P.L. Stabilization of cell–cell junctions by active vitamin D ameliorates uraemia-induced loss of human endothelial barrier function. Nephrol. Dial. Transplant. 2019, 34, 252–264. [Google Scholar] [CrossRef]
- Peng, Y.S.; Lin, Y.T.; Chen, Y.; Hung, K.Y.; Wang, S.M. Effects of indoxyl sulfate on adherens junctions of endothelial cells and the underlying signaling mechanism. J. Cell. Biochem. 2012, 113, 1034–1043. [Google Scholar] [CrossRef]
- Orsenigo, F.; Giampietro, C.; Ferrari, A.; Corada, M.; Galaup, A.; Sigismund, S.; Ristagno, G.; Maddaluno, L.; Koh, G.Y.; Franco, D.; et al. Phosphorylation of VE-cadherin is modulated by haemodynamic forces and contributes to the regulation of vascular permeability in vivo. Nat. Commun. 2012, 3, 1–15. [Google Scholar] [CrossRef]
- Lampugnani, M.G.; Dejana, E. Adherens junctions in endothelial cells regulate vessel maintenance and angiogenesis. Thromb. Res. 2007, 120, S1–S6. [Google Scholar] [CrossRef]
- Tornavaca, O.; Chia, M.; Dufton, N.; Almagro, L.O.; Conway, D.E.; Randi, A.M.; Schwartz, M.A.; Matter, K.; Balda, M.S. ZO-1 controls endothelial adherens junctions, cell-cell tension, angiogenesis, and barrier formation. J. Cell Biol. 2015, 208, 821–838. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Endothelial barrier and its abnormalities in cardiovascular disease. Front. Physiol. 2015, 6, 365. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.J.; Wu, C.J.; Wu, P.C.; Pan, C.F.; Wang, T.J.; Sun, F.J.; Liu, H.L.; Chen, H.H.; Yeh, H.I. Indoxyl Sulfate Impairs Endothelial Progenitor Cells and Might Contribute to Vascular Dysfunction in Patients with Chronic Kidney Disease. Kidney Blood Press. Res. 2016, 41, 1025–1036. [Google Scholar] [CrossRef]
- Choi, J.H.; Kim, K.L.; Huh, W.; Kim, B.; Byun, J.; Suh, W.; Sung, J.; Jeon, E.S.; Oh, H.Y.; Kim, D.K. Decreased number and impaired angiogenic function of endothelial progenitor cells in patients with chronic renal failure. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1246–1252. [Google Scholar] [CrossRef]
- Jourde-Chiche, N.; Dou, L.; Sabatier, F.; Calaf, R.; Cerini, C.; Robert, S.; Camoin-Jau, L.; Charpiot, P.; Argiles, A.; Dignat-George, F.; et al. Levels of circulating endothelial progenitor cells are related to uremic toxins and vascular injury in hemodialysis patients. J. Thromb. Haemost. 2009, 7, 1576–1584. [Google Scholar] [CrossRef]
- De Groot, K.; Hermann Bahlmann, F.; Sowa, J.; Koenig, J.; Menne, J.; Haller, H.; Fliser, D. Uremia causes endothelial progenitor cell deficiency. Kidney Int. 2004, 66, 641–646. [Google Scholar] [CrossRef]
- Jie, K.E.; Zaikova, M.A.; Bergevoet, M.W.T.; Westerweel, P.E.; Rastmanesh, M.; Blankestijn, P.J.; Boer, W.H.; Braam, B.; Verhaar, M.C. Progenitor cells and vascular function are impaired in patients with chronic kidney disease. Nephrol. Dial. Transplant. 2010, 25, 1875–1882. [Google Scholar] [CrossRef]
- Saum, K.; Campos, B.; Celdran-Bonafonte, D.; Nayak, L.; Sangwung, P.; Thakar, C.; Roy-Chaudhury, P.; Owens, A.P. Uremic Advanced Glycation End Products and Protein-Bound Solutes Induce Endothelial Dysfunction Through Suppression of Krüppel-Like Factor 2. J. Am. Heart Assoc. 2018, 7, e007566. [Google Scholar] [CrossRef]
- Shang, F.; Wang, S.-C.; Hsu, C.-Y.; Miao, Y.; Martin, M.; Yin, Y.; Wu, C.-C.; Wang, Y.-T.; Wu, G.; Chien, S.; et al. MicroRNA-92a Mediates Endothelial Dysfunction in CKD. J. Am. Soc. Nephrol. 2017, 28, 3251–3261. [Google Scholar] [CrossRef]
- Six, I.; Okazaki, H.; Gross, P.; Cagnard, J.; Boudot, C.; Maizel, J.; Drueke, T.B.; Massy, Z.A. Direct, acute effects of Klotho and FGF23 on vascular smooth muscle and endothelium. PLoS ONE 2014, 9, e93423. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, Y.; Ishikawa, K.; Yasuda, O.; Oguro, R.; Hanasaki, H.; Kida, I.; Takemura, Y.; Ohishi, M.; Katsuya, T.; Rakugi, H. Klotho suppresses TNF-α-induced expression of adhesion molecules in the endothelium and attenuates NF-κB activation. Endocrine 2009, 35, 341–346. [Google Scholar] [CrossRef]
