Uremic Toxins and Vascular Dysfunction





Abstract
1. Introduction
2. The Vascular Function
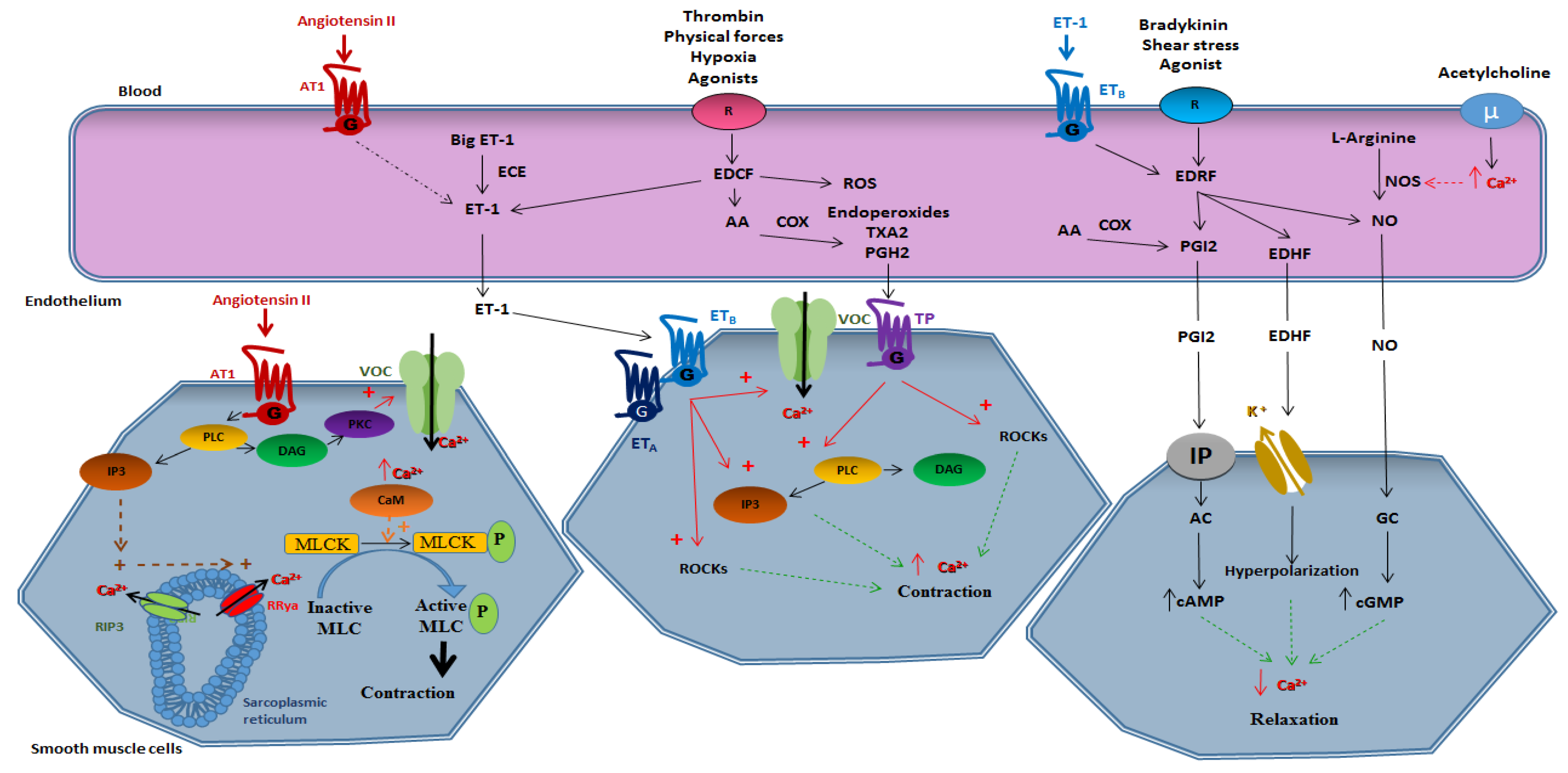
2.1. Smooth Muscle-Dependent Vasomotricity
2.1.1. Vasoconstriction
2.1.2. Vasorelaxation
2.2. Endothelium-Dependent Vasomotricity
2.2.1. Vasoconstriction
Derivatives of Arachidonic Acid
Endothelin 1
Reactive Oxygen Species
Angiotensin II
2.2.2. Vasorelaxation
Prostacycline PGI2
Nitric Oxide (NO)
Endothelium-Derived Hyperpolarizing Factor (EDHF)
3. Vascular Dysfunction in Chronic Kidney Disease
3.1. Nitric Oxide (NO)-Related Dysfunction
3.2. The Role of Reactive Oxygen Species (ROS) in Vascular Dysfunction
3.3. Vascular Dysfunction in Chronic Kidney Disease
4. The Role of Uremic Toxin in Vascular Dysfunction
- Small water-soluble compounds with a maximum molecular weight (MW) of 500 Daltons (Da). The main molecules in this group include urea, creatinine, Pi, ADMA, and guanidine compounds, which are easily removed by dialysis.
- Middle molecule compounds of moderately elevated MW (>500 Da). Many of these compounds are peptides, such as FGF-23 and PTH. They can only be removed by dialysis membranes with pores large enough to allow their passage.
- Protein-bound compounds which are, generally, of low MW. The main molecules in this group are phenols, indoles (i.e., IS), and cresols (PCS). They are difficult to remove by dialysis.
4.1. Phosphate and Vascular Dysfunction
4.2. Para-Cresyl Sulfate and Vascular Dysfunction
4.3. Indoxyl Sulfate and Vascular Dysfunction
4.4. Klotho and FGF23 and Vascular Dysfunction
4.5. Other Uremic Toxins: ADMA, SDMA, AGEs, Urea and Vascular Dysfunction
5. Conclusions
Funding
Conflicts of Interest
References
- Touyz, R.M.; Alves-Lopes, R.; Rios, F.J.; Camargo, L.L.; Anagnostopoulou, A.; Arner, A.; Montezano, A.C. Vascular smooth muscle contraction in hypertension. Cardiovasc. Res. 2018, 114, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Vanhoutte, P.M. Say NO to ET. J. Auton. Nerv. Syst. 2000, 81, 271–277. [Google Scholar] [CrossRef]
- Schiffrin, E.L.; Lipman, M.L.; Mann, J.F. Chronic kidney disease: Effects on the cardiovascular system. Circulation 2007, 116, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Bang, C.A.; Bro, S.; Bartels, E.D.; Pedersen, T.X.; Nielsen, L.B. Effect of uremia on HDL composition, vascular inflammation, and atherosclerosis in wild-type mice. Am. J. Physiol. Renal Physiol. 2007, 293, F1325–F1331. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Intengan, H.D.; Schiffrin, E.L. Vascular remodeling in hypertension: Roles of apoptosis, inflammation, and fibrosis. Hypertension 2001, 38, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Annuk, M.; Zilmer, M.; Lind, L.; Linde, T.; Fellström, B. Oxidative stress and endothelial function in chronic renal failure. J. Am. Soc. Nephrol. 2001, 12, 2747–2752. [Google Scholar]
- Lahera, V.; Goicoechea, M.; De Vinuesa, S.G.; Oubina, P.; Cachofeiro, V.; Gomez-Campdera, F.; Amann, R.; Luno, J. Oxidative stress in uremia: The role of anemia correction. J. Am. Soc. Nephrol. 2006, 17, S174–S177. [Google Scholar] [CrossRef] [PubMed]
- Touyz, R.M.; Tabet, F.; Schiffrin, E.L. Redox-dependent signalling by angiotensin II and vascular remodelling in hypertension. Clin. Exp. Pharmacol. Physiol. 2003, 30, 860–866. [Google Scholar] [CrossRef]
- Tatematsu, S.; Wakino, S.; Kanda, T.; Homma, K.; Yoshioka, K.; Hasegawa, K.; Sugano, N.; Kimoto, M.; Saruta, T.; Hayashi, K. Role of nitric oxide-producing and -degrading pathways in coronary endothelial dysfunction in chronic kidney disease. J. Am. Soc. Nephrol. 2007, 18, 741–749. [Google Scholar] [CrossRef]
- Bonomini, M.; Pandolfi, A.; Di Pietro, N.; Sirolli, V.; Giardinelli, A.; Consoli, A.; Amoroso, L.; Gizzi, F.; De Lutiis, M.A.; Felaco, M. Adherence of uremic erythrocytes to vascular endothelium decreases endothelial nitric oxide synthase expression. Kidney Int. 2005, 67, 1899–1906. [Google Scholar] [CrossRef]
- Vallance, P.; Leone, A.; Calver, A.; Collier, J.; Moncada, S. Accumulation of an endogenous inhibitor of nitric oxide synthesis in chronic renal failure. Lancet 1992, 339, 572–575. [Google Scholar]
- Orallo, F. Regulation of cytosolic calcium levels in vascular smooth muscle. Pharmacol. Ther. 1996, 69, 153–171. [Google Scholar] [CrossRef]
- Lüscher, T.F. The endothelium. Target and promoter of hypertension? Hypertension 1990, 15, 482–485. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, M.; Kurihara, H.; Kimura, S.; Tomobe, Y.; Kobayashi, M.; Mitsui, Y.; Yazaki, Y.; Goto, K.; Masaki, T. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 1988, 332, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Channick, R.; Rubin, L. Endothelin Receptor Antagonism: A New Era in the Treatment of Pulmonary Arterial Hypertension. Adv. Pulm. Hypertens. 2002, 1, 13–17. [Google Scholar] [CrossRef]
- Miyagawa, K.; Ohashi, M.; Yamashita, S.; Kojima, M.; Sato, K.; Ueda, R.; Dohi, Y. Increased oxidative stress impairs endothelial modulation of contractions in arteries from spontaneously hypertensive rats. J. Hypertens. 2007, 25, 415–421. [Google Scholar] [CrossRef]
- Rubanyi, G.M.; Romero, J.C.; Vanhoutte, P.M. Flow-induced release of endothelium-derived relaxing factor. Am. J. Physiol. 1986, 250, H1145–H1149. [Google Scholar] [CrossRef]
- Macarthur, H.; Westfall, T.C.; Wilken, G.H. Oxidative stress attenuates NO-induced modulation of sympathetic neurotransmission in the mesenteric arterial bed of spontaneously hypertensive rats. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H183–H189. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Auch-Schwelk, W.; Katusic, Z.S.; Vanhoutte, P.M. Contractions to oxygen-derived free radicals are augmented in aorta of the spontaneously hypertensive rat. Hypertension 1989, 13, 859–864. [Google Scholar] [CrossRef] [PubMed]
- Katusic, Z.S.; Vanhoutte, P.M. Superoxide anion is an endothelium-derived contracting factor. Am. J. Physiol. 1989, 257, H33–H37. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Martinez, M.A.; Garcia-Cohen, E.C.; Baena, A.B.; Gonzalez, R.; Salaices, M.; Marin, J. Contractile responses elicited by hydrogen peroxide in aorta from normotensive and hypertensive rats. Endothelial modulation and mechanism involved. Br. J. Pharmacol. 1998, 125, 1329–1335. [Google Scholar] [CrossRef] [PubMed]
- Gil-Longo, J.; Gonzalez-Vazquez, C. Characterization of four different effects elicited by H2O2 in rat aorta. Vascul. Pharmacol. 2005, 43, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.W.; Zheng, T.; Zhang, A.; Altura, B.T.; Altura, B.M. Mechanisms of hydrogen peroxide-induced contraction of rat aorta. Eur. J. Pharmacol. 1998, 344, 169–181. [Google Scholar] [CrossRef]
- Li, J.; Li, W.; Liu, W.; Altura, B.T.; Altura, B.M. Mechanisms of hydroxyl radical-induced contraction of rat aorta. Eur. J. Pharmacol. 2004, 499, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Moncada, S.; Korbut, R.; Bunting, S.; Vane, J.R. Prostacyclin is a circulating hormone. Nature 1978, 273, 767–768. [Google Scholar] [CrossRef]
- Furchgott, R.F.; Zawadzki, J.V. The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 1980, 288, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Loscalzo, J.; Welch, G. Nitric oxide and its role in the cardiovascular system. Prog. Cardiovasc. Dis. 1995, 38, 87–104. [Google Scholar] [CrossRef]
- Forstermann, U.; Gorsky, L.D.; Pollock, J.S.; Schmidt, H.H.; Heller, M.; Murad, F. Regional distribution of EDRF/NO-synthesizing enzyme(s) in rat brain. Biochem. Biophys. Res. Commun. 1990, 168, 727–732. [Google Scholar] [CrossRef]
- Moncada, S.; Martin, J.F. Evolution of nitric oxide. Lancet 1993, 341, 1511. [Google Scholar] [CrossRef]
- Mount, P.; Kemp, B.E.; Power, D. Regulation of endothelial and myocardial NO synthesis by multisite eNOS phosphorylation. J. Mol. Cell Cardiol. 2007, 42, 271–279. [Google Scholar] [CrossRef]
- Chen, G.F.; Suzuki, H. Calcium dependency of the endothelium-dependent hyperpolarization in smooth muscle cells of the rabbit carotid artery. J. Physiol. 1990, 421, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Nagao, T.; Vanhoutte, P.M. Hyperpolarization as a mechanism for endothelium-dependent relaxations in the porcine coronary artery. J. Physiol. 1992, 445, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Chaytor, A.T.; Evans, W.H.; Griffith, T.M. Central role of heterocellular gap junctional communication in endothelium-dependent relaxations of rabbit arteries. J. Physiol. 1998, 508 Pt 2, 561–573. [Google Scholar] [CrossRef]
- Sandow, S.L.; Tare, M.; Coleman, H.A.; Hill, C.E.; Parkington, H.C. Involvement of myoendothelial gap junctions in the actions of endothelium-derived hyperpolarizing factor. Circ. Res. 2002, 90, 1108–1113. [Google Scholar] [CrossRef] [PubMed]
- Matoba, T.; Shimokawa, H.; Nakashima, M.; Hirakawa, Y.; Mukai, Y.; Hirano, K.; Kanaide, H.; Takeshita, A. Hydrogen peroxide is an endothelium-derived hyperpolarizing factor in mice. J. Clin. Investig. 2000, 106, 1521–1530. [Google Scholar] [CrossRef]
- Morikawa, K.; Shimokawa, H.; Matoba, T.; Kubota, H.; Akaike, T.; Talukder, M.A.; Hatanaka, M.; Fujiki, T.; Maeda, H.; Takahashi, S.; et al. Pivotal role of Cu, Zn-superoxide dismutase in endothelium-dependent hyperpolarization. J. Clin. Investig. 2003, 112, 1871–1879. [Google Scholar] [CrossRef]
- Edwards, G.; Dora, K.A.; Gardener, M.J.; Garland, C.J.; Weston, A.H. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature 1998, 396, 269–272. [Google Scholar] [CrossRef]
- Quignard, J.F.; Félétou, M.; Thollon, C.; Vilaine, J.P.; Duhault, J.; Vanhoutte, P.M. Potassium ions and endothelium-derived hyperpolarizing factor in guinea-pig carotid and porcine coronary arteries. Br. J. Pharmacol. 1999, 127, 27–34. [Google Scholar] [CrossRef]
- Hecker, M.; Bara, A.T.; Bauersachs, J.; Busse, R. Characterization of endothelium-derived hyperpolarizing factor as a cytochrome P450-derived arachidonic acid metabolite in mammals. J. Physiol. 1994, 481, 407–414. [Google Scholar] [CrossRef]
- Félétou, M.; Vanhoutte, P.M. EDHF: An update. Clin. Sci. (Lond.) 2009, 117, 139–155. [Google Scholar] [CrossRef]
- Pino, R.Z.; Feelisch, M. Bioassay discrimination between nitric oxide (NO.) and nitroxyl (NO-) using L-cysteine. Biochem. Biophys. Res. Commun. 1994, 201, 54–62. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Irvine, J.C.; Favaloro, J.L.; Widdop, R.E.; Kemp-Harper, B.K. Nitroxyl anion donor, Angeli’s salt, does not develop tolerance in rat isolated aortae. Hypertension 2007, 49, 885–892. [Google Scholar] [CrossRef]
- Andrews, K.L.; Irvine, J.C.; Tare, M.; Apostolopoulos, J.; Favaloro, J.L.; Triggle, C.R.; Kemp-Harper, B.K. A role for nitroxyl (HNO) as an endothelium-derived relaxing and hyperpolarizing factor in resistance arteries. Br. J. Pharmacol. 2009, 157, 540–550. [Google Scholar] [CrossRef]
- Xu, W.; Kaneko, F.T.; Zheng, S.; Comhair, S.A.; Janocha, A.J.; Goggans, T.; Thunnissen, F.B.; Farver, C.; Hazen, S.L.; Jennings, C.; et al. Increased arginase II and decreased NO synthesis in endothelial cells of patients with pulmonary arterial hypertension. FASEB J. 2004, 18, 1746–1748. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Hein, T.W.; Wang, W.; Miller, M.W.; Fossum, T.W.; McDonald, M.M.; Humphrey, J.D.; Kuo, L. Upregulation of vascular arginase in hypertension decreases nitric oxide-mediated dilation of coronary arterioles. Hypertension 2004, 44, 935–943. [Google Scholar] [CrossRef] [PubMed]
- Vasquez-Vivar, J.; Kalyanaraman, B.; Martasek, P. The role of tetrahydrobiopterin in superoxide generation from eNOS: Enzymology and physiological implications. Free Radic. Res. 2003, 37, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.H.; Shi, C.; Cohen, R.A. Oxidation of the zinc-thiolate complex and uncoupling of endothelial nitric oxide synthase by peroxynitrite. J. Clin. Investig. 2002, 109, 817–826. [Google Scholar] [CrossRef]
- Cooke, J.P. Does ADMA cause endothelial dysfunction? Arterioscler. Thromb. Vasc. Biol. 2000, 20, 2032–2037. [Google Scholar] [CrossRef]
- Boerrigter, G.; Burnett, J.C., Jr. Soluble guanylate cyclase: Not a dull enzyme. Circulation 2009, 119, 2752–2754. [Google Scholar] [CrossRef]
- Zou, M.H.; Cohen, R.; Ullrich, V. Peroxynitrite and vascular endothelial dysfunction in diabetes mellitus. Endothelium 2004, 11, 89–97. [Google Scholar] [CrossRef]
- Münzel, T.; Daiber, A.; Ullrich, V.; Mülsch, A. Vascular consequences of endothelial nitric oxide synthase uncoupling for the activity and expression of the soluble guanylyl cyclase and the cGMP-dependent protein kinase. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- Zweier, J.L.; Chen, C.A.; Druhan, L.J. S-glutathionylation reshapes our understanding of endothelial nitric oxide synthase uncoupling and nitric oxide/reactive oxygen species-mediated signaling. Antioxid. Redox Signal. 2011, 14, 1769–1775. [Google Scholar] [CrossRef] [PubMed]
- Antoniades, C.; Shirodaria, C.; Crabtree, M.; Rinze, R.; Alp, N.; Cunnington, C.; Diesch, J.; Tousoulis, D.; Stefanadis, C.; Leeson, P.; et al. Altered plasma versus vascular biopterins in human atherosclerosis reveal relationships between endothelial nitric oxide synthase coupling, endothelial function, and inflammation. Circulation 2007, 116, 2851–2859. [Google Scholar] [CrossRef] [PubMed]