- Kitagawa, M.; Sugiyama, H.; Morinaga, H.; Inoue, T.; Takiue, K.; Ogawa, A.; Yamanari, T.; Kikumoto, Y.; Uchida, H.A.; Kitamura, S.; et al. A Decreased Level of Serum Soluble Klotho Is an Independent Biomarker Associated with Arterial Stiffness in Patients with Chronic Kidney Disease. PLoS ONE 2013, 8, e56695. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.-Y.; Chang, S.-C.; Wu, M.-S. Suppression of Klotho expression by protein-bound uremic toxins is associated with increased DNA methyltransferase expression and DNA hypermethylation. Kidney Int. 2012, 81, 640–650. [Google Scholar] [CrossRef] [PubMed]
- Burckhardt, G. Drug transport by Organic Anion Transporters (OATs). Pharmacol. Ther. 2012, 136, 106–130. [Google Scholar] [CrossRef]
- Nigam, S.K.; Bush, K.T.; Martovetsky, G.; Ahn, S.-Y.; Liu, H.C.; Richard, E.; Bhatnagar, V.; Wu, W. The Organic Anion Transporter (OAT) Family: A Systems Biology Perspective. Physiol. Rev. 2015, 95, 83–123. [Google Scholar] [CrossRef]
- Deanfield, J.E.; Halcox, J.P.; Rabelink, T.J. Endothelial function and dysfunction: Testing and clinical relevance. Circulation 2007, 115, 1285–1295. [Google Scholar] [CrossRef]
- Yazdani, S.; Jaldin-Fincati, J.R.; Pereira, R.V.S.; Klip, A. Endothelial cell barriers: Transport of molecules between blood and tissues. Traffic 2019, 20, 390–403. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Watanabe, H.; Noguchi, T.; Kotani, S.; Nakajima, M.; Kadowaki, D.; Otagiri, M.; Maruyama, T. Organic anion transporters play an important role in the uptake of p-cresyl sulfate, a uremic toxin, in the kidney. Nephrol. Dial. Transplant. 2011, 26, 2498–2502. [Google Scholar] [CrossRef]
- Deguchi, T.; Kusuhara, H.; Takadate, A.; Endou, H.; Otagiri, M.; Sugiyama, Y. Characterization of uremic toxin transport by organic anion transporters in the kidney. Kidney Int. 2004, 65, 162–174. [Google Scholar] [CrossRef]
- Uchiyama, H.; Tsujimoto, M.; Kimura, A.; Yuki, E.; Saiki, T.; Yoshida, T.; Furukubo, T.; Izumi, S.; Yamakawa, T.; Tachiki, H.; et al. Effects of Uremic Serum Residue on OATP1B1- and OATP1B3-Mediated Pravastatin Uptake in OATP-Expressing HEK293 Cells and Human Hepatocytes. Ther. Apher. Dial. 2019, 23, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Hagenbuch, B.; Stieger, B. The SLCO (former SLC21) superfamily of transporters. Mol. Aspects Med. 2013, 34, 396–412. [Google Scholar] [CrossRef] [PubMed]
- Grube, M.; Kock, K.; Oswald, S.; Draber, K.; Meissner, K.; Eckel, L.; Bohm, M.; Felix, S.; Vogelgesang, S.; Jedlitschky, G.; et al. Organic anion transporting polypeptide 2B1 is a high-affinity transporter for atorvastatin and is expressed in the human heart. Clin. Pharmacol. Ther. 2006, 80, 607–620. [Google Scholar] [CrossRef]
- Bronger, H.; König, J.; Kopplow, K.; Steiner, H.-H.; Ahmadi, R.; Herold-Mende, C.; Keppler, D.; Nies, A.T. ABCC Drug Efflux Pumps and Organic Anion Uptake Transporters in Human Gliomas and the Blood-Tumor Barrier. Cancer Res. 2005, 65, 11419–11428. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Katsuki, S.; Chen, M.; Decano, J.L.; Halu, A.; Lee, L.H.; Pestana, D.V.S.; Kum, A.S.T.; Kuromoto, R.K.; Golden, W.S.; et al. Uremic Toxin Indoxyl Sulfate Promotes Proinflammatory Macrophage Activation Via the Interplay of OATP2B1 and Dll4-Notch Signaling. Circulation 2019, 139, 78–96. [Google Scholar] [CrossRef] [PubMed]
- Van Aubel, R.A.M.H.; Smeets, P.H.E.; van den Heuvel, J.J.M.W.; Russel, F.G.M. Human organic anion transporter MRP4 (ABCC4) is an efflux pump for the purine end metabolite urate with multiple allosteric substrate binding sites. Am. J. Physiol. Physiol. 2005, 288, F327–F333. [Google Scholar] [CrossRef]
- Mutsaers, H.A.M.; van den Heuvel, L.P.; Ringens, L.H.J.; Dankers, A.C.A.; Russel, F.G.M.; Wetzels, J.F.M.; Hoenderop, J.G.; Masereeuw, R. Uremic toxins inhibit transport by breast cancer resistance protein and multidrug resistance protein 4 at clinically relevant concentrations. PLoS ONE 2011, 6, e18438. [Google Scholar] [CrossRef]