- Kusama, N.; Kajikuri, J.; Yamamoto, T.; Watanabe, Y.; Suzuki, Y.; Katsuya, H.; Itoh, T. Reduced hyperpolarization in endothelial cells of rabbit aortic valve following chronic nitroglycerine administration. Br. J. Pharmacol. 2005, 146, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Griffith, T.M.; Chaytor, A.T.; Bakker, L.M.; Edwards, D.H. 5-Methyltetrahydrofolate and tetrahydrobiopterin can modulate electrotonically mediated endothelium-dependent vascular relaxation. Proc. Natl. Acad. Sci. USA 2005, 102, 7008–7013. [Google Scholar] [CrossRef]
- Vanhoutte, P.M.; Feletou, M.; Taddei, S. Endothelium-dependent contractions in hypertension. Br. J. Pharmacol. 2005, 144, 449–458. [Google Scholar] [CrossRef]
- Morrow, J.D.; Awad, J.A.; Boss, H.J.; Blair, I.A.; Roberts, L.J., 2nd. Non-cyclooxygenase-derived prostanoids (F2-isoprostanes) are formed in situ on phospholipids. Proc. Natl. Acad. Sci. USA 1992, 89, 10721–10725. [Google Scholar] [CrossRef]
- Hoffman, S.W.; Moore, S.; Ellis, E.F. Isoprostanes: Free radical-generated prostaglandins with constrictor effects on cerebral arterioles. Stroke 1997, 28, 844–849. [Google Scholar] [CrossRef]
- Janssen, L.J.; Premji, M.; Netherton, S.; Catalli, A.; Cox, G.; Keshavjee, S.; Crankshaw, D.J. Excitatory and inhibitory actions of isoprostanes in human and canine airway smooth muscle. J. Pharmacol. Exp. Ther. 2000, 295, 506–511. [Google Scholar]
- Mezzano, D.; Tagle, R.; Pais, E.; Panes, O.; Perez, M.; Downey, P.; Munoz, B.; Aranda, E.; Barja, P.; Thambo, S.; et al. Endothelial cell markers in chronic uremia: Relationship with hemostatic defects and severity of renal failure. Thromb. Res. 1997, 88, 465–472. [Google Scholar] [CrossRef]
- Bonomini, M.; Reale, M.; Santarelli, P.; Stuard, S.; Settefrati, N.; Albertazzi, A. Serum levels of soluble adhesion molecules in chronic renal failure and dialysis patients. Nephron 1998, 79, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Stam, F.; van Guldener, C.; Becker, A.; Dekker, J.M.; Heine, R.J.; Bouter, L.M.; Stehouwer, C.D. Endothelial dysfunction contributes to renal function-associated cardiovascular mortality in a population with mild renal insufficiency: The Hoorn study. J. Am. Soc. Nephrol. JASN 2006, 17, 537–545. [Google Scholar] [CrossRef]
- Maizel, J.; Six, I.; Slama, M.; Tribouilloy, C.; Sevestre, H.; Poirot, S.; Giummelly, P.; Atkinson, J.; Choukroun, G.; Andrejak, M.; et al. Mechanisms of aortic and cardiac dysfunction in uremic mice with aortic calcification. Circulation 2009, 119, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Goligorsky, M.S.; Yasuda, K.; Ratliff, B. Dysfunctional endothelial progenitor cells in chronic kidney disease. J. Am. Soc. Nephrol. 2010, 21, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Luksha, L.; Stenvinkel, P.; Hammarqvist, F.; Carrero, J.J.; Davidge, S.T.; Kublickiene, K. Mechanisms of endothelial dysfunction in resistance arteries from patients with end-stage renal disease. PLoS ONE 2012, 7, e36056. [Google Scholar] [CrossRef] [PubMed]
- Kao, M.P.; Ang, D.S.; Pall, A.; Struthers, A.D. Oxidative stress in renal dysfunction: Mechanisms, clinical sequelae and therapeutic options. J. Hum. Hypertens. 2010, 24, 1–8. [Google Scholar] [CrossRef]
- Koç, M.; Bihorac, A.; Segal, M.S. Circulating endothelial cells as potential markers of the state of the endothelium in hemodialysis patients. Am. J. Kidney Dis. 2003, 42, 704–712. [Google Scholar] [CrossRef]
- Al-Massarani, G.; Vacher-Coponat, H.; Paul, P.; Widemann, A.; Arnaud, L.; Loundou, A.; Robert, S.; Berland, Y.; Dignat-George, F.; Camoin-Jau, L. Impact of immunosuppressive treatment on endothelial biomarkers after kidney transplantation. Am. J. Transplant. 2008, 8, 2360–2367. [Google Scholar] [CrossRef]
- Amabile, N.; Guerin, A.P.; Leroyer, A.; Mallat, Z.; Nguyen, C.; Boddaert, J.; London, G.M.; Tedgui, A.; Boulanger, C.M. Circulating endothelial microparticles are associated with vascular dysfunction in patients with end-stage renal failure. J. Am. Soc. Nephrol. 2005, 16, 3381–3388. [Google Scholar] [CrossRef]
- Vanholder, R.; De Smet, R.; Glorieux, G.; Argiles, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; De Deyn, P.P.; Deppisch, R.; et al. Review on uremic toxins: Classification, concentration, and interindividual variability. Kidney Int. 2003, 63, 1934–1943. [Google Scholar] [CrossRef]
- Vanholder, R.; Pletinck, A.; Schepers, E.; Glorieux, G. Biochemical and clinical impact of organic uremic retention solutes: A comprehensive update. Toxins (Basel) 2018, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Zafeiropoulou, K.; Bita, T.; Polykratis, A.; Karabina, S.; Vlachojannis, J.; Katsoris, P. Hemodialysis removes uremic toxins that alter the biological actions of endothelial cells. PLoS ONE 2012, 7, e30975. [Google Scholar] [CrossRef] [PubMed]
- Burke, S.K. Phosphate Is a Uremic Toxin. J. Ren. Nutr. 2008, 18, 27–32. [Google Scholar] [CrossRef]
- Chertow, G.M.; Burke, S.K.; Raggi, P. Sevelamer attenuates the progression of coronary and aortic calcification in hemodialysis patients. Kidney Int. 2002, 62, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Block, G.A.; Spiegel, D.M.; Ehrlich, J.; Mehta, R.; Lindbergh, J.; Dreisbach, A.; Raggi, P. Effects of sevelamer and calcium on coronary artery calcification in patients new to hemodialysis. Kidney Int. 2005, 68, 1815–1824. [Google Scholar] [CrossRef] [PubMed]
- Qunibi, W.; Moustafa, M.; Muenz, L.R.; He, D.Y.; Kessler, P.D.; Diaz-Buxo, J.A.; Budoff, M. CARE-2 Investigators. A 1-year randomized trial of calcium acetate versus sevelamer on progression of coronary artery calcification in hemodialysis patients with comparable lipid control: The Calcium Acetate Renagel Evaluation-2 (CARE-2) study. Am. J. Kidney Dis. 2008, 51, 952–965. [Google Scholar] [CrossRef]
- Block, G.A.; Raggi, P.; Bellasi, A.; Kooienga, L.; Spiegel, D.M. Mortality effect of coronary calcification and phosphate binder choice in incident hemodialysis patients. Kidney Int. 2007, 71, 438–441. [Google Scholar] [CrossRef]
- Six, I.; Maizel, J.; Barreto, F.C.; Rangrez, A.Y.; Dupont, S.; Slama, M.; Tribouilloy, C.; Choukroun, G.; Maziere, J.C.; Bode-Boeger, S.; et al. Effects of phosphate on vascular function under normal conditions and influence of the uraemic state. Cardiovasc. Res. 2012, 96, 130–139. [Google Scholar] [CrossRef]
- Shuto, E.; Taketani, Y.; Tanaka, R.; Harada, N.; Isshiki, M.; Sato, M.; Nashiki, K.; Amo, K.; Yamamoto, H.; Higashi, Y.; et al. Dietary phosphorus acutely impairs endothelial function. J. Am. Soc. Nephrol. 2009, 20, 1504–1512. [Google Scholar] [CrossRef]
- Caglar, K.; Yilmaz, M.I.; Saglam, M.; Cakir, E.; Acikel, C.; Eyileten, T.; Yenicesu, M.; Oguz, Y.; Vural, A.; Carrero, J.J.; et al. Short-term treatment with sevelamer increases serum fetuin-A concentration and improves endothelial dysfunction in chronic kidney disease stage 4 patients. Clin. J. Am. Soc. Nephrol. 2008, 3, 61–68. [Google Scholar] [CrossRef]
- Peng, A.; Wu, T.; Zeng, C.; Rakheja, D.; Zhu, J.; Ye, T.; Hutcheson, J.; Vaziri, N.D.; Liu, Z.; Mohan, C.; et al. Adverse effects of simulated hyper- and hypo-phosphatemia on endothelial cell function and viability. PLoS ONE 2011, 6, e23268. [Google Scholar] [CrossRef] [PubMed]
- Takeda, E.; Taketani, Y.; Nashiki, K.; Nomoto, M.; Shuto, E.; Sawada, N.; Yamamoto, H.; Isshiki, M. A novel function of phosphate-mediated intracellular signal transduction pathways. Adv. Enzyme Regul. 2006, 46, 154–161. [Google Scholar] [CrossRef]
- Di Marco, G.S.; Hausberg, M.; Hillebrand, U.; Rustemeyer, P.; Wittkowski, W.; Lang, D.; Pavenstädt, H. Increased inorganic phosphate induces human endothelial cell apoptosis in vitro. Am. J. Physiol. Renal Physiol. 2008, 294, F1381–F1387. [Google Scholar] [CrossRef] [PubMed]
- Di Marco, G.S.; König, M.; Stock, C.; Wiesinger, A.; Hillebrand, U.; Reiermann, S.; Reuter, S.; Amler, S.; Köhler, G.; Buck, F.; et al. High phosphate directly affects endothelial function by downregulating annexin II. Kidney Int. 2013, 83, 213–222. [Google Scholar] [CrossRef]
- Nikolov, I.G.; Joki, N.; Maizel, J.; Lacour, B.; Drüeke, T.B.; Massy, Z.A. Pleiotropic effects of the non-calcium phosphate binder sevelamer. Kidney Int. Suppl. 2006, 105, S16–S23. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Maizel, J.; Six, I.; Dupont, S.; Secq, E.; Dehedin, B.; Barreto, F.C.; Benchitrit, J.; Poirot, S.; Slama, M.; Tribouilloy, C.; et al. Effects of sevelamer treatment on cardiovascular abnormalities in mice with chronic renal failure. Kidney Int. 2013, 84, 491–500. [Google Scholar] [CrossRef]
- Burchell, B.; Coughtrie, M.W. Genetic and environmental factors associated with variation of human xenobiotic glucuronidation and sulfation. Environ. Health Perspect. 1997, 105, 739–747. [Google Scholar]
- Vanholder, R.; Bammens, B.; de Loor, H.; Glorieux, G.; Meijers, B.; Schepers, E.; Massy, Z.A.; Evenepoel, P. Warning: The unfortunate end of p-cresol as a uraemic toxin. Nephrol. Dial. Transplant. 2011, 26, 1464–1467. [Google Scholar] [CrossRef]
- Meyer, T.W.; Hostetter, T.H. Uremic solutes from colon microbes. Kidney Int. 2012, 81, 949–954. [Google Scholar] [CrossRef]
- Gryp, T.; Vanholder, R.; Vaneechoutte, M.; Glorieux, G. P-Cresyl Sulfate. Toxins 2017, 9, 52. [Google Scholar] [CrossRef]
- Deltombe, O.; Van Biesen, W.; Glorieux, G.; Massy, Z.A.; Dhondt, A.; Eloot, S.; Yun, C.C. Exploring protein binding of uremic toxins in patients with different stages of chronic kidney disease and during hemodialysis. Toxins 2015, 7, 3933–3946. [Google Scholar] [CrossRef] [PubMed]
- Pretorius, C.J.; McWhinney, B.C.; Sipinkoski, B.; Johnson, L.A.; Rossi, M.; Campbell, K.L.; Ungerer, J.P. Reference ranges and biological variation of free and total serum indoxyl- and p-cresyl sulphate measured with a rapid UPLC fluorescence detection method. Clin. Chim. Acta 2013, 419, 122–126. [Google Scholar] [CrossRef]
- Boelaert, J.; Lynen, F.; Glorieux, G.; Eloot, S.; Van Landschoot, M.; Waterloos, M.A.; Sandra, P.; Vanholder, R.C. A novel UPLC-MS-MS method for simultaneous determination of seven uremic retention toxins with cardiovascular relevance in chronic kidney disease patients. Anal. Bioanal. Chem. 2013, 405, 1937–1947. [Google Scholar] [CrossRef] [PubMed]
- Cuoghi, A.; Caiazzo, M.; Bellei, E.; Monari, E.; Bergamini, S.; Palladino, G.; Ozben, T.; Tomasi, A. Quantification of p-cresol sulphate in human plasma by selected reaction monitoring. Anal. Bioanal. Chem. 2012, 404, 2097–2104. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Sakaguchi, Y.; Sugimoto, R.; Kaneko, K.; Iwata, H.; Kotani, S.; Nakajima, M.; Ishima, Y.; Otagiri, M.; Maruyama, T. Human organic anion transporters function as a high-capacity transporter for p-cresyl sulfate, a uremic toxin. Clin. Exp. Nephrol. 2014, 18, 814–820. [Google Scholar] [CrossRef]
- Gross, P.; Massy, Z.A.; Henaut, L.; Boudot, C.; Cagnard, J.; March, C.; Kamel, S.; Drueke, T.B.; Six, I. Para-Cresyl Sulfate Acutely Impairs Vascular Reactivity and Induces Vascular Remodeling. J. Cell Physiol. 2015, 230, 2927–2935. [Google Scholar] [CrossRef]
- Han, H.; Chen, Y.; Zhu, J.; Ni, T.; Sun, J.; Zhang, R. Atorvastatin attenuates p-cresyl sulfate-induced atherogenesis and plaque instability in ApoE knockout mice. Mol. Med. Rep. 2016, 14, 3122–3128. [Google Scholar] [CrossRef][Green Version]
- Tang, W.H.; Wang, C.P.; Yu, T.H.; Tai, P.Y.; Liang, S.S.; Hung, W.C.; Wu, C.C.; Huang, S.H.; Lee, Y.J.; Chen, S.C. Protein-bounded uremic toxin p-cresylsulfate induces vascular permeability alternations. Histochem. Cell Biol. 2018, 149, 607–617. [Google Scholar] [CrossRef]
- Meijers, B.K.; Van Kerckhoven, S.; Verbeke, K.; Dehaen, W.; Vanrenterghem, Y.; Hoylaerts, M.F.; Evenepoel, P. The uremic retention solute p-cresyl sulfate and markers of endothelial damage. Am. J. Kidney Dis. 2009, 54, 891–901. [Google Scholar] [CrossRef]
- Watanabe, H.; Miyamoto, Y.; Enoki, Y.; Ishima, Y.; Kadowaki, D.; Kotani, S.; Nakajima, M.; Tanaka, M.; Matsushita, K.; Mori, Y.; et al. P-Cresyl sulfate, a uremic toxin, causes vascular endothelial and smooth muscle cell damages by inducing oxidative stress. Pharmacol. Res. Perspect. 2015, 3, e00092. [Google Scholar] [CrossRef]
- Sato, E.; Hosomi, K.; Sekimoto, A.; Mishima, E.; Oe, Y.; Saigusa, D.; Ito, S.; Abe, T.; Sato, H.; Kunisawa, J.; et al. Effects of the oral adsorbent AST-120 on fecal p-cresol and indole levels and on the gut microbiota composition. Biochem. Biophys. Res. Commun. 2020, 525, 773–779. [Google Scholar] [CrossRef] [PubMed]
- Cosola, C.; De Angelis, M.; Rocchetti, M.T.; Montemurno, E.; Maranzano, V.; Dalfino, G.; Manno, C.; Zito, A.; Gesualdo, M.; Ciccone, M.M.; et al. Beta-Glucans Supplementation Associates with Reduction in P-Cresyl Sulfate Levels and Improved Endothelial Vascular Reactivity in Healthy Individuals. PLoS ONE 2017, 12, e0169635. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.J.; Chen, H.H.; Pan, C.F.; Chuang, C.K.; Wang, T.J.; Sun, F.J.; Wu, C.J. p-Cresylsulfate and indoxyl sulfate level at different stages of chronic kidney disease. J. Clin. Lab. Anal. 2011, 25, 191–197. [Google Scholar] [CrossRef]
- Barreto, F.C.; Barreto, D.V.; Liabeuf, S.; Meert, N.; Glorieux, G.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A. European Uremic Toxin Work Group (EUTox). Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 1551–1558. [Google Scholar] [CrossRef] [PubMed]
- Fan, P.C.; Chang, J.C.H.; Lin, C.N.; Lee, C.C.; Chen, Y.T.; Chu, P.H.; Kou, G.; Lu, Y.A.; Yang, C.W.; Chen, Y.C. Serum indoxyl sulfate predicts adverse cardiovascular events in patients with chronic kidney disease. J. Formos. Med. Assoc. 2019, 118, 1099–1106. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.J.; Wu, V.; Wu, P.C.; Wu, C.J. Meta-Analysis of the Associations of p-Cresyl Sulfate (PCS) and Indoxyl Sulfate (IS) with Cardiovascular Events and All-Cause Mortality in Patients with Chronic Renal Failure. PLoS ONE 2015, 10, e0132589. [Google Scholar] [CrossRef]