- Morimoto, K.; Tominaga, Y.; Agatsuma, Y.; Miyamoto, M.; Kashiwagura, S.; Takahashi, A.; Sano, Y.; Yano, K.; Kakinuma, C.; Ogihara, T.; et al. Intestinal secretion of indoxyl sulfate as a possible compensatory excretion pathway in chronic kidney disease. Biopharm. Drug Dispos. 2018, 39, 328–334. [Google Scholar] [CrossRef]
- Voormolen, N.; Noordzij, M.; Grootendorst, D.C.; Beetz, I.; Sijpkens, Y.W.; Van Manen, J.G.; Boeschoten, E.W.; Huisman, R.M.; Krediet, R.T.; Dekker, F.W.; et al. High plasma phosphate as a risk factor for decline in renal function and mortality in pre-dialysis patients. Nephrol. Dial. Transplant. 2007, 22, 2909–2916. [Google Scholar] [CrossRef]
- Inden, M.; Iriyama, M.; Takagi, M.; Kaneko, M.; Hozumi, I. Localization of type-III sodium-dependent phosphate transporter 2 in the mouse brain. Brain Res. 2013, 1531, 75–83. [Google Scholar] [CrossRef]
- Inden, M.; Iriyama, M.; Zennami, M.; Sekine, S.I.; Hara, A.; Yamada, M.; Hozumi, I. The type III transporters (PiT-1 and PiT-2) are the major sodium-dependent phosphate transporters in the mice and human brains. Brain Res. 2016, 1637, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Abbasian, N.; Burton, J.O.; Herbert, K.E.; Tregunna, B.-E.; Brown, J.R.; Ghaderi-Najafabadi, M.; Brunskill, N.J.; Goodall, A.H.; Bevington, A. Hyperphosphatemia, Phosphoprotein Phosphatases, and Microparticle Release in Vascular Endothelial Cells. J. Am. Soc. Nephrol. 2015, 26, 2152–2162. [Google Scholar] [CrossRef] [PubMed]
- Gross, P.; Six, I.; Kamel, S.; Massy, Z.A. Vascular Toxicity of Phosphate in Chronic Kidney Disease. Circ. J. 2014, 78, 2339–2346. [Google Scholar] [CrossRef] [PubMed]
- Rempel, L.; Finco, A.; Maciel, R.; Bosquetti, B.; Alvarenga, L.; Souza, W.; Pecoits-Filho, R.; Stinghen, A. Effect of PKC-β Signaling Pathway on Expression of MCP-1 and VCAM-1 in Different Cell Models in Response to Advanced Glycation End Products (AGEs). Toxins 2015, 7, 1722–1737. [Google Scholar] [CrossRef] [PubMed]
- Stinghen, A.E.M.; Massy, Z.A.; Vlassara, H.; Striker, G.E.; Boullier, A. Uremic Toxicity of Advanced Glycation End Products in CKD. J. Am. Soc. Nephrol. 2015, 27, 354–370. [Google Scholar] [CrossRef]
- Lin, C.J.; Lin, J.; Pan, C.F.; Chuang, C.K.; Liu, H.L.; Sun, F.J.; Wang, T.J.; Chen, H.H.; Wu, C.J. Indoxyl sulfate, not P-cresyl sulfate, is associated with advanced glycation end products in patients on long-term hemodialysis. Kidney Blood Press. Res. 2015, 40, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Finco, A.B.; Machado-de-Ávila, R.A.; Maciel, R.; De Moura, J.; Billiald, P.; Stinghen, A.E.M.; Alvarenga, L.M. Generation and characterization of monoclonal antibody against Advanced Glycation End Products in chronic kidney disease. Biochem. Biophys. Rep. 2016, 6, 142–148. [Google Scholar] [CrossRef]
- Ren, X.; Ren, L.; Wei, Q.; Shao, H.; Chen, L.; Liu, N. Advanced glycation end-products decreases expression of endothelial nitric oxide synthase through oxidative stress in human coronary artery endothelial cells. Cardiovasc. Diabetol. 2017, 16, 52. [Google Scholar] [CrossRef]
- Martens, R.J.H.; Broers, N.J.H.; Canaud, B.; Christiaans, M.H.L.; Cornelis, T.; Gauly, A.; Hermans, M.M.H.; Konings, C.J.A.M.; Van Der Sande, F.M.; Scheijen, J.L.J.M.; et al. Relations of advanced glycation endproducts and dicarbonyls with endothelial dysfunction and low-grade inflammation in individuals with end-stage renal disease in the transition to renal replacement therapy: A cross-sectional observational study. PLoS ONE 2019, 14, 1–18. [Google Scholar] [CrossRef]
- Chuah, Y.K.; Basir, R.; Talib, H.; Tie, T.H.; Nordin, N. Receptor for advanced glycation end products and its involvement in inflammatory diseases. Int. J. Inflam. 2013, 2013. [Google Scholar] [CrossRef]
- Alvarez, E.; Paradela-Dobarro, B.; González-Peteiro, M.; González-Juanatey, J.R. Impact of Advanced Glycation End Products on Endothelial Function and Their Potential Link to Atherosclerosis. Endothel. Dysfunct. Old Concepts New Chall. 2018, 2018, 211. [Google Scholar] [CrossRef]