- Vanholder, R.; Schepers, E.; Pletinck, A.; Nagler, E.V.; Glorieux, G. The uremic toxicity of indoxyl sulfate and p-cresyl sulfate: A systematic review. J. Am. Soc. Nephrol. 2014, 25, 1897–1907. [Google Scholar] [CrossRef]
- Wang, C.H.; Lai, Y.H.; Kuo, C.H.; Lin, Y.L.; Tsai, J.P.; Hsu, B.G. Association between Serum Indoxyl Sulfate Levels and Endothelial Function in Non-Dialysis Chronic Kidney Disease. Toxins (Basel) 2019, 11, 589. [Google Scholar] [CrossRef]
- Six, I.; Gross, P.; Remond, M.C.; Chillon, J.M.; Poirot, S.; Drueke, T.B.; Massy, Z.A. Deleterious vascular effects of indoxyl sulfate and reversal by oral adsorbent AST-120. Atherosclerosis 2015, 243, 248–256. [Google Scholar] [CrossRef]
- Lee, W.C.; Li, L.C.; Chen, J.B.; Chang, H.W. Indoxyl Sulfate-Induced Oxidative Stress, Mitochondrial Dysfunction, and Impaired Biogenesis Are Partly Protected by Vitamin C and N-Acetylcysteine. Sci. World J. 2015, 2015, 1–6. [Google Scholar] [CrossRef]
- Tumur, Z.; Shimizu, H.; Enomoto, A.; Miyazaki, H.; Niwa, T. Indoxyl sulfate upregulates expression of ICAM-1 and MCP-1 by oxidative stress-induced NF-kappaB activation. Am. J. Nephrol. 2010, 31, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Tumur, Z.; Niwa, T. Indoxyl sulfate inhibits nitric oxide production and cell viability by inducing oxidative stress in vascular endothelial cells. Am. J. Nephrol. 2009, 29, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Kharait, S.; Haddad, D.J.; Springer, M.L. Nitric oxide counters the inhibitory effects of uremic toxin indoxyl sulfate on endothelial cells by governing ERK MAP kinase and myosin light chain activation. Biochem. Biophys. Res. Commun. 2011, 409, 758–763. [Google Scholar] [CrossRef]
- Yu, M.; Kim, Y.J.; Kang, D.H. Indoxyl sulfate-induced endothelial dysfunction in patients with chronic kidney disease via an induction of oxidative stress. Clin. J. Am. Soc. Nephrol. 2011, 6, 30–39. [Google Scholar] [CrossRef]
- Dou, L.; Jourde-Chiche, N.; Faure, V.; Cerini, C.; Berland, Y.; Dignat-George, F.; Brunet, P. The uremic solute indoxyl sulfate induces oxidative stress in endothelial cells. J. Thromb. Haemost. 2007, 5, 1302–1308. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, T.; Takayanagi, K.; Kojima, M.; Katome, T.; Taguchi, K.; Kobayashi, T. Direct Impairment of the Endothelial Function by Acute Indoxyl Sulfate through Declined Nitric Oxide and Not Endothelium-Derived Hyperpolarizing Factor or Vasodilator Prostaglandins in the Rat Superior Mesenteric Artery. Biol. Pharm. Bull. 2019, 42, 1236–1242. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.C.; Liang, C.J.; Huang, T.M.; Liu, C.W.; Wang, S.H.; Young, G.H.; Tsai, J.S.; Tseng, Y.C.; Peng, Y.S.; Wu, V.C.; et al. Indoxyl sulfate enhances IL-1β-induced E-selectin expression in endothelial cells in acute kidney injury by the ROS/MAPKs/NFκB/AP-1 pathway. Arch. Toxicol. 2016, 90, 2779–2792. [Google Scholar] [CrossRef]
- Masai, N.; Tatebe, J.; Yoshino, G.; Morita, T. Indoxyl sulfate stimulates monocyte chemoattractant protein-1 expression in human umbilical vein endothelial cells by inducing oxidative stress through activation of the NADPH oxidase-nuclear factor-κB pathway. Circ. J. 2010, 74, 2216–2224. [Google Scholar] [CrossRef]
- Maciel, R.A.P.; Cunha, R.S.; Busato, V.; Franco, C.R.C.; Gregorio, P.C.; Dolenga, C.J.R.; Nakao, L.S.; Massy, Z.A.; Boullier, A.; Pecoits-Filho, R.; et al. Uremia Impacts VE-Cadherin and ZO-1 Expression in Human Endothelial Cell-to-Cell Junctions. Toxins (Basel) 2018, 10, 404. [Google Scholar] [CrossRef]
- Carmona, A.; Guerrero, F.; Buendia, P.; Obrero, T.; Aljama, P.; Carracedo, J. Microvesicles Derived from Indoxyl Sulfate Treated Endothelial Cells Induce Endothelial Progenitor Cells Dysfunction. Front. Physiol. 2017, 8, 666. [Google Scholar] [CrossRef]
- Lin, C.J.; Wu, C.J.; Wu, P.C.; Pan, C.F.; Wang, T.J.; Sun, F.J.; Liu, H.L.; Chen, H.H.; Yeh, H.I. Indoxyl Sulfate Impairs Endothelial Progenitor Cells and Might Contribute to Vascular Dysfunction in Patients with Chronic Kidney Disease. Kidney Blood Press Res. 2016, 41, 1025–1036. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Kawagoe, Y.; Matsuda, T.; Ueda, Y.; Shimada, N.; Ebihara, I.; Koide, H. Oral Adsorbent AST-120 Decreases Carotid Intima-Media Thickness and Arterial Stiffness in Patients with Chronic Renal Failure. Kidney Blood Press Res. 2004, 27, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.H.; Yu, M.; Lee, S.; Ryu, D.R.; Kim, S.J.; Kang, D.H.; Choi, K.B. AST-120 Improves Microvascular Endothelial Dysfunction in End-Stage Renal Disease Patients Receiving Hemodialysis. Yonsei Med. J. 2016, 57, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Hasegawa, H.; Yamazaki, Y.; Muto, T.; Hino, R.; Takeuchi, Y.; Fujita, T.; Nakahara, K.; Fukumoto, S.; Yamashita, T. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J. Bone Miner. Res. 2004, 19, 429–435. [Google Scholar] [CrossRef]
- Shimada, T.; Kakitani, M.; Yamazaki, Y.; Hasegawa, H.; Takeuchi, Y.; Fujita, T.; Fukumoto, S.; Tomizuka, K.; Yamashita, T. Targeted ablation of Fgf23 demonstrates an essential physiological role of FGF23 in phosphate and vitamin D metabolism. J. Clin. Investig. 2004, 113, 561–568. [Google Scholar] [CrossRef]
- Shimada, T.; Urakawa, I.; Yamazaki, Y.; Hasegawa, H.; Hino, R.; Yoneya, T.; Takeuchi, Y.; Fujita, T.; Fukumoto, S.; Yamashita, T. FGF-23 transgenic mice demonstrate hypophosphatemic rickets with reduced expression of sodium phosphate cotransporter type IIa. Biochem. Biophys. Res. Commun. 2004, 314, 409–414. [Google Scholar] [CrossRef]
- Shimada, T.; Yamazaki, Y.; Takahashi, M.; Hasegawa, H.; Urakawa, I.; Oshima, T.; Ono, K.; Kakitani, M.; Tomizuka, K.; Fujita, T.; et al. Vitamin D receptor-independent FGF23 actions in regulating phosphate and vitamin D metabolism. Am. J. Physiol. Renal Physiol. 2005, 289, F1088–F1095. [Google Scholar] [CrossRef]
- Ben-Dov, I.Z.; Galitzer, H.; Lavi-Moshayoff, V.; Goetz, R.; Kuro-o, M.; Mohammadi, M.; Sirkis, R.; Naveh-Many, T.; Silver, J. The parathyroid is a target organ for FGF23 in rats. J. Clin. Investig. 2007, 117, 4003–4008. [Google Scholar] [CrossRef]
- Urakawa, I.; Yamazaki, Y.; Shimada, T.; Iijima, K.; Hasegawa, H.; Okawa, K.; Fujita, T.; Fukumoto, S.; Yamashita, T. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 2006, 444, 770–774. [Google Scholar] [CrossRef]
- Kuro-o, M.; Matsumura, Y.; Aizawa, H.; Kawaguchi, H.; Suga, T.; Utsugi, T.; Ohyama, Y.; Kurabayashi, M.; Kaname, T.; Kume, E.; et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 1997, 390, 45–51. [Google Scholar] [CrossRef]
- Semba, R.D.; Cappola, A.R.; Sun, K.; Bandinelli, S.; Dalal, M.; Crasto, C.; Guralnik, J.M.; Ferrucci, L. Plasma klotho and cardiovascular disease in adults. J. Am. Geriatr. Soc. 2011, 59, 1596–1601. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.; Lu, T.S.; Molostvov, G.; Lee, C.; Lam, F.T.; Zehnder, D.; Hsiao, L.L. Vascular Klotho deficiency potentiates the development of human artery calcification and mediates resistance to fibroblast growth factor 23. Circulation 2012, 125, 2243–2255. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, Y.; Aizawa, H.; Shiraki-Iida, T.; Nagai, R.; Kuro-o, M.; Nabeshima, Y. Identification of the human klotho gene and its two transcripts encoding membrane and secreted klotho protein. Biochem. Biophys. Res. Commun. 1998, 242, 626–630. [Google Scholar] [CrossRef]
- Fliser, D.; Seiler, S.; Heine, G.H.; Ketteler, M. Measurement of serum soluble Klotho levels in CKD 5D patients: Useful tool or dispensable biomarker? Nephrol. Dial. Transplant. 2012, 27, 1702–1703. [Google Scholar] [CrossRef][Green Version]
- Kim, H.R.; Nam, B.Y.; Kim, D.W.; Kang, M.W.; Han, J.H.; Lee, M.J.; Shin, D.H.; Doh, F.M.; Koo, H.M.; Ko, K.I.; et al. Circulating α-klotho levels in CKD and relationship to progression. Am. J. Kidney Dis. 2013, 61, 899–909. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, M.; Sugiyama, H.; Morinaga, H.; Inoue, T.; Takiue, K.; Ogawa, A.; Yamanari, T.; Kikumoto, Y.; Uchida, H.A.; Kitamura, S.; et al. A decreased level of serum soluble Klotho is an independent biomarker associated with arterial stiffness in patients with chronic kidney disease. PLoS ONE 2013, 8, e56695. [Google Scholar] [CrossRef]
- Hu, M.C.; Shi, M.; Zhang, J.; Addo, T.; Cho, H.J.; Barker, S.L.; Ravikumar, P.; Gillings, N.; Bian, A.; Sidhu, S.S.; et al. Renal production, uptake, and handling of circulating αKlotho. J. Am. Soc. Nephrol. 2016, 27, 79–90. [Google Scholar] [CrossRef]
- Chen, C.D.; Tung, T.Y.; Liang, J.; Zeldich, E.; Tucker Zhou, T.B.; Turk, B.E.; Abraham, C.R. Identification of cleavage sites leading to the shed form of the anti-aging protein klotho. Biochemistry 2014, 53, 5579–5587. [Google Scholar] [CrossRef]
- Arking, D.E.; Atzmon, G.; Arking, A.; Barzilai, N.; Dietz, H.C. Association between a functional variant of the KLOTHO gene and high-density lipoprotein cholesterol, blood pressure, stroke, and longevity. Circ. Res. 2005, 96, 412–418. [Google Scholar] [CrossRef]
- Arking, D.E.; Krebsova, A.; Macek, M.; Macek, M.; Arking, A.; Mian, I.S.; Fried, L.; Hamosh, A.; Dey, S.; McIntosh, I.; et al. Association of human aging with a functional variant of klotho. Proc. Natl. Acad. Sci. USA 2002, 99, 856–861. [Google Scholar] [CrossRef]
- Yokoyama, J.S.; Marx, G.; Brown, J.A.; Bonham, L.W.; Wang, D.; Coppola, G.; Seeley, W.W.; Rosen, H.J.; Miller, B.L.; Kramer, J.H.; et al. Systemic klotho is associated with KLOTHO variation and predicts intrinsic cortical connectivity in healthy human aging. Brain Imaging Behav. 2017, 11, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, V.; Nagaraja, D.; Christopher, R. Association of the functional KL-VS variant of Klotho gene with early-onset ischemic stroke. Biochem. Biophys. Res. Commun. 2010, 403, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Kurosu, H.; Yamamoto, M.; Clark, J.D.; Pastor, J.V.; Nandi, A.; Gurnani, P.; McGuinness, O.P.; Chikuda, H.; Yamaguchi, M.; Kawaguchi, H.; et al. Suppression of aging in mice by the hormone Klotho. Science 2005, 309, 1829–1833. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.C.; Kuro-o, M.; Rosenblatt, K.P.; Brobey, R.; Papaconstantinou, J. The ASK1-Signalosome regulates p38 MAPK activity in response to levels of endogenous oxidative stress in the Klotho mouse models of aging. Aging (Albany N. Y.) 2010, 2, 597–611. [Google Scholar] [CrossRef]
- Mirza, M.A.; Hansen, T.; Johansson, L.; Ahlstrom, H.; Larsson, A.; Lind, L.; Larsson, T.E. Relationship between circulating FGF23 and total body atherosclerosis in the community. Nephrol. Dial. Transplant. 2009, 24, 3125–3131. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.A.; Larsson, A.; Lind, L.; Larsson, T.E. Circulating fibroblast growth factor-23 is associated with vascular dysfunction in the community. Atherosclerosis 2009, 205, 385–390. [Google Scholar] [CrossRef]
- Mirza, M.A.; Larsson, A.; Melhus, H.; Lind, L.; Larsson, T.E. Serum intact FGF23 associate with left ventricular mass, hypertrophy and geometry in an elderly population. Atherosclerosis 2009, 207, 546–551. [Google Scholar] [CrossRef]
- Gutierrez, O.M.; Januzzi, J.L.; Isakova, T.; Laliberte, K.; Smith, K.; Collerone, G.; Sarwar, A.; Hoffmann, U.; Coglianese, E.; Christenson, R.; et al. Fibroblast growth factor 23 and left ventricular hypertrophy in chronic kidney disease. Circulation 2009, 119, 2545–2552. [Google Scholar] [CrossRef]
- Sitara, D.; Razzaque, M.S.; Hesse, M.; Yoganathan, S.; Taguchi, T.; Erben, R.G.; Juppner, H.; Lanske, B. Homozygous ablation of fibroblast growth factor- 23 results in hyperphosphatemia and impaired skeletogenesis, and reverses hypophosphatemia in Phex-deficient mice. Matrix Biol. 2004, 23, 421–432. [Google Scholar] [CrossRef]
- Ichikawa, S.; Imel, E.A.; Kreiter, M.L.; Yu, X.; Mackenzie, D.S.; Sorenson, A.H.; Goetz, R.; Mohammadi, M.; White, K.E.; Econs, M.J. A homozygous missense mutation in human KLOTHO causes severe tumoral calcinosis. J. Clin. Investig. 2007, 117, 2684–2691. [Google Scholar] [CrossRef]
- Xie, J.; Cha, S.K.; An, S.W.; Kuro-O, M.; Birnbaumer, L.; Huang, C.L. Cardioprotection by Klotho through downregulation of TRPC6 channels in the mouse heart. Nat. Commun. 2012, 3, 1238. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.C.; Kuro-o, M.; Moe, O.W. Klotho and chronic kidney disease. Contrib. Nephrol. 2013, 180, 47–63. [Google Scholar] [PubMed]
- Koh, N.; Fujimori, T.; Nishiguchi, S.; Tamori, A.; Shiomi, S.; Nakatani, T.; Sugimura, K.; Kishimoto, T.; Kinoshita, S.; Kuroki, T.; et al. Severely reduced production of Klotho in human chronic renal failure kidney. Biochem. Biophys. Res. Commun. 2001, 280, 1015–1020. [Google Scholar] [CrossRef] [PubMed]
- Portale, A.A.; Wolf, M.; Jüppner, H.; Messinger, S.; Kumar, J.; WesselingPerry, K.; Schwartz, G.J.; Furth, S.L.; Warady, B.A.; Salusky, I.B. Disordered FGF23 and mineral metabolism in children with CKD. Clin. J. Am. Soc. Nephrol. 2014, 9, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, L.; Liabeuf, S.; Renard, C.; Lenglet, A.; Lemke, H.D.; Choukroun, G.; Drueke, T.B.; Massy, Z.A. European Uremic Toxin (EUTox) Work Group. FGF23 is independently associated with vascular calcification but not bone mineral density in patients at various CKD stages. Osteoporos. Int. 2012, 23, 2017–2025. [Google Scholar] [CrossRef] [PubMed]