- Burr, S.D.; Harmon, M.B.; Stewart , J.A., Jr. The Impact of Diabetic Conditions and AGE/RAGE Signaling on Cardiac Fibroblast Migration. Front. Cell Dev. Biol. 2020, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-C.; Lee, A.-S.; Liu, S.-H.; Chang, K.-C.; Shen, M.-Y.; Chang, C.-T. Spironolactone ameliorates endothelial dysfunction through inhibition of the AGE/RAGE axis in a chronic renal failure rat model. BMC Nephrol. 2019, 20, 351. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Thornalley, P.J. Advanced glycation end products in the pathogenesis of chronic kidney disease. Kidney Int. 2018, 93, 803–813. [Google Scholar] [CrossRef]
- Belmokhtar, K.; Ortillon, J.; Jaisson, S.; Massy, Z.A.; Boulagnon Rombi, C.; Doué, M.; Maurice, P.; Fritz, G.; Gillery, P.; Schmidt, A.M.; et al. Receptor for advanced glycation end products: A key molecule in the genesis of chronic kidney disease vascular calcification and a potential modulator of sodium phosphate co-transporter PIT-1 expression. Nephrol. Dial. Transplant. 2019, 34, 2018–2030. [Google Scholar] [CrossRef]
- Taguchi, K.; Elias, B.C.; Brooks, C.R.; Ueda, S.; Fukami, K. Uremic toxin-targeting as a therapeutic strategy for preventing cardiorenal syndrome. Circ. J. 2019, 84, 2–8. [Google Scholar] [CrossRef]
- Gregório, P.C.; Favretto, G.; Sassaki, G.L.; Cunha, R.S.; Becker-Finco, A.; Pecoits-Filho, R.; Souza, W.M.; Barreto, F.C.; Stinghen, A.E.M. Sevelamer reduces endothelial inflammatory response to advanced glycation end products. Clin. Kidney J. 2017, 10, 89–98. [Google Scholar] [CrossRef]
- Gao, X.; Zhang, H.; Schmidt, A.M.; Zhang, C. AGE/RAGE produces endothelial dysfunction in coronary arterioles in Type 2 diabetic mice. Am. J. Physiol. Hear. Circ. Physiol. 2008, 295, 491–499. [Google Scholar] [CrossRef]
- Kay, A.M.; Simpson, C.L.; Stewart, J.A. The Role of AGE/RAGE Signaling in Diabetes-Mediated Vascular Calcification. J. Diabetes Res. 2016, 2016, 6809703. [Google Scholar] [CrossRef]
- Cai, W.; Duan, X.M.; Liu, Y.; Yu, J.; Tang, Y.L.; Liu, Z.L.; Jiang, S.; Zhang, C.P.; Liu, J.Y.; Xu, J.X. Uric Acid Induces Endothelial Dysfunction by Activating the HMGB1/RAGE Signaling Pathway. BioMed Res. Int. 2017. [Google Scholar] [CrossRef]
- Wang, Z.; Zhang, J.; Chen, L.; Li, J.; Zhang, H.; Guo, X. Glycine suppresses AGE/RAGE signaling pathway and subsequent oxidative stress by restoring Glo1 function in the aorta of diabetic rats and in HUVECs. Oxid. Med. Cell. Longev. 2019. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.-Y.; Young, G.-H.; Hsieh, Y.-T.; Chen, Y.-H.; Wu, M.-S.; Wu, V.-C.; Lee, J.-H.; Lee, C.-C. Protein-Bound Uremic Toxins Induce Tissue Remodeling by Targeting the EGF Receptor. J. Am. Soc. Nephrol. 2015, 26, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Hirose, Y.; Goto, S.; Nishijima, F.; Zrelli, H.; Zghonda, N.; Niwa, T.; Miyazaki, H. Indoxyl sulfate enhances angiotensin II signaling through upregulation of epidermal growth factor receptor expression in vascular smooth muscle cells. Life Sci. 2012, 91, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Soshilov, A.A.; Motta, S.; Bonati, L.; Denison, M.S. Transitional States in Ligand-Dependent Transformation of the Aryl Hydrocarbon Receptor into Its DNA-Binding Form. Int. J. Mol. Sci. 2020, 21, 2474. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, I.; Tatebe, J.; Namba, S.; Koizumi, M.; Yamazaki, J.; Morita, T. Activation of Aryl Hydrocarbon Receptor Mediates Indoxyl Sulfate-Induced Monocyte Chemoattractant Protein-1 Expression in Human Umbilical Vein Endothelial Cells. Circ. J. 2013, 77, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Ambolet-Camoit, A.; Ottolenghi, C.; Leblanc, A.; Kim, M.J.; Letourneur, F.; Jacques, S.; Cagnard, N.; Guguen-Guillouzo, C.; Barouki, R.; Aggerbeck, M. Two persistent organic pollutants which act through different xenosensors (alpha-endosulfan and 2,3,7,8 tetrachlorodibenzo-p-dioxin) interact in a mixture and downregulate multiple genes involved in human hepatocyte lipid and glucose metabolism. Biochimie 2015, 116, 79–91. [Google Scholar] [CrossRef]
- Han, S.G.; Han, S.S.; Toborek, M.; Hennig, B. EGCG protects endothelial cells against PCB 126-induced inflammation through inhibition of AhR and induction of Nrf2-regulated genes. Toxicol. Appl. Pharmacol. 2012, 261, 181–188. [Google Scholar] [CrossRef]
- Asai, H.; Hirata, J.; Watanabe-Akanuma, M. Indoxyl glucuronide, a protein-bound uremic toxin, inhibits hypoxia-inducible factor‒dependent erythropoietin expression through activation of aryl hydrocarbon receptor. Biochem. Biophys. Res. Commun. 2018, 504, 538–544. [Google Scholar] [CrossRef]
- Kolachalama, V.B.; Shashar, M.; Alousi, F.; Shivanna, S.; Rijal, K.; Belghasem, M.E.; Walker, J.; Matsuura, S.; Chang, G.H.; Gibson, C.M.; et al. Uremic Solute-Aryl Hydrocarbon Receptor-Tissue Factor Axis Associates with Thrombosis after Vascular Injury in Humans. J. Am. Soc. Nephrol. 2018, 29, 1063–1072. [Google Scholar] [CrossRef]
- Larigot, L.; Juricek, L.; Dairou, J.; Coumoul, X. AhR signaling pathways and regulatory functions. Biochim. Open 2018, 7, 1–9. [Google Scholar] [CrossRef]
- Jansen, J.; Jansen, K.; Neven, E.; Poesen, R.; Othman, A.; van Mil, A.; Sluijter, J.; Sastre Torano, J.; Zaal, E.A.; Berkers, C.R.; et al. Remote sensing and signaling in kidney proximal tubules stimulates gut microbiome-derived organic anion secretion. Proc. Natl. Acad. Sci. USA 2019, 116, 16105–16110. [Google Scholar] [CrossRef] [PubMed]
- Machado, T.S.; Poitevin, S.; Paul, P.; McKay, N.; Jourde-Chiche, N.; Legris, T.; Mouly-Bandini, A.; Dignat-George, F.; Brunet, P.; Masereeuw, R.; et al. Indoxyl sulfate upregulates liver P-glycoprotein expression and activity through aryl hydrocarbon receptor signaling. J. Am. Soc. Nephrol. 2018, 29, 906–918. [Google Scholar]
- Dou, L.; Poitevin, S.; Sallée, M.; Addi, T.; Gondouin, B.; McKay, N.; Denison, M.S.; Jourde-Chiche, N.; Duval-Sabatier, A.; Cerini, C.; et al. Aryl hydrocarbon receptor is activated in patients and mice with chronic kidney disease. Kidney Int. 2018, 93, 986–999. [Google Scholar] [CrossRef] [PubMed]
- Shivanna, S.; Kolandaivelu, K.; Shashar, M.; Belghasim, M.; Al-Rabadi, L.; Balcells, M.; Zhang, A.; Weinberg, J.; Francis, J.; Pollastri, M.P.; et al. The Aryl Hydrocarbon Receptor is a Critical Regulator of Tissue Factor Stability and an Antithrombotic Target in Uremia. J. Am. Soc. Nephrol. 2016, 27, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Lano, G.; Laforêt, M.; Von Kotze, C.; Perrin, J.; Addi, T.; Brunet, P.; Poitevin, S.; Burtey, S.; Dou, L. Aryl Hydrocarbon Receptor Activation and Tissue Factor Induction by Fluid Shear Stress and Indoxyl Sulfate in Endothelial Cells. Int. J. Mol. Sci. 2020, 21, 2392. [Google Scholar] [CrossRef]
- Mussbacher, M.; Salzmann, M.; Brostjan, C.; Hoesel, B.; Schoergenhofer, C.; Datler, H.; Hohensinner, P.; Basílio, J.; Petzelbauer, P.; Assinger, A.; et al. Cell Type-Specific Roles of NF-κB Linking Inflammation and Thrombosis. Front. Immunol. 2019, 10, 85. [Google Scholar] [CrossRef]
- Sun, S.-C. The non-canonical NF-κB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. [Google Scholar] [CrossRef]
- Tumur, Z.; Shimizu, H.; Enomoto, A.; Miyazaki, H.; Niwa, T. Indoxyl Sulfate Upregulates Expression of ICAM-1 and MCP-1 by Oxidative Stress-Induced NF-ĸB Activation. Am. J. Nephrol. 2010, 31, 435–441. [Google Scholar] [CrossRef]
- Buendía, P.; Carracedo, J.; Soriano, S.; Madueño, J.A.; Ortiz, A.; Martín-Malo, A.; Aljama, P.; Ramírez, R. Klotho Prevents NFκB Translocation and Protects Endothelial Cell From Senescence Induced by Uremia. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2015, 70, 1198–1209. [Google Scholar] [CrossRef]
- Coulthard, L.R.; White, D.E.; Jones, D.L.; McDermott, M.F.; Burchill, S.A. p38MAPK: Stress responses from molecular mechanisms to therapeutics. Trends Mol. Med. 2009, 15, 369–379. [Google Scholar] [CrossRef]
- Morrison, D.K. MAP Kinase Pathways. Cold Spring Harb. Perspect. Biol. 2012, 4, a011254. [Google Scholar] [CrossRef] [PubMed]
- Cargnello, M.; Roux, P.P. Activation and Function of the MAPKs and Their Substrates, the MAPK-Activated Protein Kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Kim, C.W.; Simmons, R.D.; Jo, H. Role of Flow-Sensitive microRNAs in Endothelial Dysfunction and Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2206–2216. [Google Scholar] [CrossRef] [PubMed]