- Erben, R.G. Update on FGF23 and Klotho signaling. Mol. Cell. Endocrinol. 2016, 432, 56–65. [Google Scholar] [CrossRef]
- Olauson, H.; Larsson, T.E. FGF23 and Klotho in Chronic Kidney Disease. Curr. Opin. Nephrol. Hypertens. 2013, 22, 397–404. [Google Scholar] [CrossRef]
- Six, I.; Okazaki, H.; Gross, P.; Cagnard, J.; Boudot, C.; Maizel, J.; Drueke, T.B.; Massy, Z.A. Direct, acute effects of Klotho and FGF23 on vascular smooth muscle and endothelium. PLoS ONE 2014, 9, e93423. [Google Scholar] [CrossRef]
- Gao, D.; Zuo, Z.; Tian, J.; Ali, Q.; Lin, Y.; Lei, H.; Sun, Z. Activation of SIRT1 attenuates Klotho deficiency-induced arterial stiffness and hypertension by enhancing AMP-activated protein kinase activity. Hypertension 2016, 68, 1191–1199. [Google Scholar] [CrossRef]
- Chen, K.; Zhou, X.; Sun, Z. Haplodeficiency of Klotho gene causes arterial stiffening via upregulation of scleraxis expression and induction of autophagy. Hypertension 2015, 66, 1006–1013. [Google Scholar] [CrossRef]
- Saito, Y.; Yamagishi, T.; Nakamura, T.; Ohyama, Y.; Aizawa, H.; Suga, T.; Matsumura, Y.; Masuda, H.; Kurabayashi, M.; Kuro-o, M.; et al. Klotho protein protects against endothelial dysfunction. Biochem. Biophys. Res. Commun. 1998, 248, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Nakamura, T.; Ohyama, Y.; Suzuki, T.; Iida, A.; Shiraki-Iida, T.; Kuro-o, M.; Nabeshima, Y.; Kurabayashi, M.; Nagai, R. In vivo klotho gene delivery protects against endothelial dysfunction in multiple risk factor syndrome. Biochem. Biophys. Res. Commun. 2000, 276, 767–772. [Google Scholar] [CrossRef] [PubMed]
- Ohta, J.; Rakugi, H.; Ishikawa, K.; Yang, J.; Ikushima, M.; Chihara, Y.; Maekawa, Y.; Oguro, R.; Hanasaki, H.; Kida, I. Klotho gene delivery suppresses oxidative stress in vivo. Geriatr. Gerontol. Int. 2007, 7, 293–299. [Google Scholar] [CrossRef]
- Carracedo, J.; Buendıa, P.; Merino, A.; Madueno, J.A.; Peralbo, E.; Ortiz, A.; Martin-Malo, A.; Aljama, P.; Rodriguez, M.; Ramirez, R. Klotho modulates the stress response in human senescent endothelial cells. Mech. Ageing Dev. 2012, 133, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Richter, B.; Haller, J.; Haffner, D.; Leifheit-Nestler, M. Klotho modulates FGF23-mediated NO synthesis and oxidative stress in human coronary artery endothelial cells. Pflugers. Arch. 2016, 468, 1621–1635. [Google Scholar] [CrossRef]
- Seiler, S.; Rogacev, K.S.; Roth, H.J.; Shafein, P.; Emrich, I.; Neuhaus, S.; Floege, J.; Fliser, D.; Heine, G.H. Associations of FGF-23 and sKlotho with cardiovascular outcomes among patients with CKD stages 2–4. Clin. J. Am. Soc. Nephrol. 2014, 9, 1049–1058. [Google Scholar] [CrossRef]
- Faul, C.; Amaral, A.P.; Oskouei, B.; Hu, M.C.; Sloan, A.; Isakova, T.; Gutierrez, O.M.; Aguillon-Prada, R.; Lincoln, J.; Hare, J.M.; et al. FGF23 induces left ventricular hypertrophy. J. Clin. Investig. 2011, 121, 4393–4408. [Google Scholar] [CrossRef]
- Ix, J.H.; Katz, R.; Kestenbaum, B.R.; de Boer, I.H.; Chonchol, M.; Mukamal, K.J.; Rifkin, D.; Siscovick, D.S.; Sarnak, M.J.; Shlipak, M.G. Fibroblast growth factor-23 and death, heart failure, and cardiovascular events in community-living individuals: CHS (Cardiovascular Health Study). J. Am. Coll. Cardiol. 2012, 60, 200–207. [Google Scholar] [CrossRef]
- Arnlov, J.; Carlsson, A.C.; Sundstrom, J.; Ingelsson, E.; Larsson, A.; Lind, L.; Larsson, T.E. Higher fibroblast growth factor-23 increases the risk of all-cause and cardiovascular mortality in the community. Kidney Int. 2013, 83, 160–166. [Google Scholar] [CrossRef]
- Silswal, N.; Touchberry, C.D.; Daniel, D.R.; McCarthy, D.L.; Zhang, S.; Andresen, J.; Stubbs, J.R.; Wacker, M.J. FGF23 directly impairs endothelium-dependent vasorelaxation by increasing superoxide levels and reducing nitric oxide bioavailability. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E426–E436. [Google Scholar] [CrossRef]
- Stevens, K.K.; McQuarrie, E.P.; Sands, W.; Hillyard, D.Z.; Patel, R.K.; Mark, P.B.; Jardine, A.G. Fibroblast growth factor 23 predicts left ventricular mass and induces cell adhesion molecule formation. Int. J. Nephrol. 2011, 2011, 297070. [Google Scholar] [CrossRef]
- Arnlov, J.; Carlsson, A.C.; Sundstrom, J.; Ingelsson, E.; Larsson, A.; Lind, L.; Larsson, T.E. Serum FGF23 and risk of cardiovascular events in relation to mineral metabolism and cardiovascular pathology. Clin. J. Am. Soc. Nephrol. 2013, 8, 781–786. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.H.; Dong, C.; Elkind, M.S.; Sacco, R.L.; Mendez, A.J.; Hudson, B.I.; Silverberg, S.; Wolf, M.; Rundek, T.; Wright, C.B. Fibroblast growth factor 23 is associated with carotid plaque presence and area: The Northern Manhattan Study. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2048–2053. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lindberg, K.; Olauson, H.; Amin, R.; Ponnusamy, A.; Goetz, R.; Rebecca, F.; Taylor, R.F.; Mohammadi, M.; Canfield, A.; Kublickiene, K.; et al. Arterial Klotho Expression and FGF23 Effects on Vascular Calcification and Function. PLoS ONE 2013, 8, e60658. [Google Scholar] [CrossRef]
- Richter, B.; Faul, C. FGF23 Actions on Target Tissues—With and Without Klotho. Front. Endocrinol. 2018, 9, 189. [Google Scholar] [CrossRef] [PubMed]
- Meinitzer, A.; Seelhorst, U.; Wellnitz, B.; Halwachs-Baumann, G.; Boehm, B.O.; Winkelmann, B.R.; Marz, W. Asymmetrical dimethylarginine independently predicts total and cardiovascular mortality in individuals with angiographic coronary artery disease (the Ludwigshafen Risk and Cardiovascular Health study). Clin. Chem. 2007, 53, 273–283. [Google Scholar] [CrossRef]
- Meinitzer, A.; Kielstein, J.T.; Pilz, S.; Drechsler, C.; Ritz, E.; Boehm, B.O.; Winkelmann, B.R.; Marz, W. Symmetrical and asymmetrical dimethylarginine as predictors for mortality in patients referred for coronary angiography: The Ludwigshafen Risk and Cardiovascular Health study. Clin. Chem. 2011, 57, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Scalera, F.; Borlak, J.; Beckmann, B.; Martens-Lobenhoffer, J.; Thum, T.; Täger, M.; Bode-Boger, S.M. Endogenous nitric oxide synthesis inhibitor asymmetric dimethyl L-arginine accelerates endothelial cell senescence. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1816–1822. [Google Scholar] [CrossRef]
- Juonala, M.; Viikari, J.S.; Alfthan, G.; Marniemi, J.; Kahonen, M.; Taittonen, L.; Laitinen, T.; Raitakari, O.T. Brachial artery flow-mediated dilation and asymmetrical dimethylarginine in the cardiovascular risk in young Finns study. Circulation 2007, 116, 1367–1373. [Google Scholar] [CrossRef]