- Schober, A.; Weber, C. Mechanisms of MicroRNAs in Atherosclerosis. Annu. Rev. Pathol. Mech. Dis. 2016, 11, 583–616. [Google Scholar] [CrossRef]
- Wiese, C.B.; Zhong, J.; Xu, Z.-Q.; Zhang, Y.; Ramirez Solano, M.A.; Zhu, W.; Linton, M.F.; Sheng, Q.; Kon, V.; Vickers, K.C. Dual inhibition of endothelial miR-92a-3p and miR-489-3p reduces renal injury-associated atherosclerosis. Atherosclerosis 2019, 282, 121–131. [Google Scholar] [CrossRef]
- Kétszeri, M.; Kirsch, A.; Frauscher, B.; Moschovaki-Filippidou, F.; Mooslechner, A.A.; Kirsch, A.H.; Schabhuettl, C.; Aringer, I.; Artinger, K.; Pregartner, G.; et al. MicroRNA-142-3p improves vascular relaxation in uremia. Atherosclerosis 2019, 280, 28–36. [Google Scholar] [CrossRef]
- Li, S.; Xie, Y.; Yang, B.; Huang, S.; Zhang, Y.; Jia, Z.; Ding, G.; Zhang, A. MicroRNA-214 targets COX-2 to antagonize indoxyl sulfate (IS)-induced endothelial cell apoptosis. Apoptosis 2020, 25, 92–104. [Google Scholar] [CrossRef]
- Fourdinier, O.; Schepers, E.; Metzinger-Le Meuth, V.; Glorieux, G.; Liabeuf, S.; Verbeke, F.; Vanholder, R.; Brigant, B.; Pletinck, A.; Diouf, M.; et al. Serum levels of miR-126 and miR-223 and outcomes in chronic kidney disease patients. Sci. Rep. 2019, 9, 4477. [Google Scholar] [CrossRef]
- M’baya-Moutoula, E.; Marchand, A.; Six, I.; Bahrar, N.; Celic, T.; Mougenot, N.; Maitrias, P.; Massy, Z.A.; Lompreh, A.-M.; Metzinger, L.; et al. Inhibition of miR-223 expression using a sponge strategy decreases restenosis in rat injured carotids. Curr. Vasc. Pharmacol. 2019, 17. [Google Scholar] [CrossRef]
- Metzinger-Le Meuth, V.; Metzinger, L. miR-223 and other miRNA’s evaluation in chronic kidney disease: Innovative biomarkers and therapeutic tools. Non-Coding RNA Res. 2019, 4, 30–35. [Google Scholar] [CrossRef]
- Favretto, G.; Cunha, R.S.d.; Dalboni, M.A.; Oliveira, R.B.d.; Barreto, F.d.C.; Massy, Z.A.; Stinghen, A.E.M. Endothelial Microparticles in Uremia: Biomarkers and Potential Therapeutic Targets. Toxins 2019, 11, 267. [Google Scholar] [CrossRef]
- Carmona, A.; Guerrero, F.; Buendia, P.; Obrero, T.; Aljama, P.; Carracedo, J. Microvesicles Derived from Indoxyl Sulfate Treated Endothelial Cells Induce Endothelial Progenitor Cells Dysfunction. Front. Physiol. 2017, 8, 666. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.H.; Jeon, E.Y.; Kim, S.J. Indoxyl sulfate-induced extracellular vesicles released from endothelial cells stimulate vascular smooth muscle cell proliferation by inducing transforming growth factor-beta production. J. Vasc. Res. 2019, 56, 129–138. [Google Scholar] [CrossRef]
- Soriano, S.; Carmona, A.; Triviño, F.; Rodriguez, M.; Alvarez-Benito, M.; Martín-Malo, A.; Alvarez-Lara, M.-A.; Ramírez, R.; Aljama, P.; Carracedo, J. Endothelial damage and vascular calcification in patients with chronic kidney disease. Am. J. Physiol. Physiol. 2014, 307, F1302–F1311. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Xie, R.; Yu, C.; Ma, R.; Dong, W.; Meng, H.; Zhang, Y.; Si, Y.; Zhang, Z.; Novakovic, V.; et al. Thrombotic role of blood and endothelial cells in uremia through phosphatidylserine exposure and microparticle release. PLoS ONE 2015, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Meijers, B.K.I.; Van kerckhoven, S.; Verbeke, K.; Dehaen, W.; Vanrenterghem, Y.; Hoylaerts, M.F.; Evenepoel, P. The Uremic Retention Solute p-Cresyl Sulfate and Markers of Endothelial Damage. Am. J. Kidney Dis. 2009, 54, 891–901. [Google Scholar] [CrossRef]
- Faure, V.; Dou, L.; Sabatier, F.; Cerini, C.; Sampol, J.; Berland, Y.; Brunet, P.; Dignat-George, F. Elevation of circulating endothelial microparticles in patients with chronic renal failure. J. Thromb. Haemost. 2006, 4, 566–573. [Google Scholar] [CrossRef]
- Di Marco, G.S.; König, M.; Stock, C.; Wiesinger, A.; Hillebrand, U.; Reiermann, S.; Reuter, S.; Amler, S.; Köhler, G.; Buck, F.; et al. High phosphate directly affects endothelial function by downregulating annexin II. Kidney Int. 2013, 83, 213–222. [Google Scholar] [CrossRef]
- Mörtberg, J.; Lundwall, K.; Mobarrez, F.; Wallén, H.; Jacobson, S.H.; Spaak, J. Increased concentrations of platelet- and endothelial-derived microparticles in patients with myocardial infarction and reduced renal function- a descriptive study. BMC Nephrol. 2019, 20, 71. [Google Scholar] [CrossRef]
- Jalal, D.; Renner, B.; Laskowski, J.; Stites, E.; Cooper, J.; Valente, K.; You, Z.; Perrenoud, L.; Le Quintrec, M.; Muhamed, I.; et al. Endothelial Microparticles and Systemic Complement Activation in Patients With Chronic Kidney Disease. J. Am. Heart Assoc. 2018, 7, e007818. [Google Scholar] [CrossRef]
- Ryu, J.-H.; Park, H.; Kim, S.-J. The effects of indoxyl sulfate-induced endothelial microparticles on neointimal hyperplasia formation in an ex vivo model. Ann. Surg. Treat. Res. 2017, 93, 11. [Google Scholar] [CrossRef] [PubMed]
- Meijers, B.K.I.; De Preter, V.; Verbeke, K.; Vanrenterghem, Y.; Evenepoel, P. p-Cresyl sulfate serum concentrations in haemodialysis patients are reduced by the prebiotic oligofructose-enriched inulin. Nephrol. Dial. Transplant. 2010, 25, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Sirich, T.L.; Plummer, N.S.; Gardner, C.D.; Hostetter, T.H.; Meyer, T.W. Effect of Increasing Dietary Fiber on Plasma Levels of Colon-Derived Solutes in Hemodialysis Patients. Clin. J. Am. Soc. Nephrol. 2014, 9, 1603–1610. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Johnson, D.W.; Morrison, M.; Pascoe, E.M.; Coombes, J.S.; Forbes, J.M.; Szeto, C.C.; McWhinney, B.C.; Ungerer, J.P.J.; Campbell, K.L. Synbiotics easing renal failure by improving gut microbiology (SYNERGY): A randomized trial. Clin. J. Am. Soc. Nephrol. 2016, 11, 223–231. [Google Scholar] [CrossRef]
- Ramos, C.I.; Armani, R.G.; Canziani, M.E.F.; Dalboni, M.A.; Dolenga, C.J.R.; Nakao, L.S.; Campbell, K.L.; Cuppari, L. Effect of prebiotic (fructooligosaccharide) on uremic toxins of chronic kidney disease patients: A randomized controlled trial. Nephrol. Dial. Transplant. 2018, 34, 1876–1884. [Google Scholar] [CrossRef]
- Kandouz, S.; Mohamed, A.S.; Zheng, Y.; Sandeman, S.; Davenport, A. Reduced protein bound uraemic toxins in vegetarian kidney failure patients treated by haemodiafiltration. Hemodial. Int. 2016, 20, 610–617. [Google Scholar] [CrossRef]
- Moe, S.M.; Zidehsarai, M.P.; Chambers, M.A.; Jackman, L.A.; Radcliffe, J.S.; Trevino, L.L.; Donahue, S.E.; Asplin, J.R. Vegetarian Compared with Meat Dietary Protein Source and Phosphorus Homeostasis in Chronic Kidney Disease. Clin. J. Am. Soc. Nephrol. 2011, 6, 257–264. [Google Scholar] [CrossRef]
- Akiyama, Y.; Takeuchi, Y.; Kikuchi, K.; Mishima, E.; Yamamoto, Y.; Suzuki, C.; Toyohara, T.; Suzuki, T.; Hozawa, A.; Ito, S.; et al. A Metabolomic Approach to Clarifying the Effect of AST-120 on 5/6 Nephrectomized Rats by Capillary Electrophoresis with Mass Spectrometry (CE-MS). Toxins 2012, 4, 1309–1322. [Google Scholar] [CrossRef]
- Yu, M.; Kim, Y.J.; Kang, D.-H. Indoxyl Sulfate–Induced Endothelial Dysfunction in Patients with Chronic Kidney Disease via an Induction of Oxidative Stress. Clin. J. Am. Soc. Nephrol. 2011, 6, 30–39. [Google Scholar] [CrossRef]
- Caglar, K.; Yilmaz, M.I.; Saglam, M.; Cakir, E.; Acikel, C.; Eyileten, T.; Yenicesu, M.; Oguz, Y.; Vural, A.; Carrero, J.J.; et al. Short-Term Treatment with Sevelamer Increases Serum Fetuin—A Concentration and Improves Endothelial Dysfunction in Chronic Kidney Disease Stage 4 Patients. Clin. J. Am. Soc. Nephrol. 2008, 3, 61–68. [Google Scholar] [CrossRef]
- Yilmaz, M.I.; Sonmez, A.; Saglam, M.; Yaman, H.; Kilic, S.; Eyileten, T.; Caglar, K.; Oguz, Y.; Vural, A.; Yenicesu, M.; et al. Comparison of Calcium Acetate and Sevelamer on Vascular Function and Fibroblast Growth Factor 23 in CKD Patients: A Randomized Clinical Trial. Am. J. Kidney Dis. 2012, 59, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Maizel, J.; Six, I.; Dupont, S.; Secq, E.; Dehedin, B.; Barreto, F.C.; Benchitrit, J.; Poirot, S.; Slama, M.; Tribouilloy, C.; et al. Effects of sevelamer treatment on cardiovascular abnormalities in mice with chronic renal failure. Kidney Int. 2013, 84, 491–500. [Google Scholar] [CrossRef] [PubMed]
- Lundwall, K.; Jörneskog, G.; Jacobson, S.H.; Spaak, J. Paricalcitol, Microvascular and Endothelial Function in Non-Diabetic Chronic Kidney Disease: A Randomized Trial. Am. J. Nephrol. 2015, 42, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Al-Badr, W.; Martin, K.J. Vitamin D and kidney disease. Clin. J. Am. Soc. Nephrol. 2008, 3, 1555–1560. [Google Scholar] [CrossRef]
- Ramirez, R.; Carracedo, J.; Merino, A.; Nogueras, S.; Alvarez-Lara, M.A.; Rodríguez, M.; Martin-Malo, A.; Tetta, C.; Aljama, P. Microinflammation induces endothelial damage in hemodialysis patients: The role of convective transport. Kidney Int. 2007, 72, 108–113. [Google Scholar] [CrossRef]
- Bellien, J.; Fréguin-Bouilland, C.; Joannidès, R.; Hanoy, M.; Rémy-Jouet, I.; Monteil, C.; Iacob, M.; Martin, L.; Renet, S.; Vendeville, C.; et al. High-efficiency on-line haemodiafiltration improves conduit artery endothelial function compared with high-flux haemodialysis in end-stage renal disease patients. Nephrol. Dial. Transplant. 2014, 29, 414–422. [Google Scholar] [CrossRef]
- Krieter, D.H.; Hackl, A.; Rodriguez, A.; Chenine, L.; Moragues, H.L.; Lemke, H.-D.; Wanner, C.; Canaud, B. Protein-bound uraemic toxin removal in haemodialysis and post-dilution haemodiafiltration. Nephrol. Dial. Transplant. 2010, 25, 212–218. [Google Scholar] [CrossRef]
- Van Gelder, M.K.; Middel, I.R.; Vernooij, R.W.M.; Bots, M.L.; Verhaar, M.C.; Masereeuw, R.; Grooteman, M.P.; Nubé, M.J.; van den Dorpel, M.A.; Blankestijn, P.J.; et al. Protein-Bound Uremic Toxins in Hemodialysis Patients Relate to Residual Kidney Function, Are Not Influenced by Convective Transport, and Do Not Relate to Outcome. Toxins 2020, 12, 234. [Google Scholar] [CrossRef]
- Meert, N.; Eloot, S.; Waterloos, M.-A.; Van Landschoot, M.; Dhondt, A.; Glorieux, G.; Ledebo, I.; Vanholder, R. Effective removal of protein-bound uraemic solutes by different convective strategies: A prospective trial. Nephrol. Dial. Transplant. 2008, 24, 562–570. [Google Scholar] [CrossRef]
- Kirsch, A.H.; Rosenkranz, A.R.; Lyko, R.; Krieter, D.H. Effects of Hemodialysis Therapy Using Dialyzers with Medium Cut-Off Membranes on Middle Molecules. In Contributions to Nephrology; Karger: Basel, Switzerland, 2017; Volume 191, pp. 158–167. [Google Scholar]
- Madero, M.; Cano, K.B.; Campos, I.; Tao, X.; Maheshwari, V.; Brown, J.; Cornejo, B.; Handelman, G.; Thijssen, S.; Kotanko, P. Removal of protein-bound uremic toxins during hemodialysis using a binding competitor. Clin. J. Am. Soc. Nephrol. 2019, 14, 394–402. [Google Scholar] [CrossRef]
- Sternkopf, M.; Thoröe-Boveleth, S.; Beck, T.; Oleschko, K.; Erlenkötter, A.; Tschulena, U.; Steppan, S.; Speer, T.; Goettsch, C.; Jankowski, V.; et al. A Bifunctional Adsorber Particle for the Removal of Hydrophobic Uremic Toxins from Whole Blood of Renal Failure Patients. Toxins 2019, 11, 389. [Google Scholar] [CrossRef] [PubMed]
- Penne, E.L.; van der Weerd, N.C.; Grooteman, M.P.C.; Mazairac, A.H.A.; van den Dorpel, M.A.; Nubé, M.J.; Bots, M.L.; Lévesque, R.; ter Wee, P.M.; Blankestijn, P.J. Role of Residual Renal Function in Phosphate Control and Anemia Management in Chronic Hemodialysis Patients. Clin. J. Am. Soc. Nephrol. 2011, 6, 281–289. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cunha, R.S.d.; Santos, A.F.; Barreto, F.C.; Stinghen, A.E.M. How do Uremic Toxins Affect the Endothelium? Toxins 2020, 12, 412. https://doi.org/10.3390/toxins12060412
Cunha RSd, Santos AF, Barreto FC, Stinghen AEM. How do Uremic Toxins Affect the Endothelium? Toxins. 2020; 12(6):412. https://doi.org/10.3390/toxins12060412
Chicago/Turabian StyleCunha, Regiane Stafim da, Andressa Flores Santos, Fellype Carvalho Barreto, and Andréa Emilia Marques Stinghen. 2020. "How do Uremic Toxins Affect the Endothelium?" Toxins 12, no. 6: 412. https://doi.org/10.3390/toxins12060412
APA StyleCunha, R. S. d., Santos, A. F., Barreto, F. C., & Stinghen, A. E. M. (2020). How do Uremic Toxins Affect the Endothelium? Toxins, 12(6), 412. https://doi.org/10.3390/toxins12060412