- Bode-Boger, S.M.; Scalera, F.; Kielstein, J.T.; Martens-Lobenhoffer, J.; Breithardt, G.; Fobker, M.; Reinecke, H. Symmetrical dimethylarginine: A new combined parameter for renal function and extent of coronary artery disease. J. Am. Soc. Nephrol. 2006, 17, 1128–1134. [Google Scholar] [CrossRef]
- Speer, T.; Rohrer, L.; Blyszczuk, P.; Shroff, R.; Kuschnerus, K.; Krankel, N.; Kania, G.; Zewinger, S.; Akhmedov, A.; Shi, Y.; et al. Abnormal high-density lipoprotein induces endothelial dysfunction via activation of Toll-like receptor-2. Immunity 2013, 38, 754–768. [Google Scholar] [CrossRef] [PubMed]
- Maillard, L.C. Action of amino acids on sugars. Formation of melanoidins in a methodical way. Compt. Rend. 1912, 154, 66–68. [Google Scholar]
- Cai, W.; He, J.C.; Zhu, L.; Chen, X.; Striker, G.E.; Vlassara, H. AGE-receptor-1 counteracts cellular oxidant stress induced by AGEs via negative regulation of p66shc-dependent FKHRL1 phosphorylation. Am. J. Physiol. Cell. Physiol. 2008, 294, C145–C152. [Google Scholar] [CrossRef] [PubMed]
- Rojas, A.; Delgado-Lopez, F.; Gonzalez, I.; Perez-Castro, R.; Romero, J.; Rojas, I. The receptor for advanced glycation end-products: A complex signaling scenario for a promiscuous receptor. Cell Signal. 2013, 25, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Rebholz, C.M.; Astor, B.C.; Grams, M.E.; Halushka, M.K.; Lazo, M.; Hoogeveen, R.C.; Ballantyne, C.M.; Coresh, J.; Selvin, E. Association of plasma levels of soluble receptor for advanced glycation end products and risk of kidney disease: The Atherosclerosis Risk in Communities study. Nephrol. Dial. Transplant. 2015, 30, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Tanikawa, T.; Okada, Y.; Tanikawa, R.; Tanaka, Y. Advanced glycation end products induce calcification of vascular smooth muscle cells through RAGE/p38 MAPK. J. Vasc. Res. 2009, 46, 572–580. [Google Scholar] [CrossRef]
- Belmokhtar, K.; Ortillon, J.; Jaisson, S.; Massy, Z.A.; Boulagnon Rombi, C.; Doué, M.; Maurice, P.; Fritz, G.; Gillery, P.; Schmidt, A.M.; et al. Receptor for advanced glycation end products: A key molecule in the genesis of chronic kidney disease vascular calcification and a potential modulator of sodium phosphate co-transporter PIT-1 expression. Nephrol. Dial. Transplant. 2019, 34, 2018–2030. [Google Scholar] [CrossRef]
- Yuan, X.; Zhang, Z.; Gong, K.; Zhao, P.; Qin, J.; Liu, N. Inhibition of reactive oxygen species/extracellular signal-regulated kinases pathway by pioglitazone attenuates advanced glycation end products-induced proliferation of vascular smooth muscle cells in rats. Biol. Pharm. Bull. 2011, 34, 618–623. [Google Scholar] [CrossRef]
- Yan, S.D.; Schmidt, A.M.; Anderson, G.M.; Zhang, J.; Brett, J.; Zou, Y.S.; Pinsky, D.; Stern, D. Enhanced cellular oxidant stress by the interaction of advanced glycation end products with their receptors/binding proteins. J. Biol. Chem. 1994, 269, 9889–9897. [Google Scholar]
- Wautier, M.P.; Chappey, O.; Corda, S.; Stern, D.M.; Schmidt, A.M.; Wautier, J.L. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E685–E694. [Google Scholar] [CrossRef]
- Schmidt, A.M.; Hori, O.; Chen, J.X.; Li, J.F.; Crandall, J.; Zhang, J.; Cao, R.; Yan, S.D.; Brett, J.; Stern, D. Advanced glycation endproducts interacting with their endothelial receptor induce expression of vascular cell adhesion molecule-1 (VCAM-1) in cultured human endothelial cells and in mice. A potential mechanism for the accelerated vasculopathy of diabetes. J. Clin. Investig. 1995, 96, 1395–1403. [Google Scholar] [CrossRef] [PubMed]
- Rashid, G.; Benchetrit, S.; Fishman, D.; Bernheim, J. Effect of advanced glycation end-products on gene expression and synthesis of TNF-alpha and endothelial nitric oxide synthase by endothelial cells. Kidney Int. 2004, 66, 1099–1106. [Google Scholar] [CrossRef]
- Quehenberger, P.; Bierhaus, A.; Fasching, P.; Muellner, C.; Klevesath, M.; Hong, M.; Stier, G.; Sattler, M.; Schleicher, E.; Speiser, W.; et al. Endothelin 1 transcription is controlled by nuclear factor-kappaB in AGE-stimulated cultured endothelial cells. Diabetes 2000, 49, 1561–1570. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Liang, C.; Ren, Y.; Zhen, Y.; He, Z.; Wang, H.; Tan, H.; Pan, X.; Wu, Z. Advanced glycation end products depress function of endothelial progenitor cells via p38 and ERK 1/2 mitogen-activated protein kinase pathways. Basic Res. Cardiol. 2009, 104, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Dong, L.; Wang, L.; Kang, L.; Xu, B. Advanced glycation end products impair function of late endothelial progenitor cells through effects on protein kinase Akt and cyclooxygenase-2. Biochem. Biophys. Res. Commun. 2009, 381, 192–197. [Google Scholar] [CrossRef]
- Liang, C.; Ren, Y.; Tan, H.; He, Z.; Jiang, Q.; Wu, J.; Zhen, Y.; Fan, M.; Wu, Z. Rosiglitazone via upregulation of Akt/eNOS pathways attenuates dysfunction of endothelial progenitor cells, induced by advanced glycation end products. Br. J. Pharmacol. 2009, 158, 1865–1873. [Google Scholar] [CrossRef] [PubMed]
- Massy, Z.A.; Pietrement, C.; Touré, F. Reconsidering the lack of urea toxicity in dialysis patients. Semin. Dial. 2016, 29, 333–337. [Google Scholar] [CrossRef]
- Vanholder, R.; Gryp, T.; Glorieux, G. Urea and chronic kidney disease: the comeback of the century? (in uraemia research). Nephrol. Dial. Transplant. 2018, 33, 4–12. [Google Scholar] [CrossRef]
- Mori, D.; Matsui, I.; Shimomura, A.; Hashimoto, N.; Matsumoto, A.; Shimada, K.; Yamaguchi, S.; Oka, T.; Kubota, K.; Yonemoto, S.; et al. Protein carbamylation exacerbates vascular calcification. Kidney Int. 2018, 94, 72–90. [Google Scholar] [CrossRef]
- Trécherel, E.; Godin, C.; Louandre, C.; Benchitrit, J.; Poirot, S.; Mazière, J.C.; Massy, Z.A.; Galmiche, A. Upregulation of BAD, a pro-apoptotic protein of the BCL2 family, in vascular smooth muscle cells exposed to uremic conditions. Biochem. Biophys. Res. Commun. 2012, 417, 479–483. [Google Scholar] [CrossRef]
- D’Apolito, M.; Du, X.; Pisanelli, D.; Pettoello-Mantovani, M.; Campanozzi, A.; Giacco, F.; Maffione, A.B.; Colia, A.L.; Brownlee, M.; Giardino, I. Urea-induced ROS cause endothelial dysfunction in chronic renal failure. Atherosclerosis 2015, 239, 393–400. [Google Scholar] [CrossRef] [PubMed]
- D’Apolito, M.; Colia, A.L.; Lasalvia, M.; Capozzi, V.; Falcone, M.P.; Pettoello-Mantovani, M.; Brownlee, M.; Maffione, A.B.; Giardino, I. Urea-induced ROS accelerate senescence in endothelial progenitor cells. Atherosclerosis 2017, 263, 127–136. [Google Scholar] [CrossRef] [PubMed]
- D’Apolito, M.; Colia, A.L.; Manca, E.; Pettoello-Mantovani, M.; Sacco, M.; Maffione, A.B.; Brownlee, M.; Giardino, I. Urea Memory: Transient Cell Exposure to Urea Causes Persistent Mitochondrial ROS Production and Endothelial Dysfunction. Toxins (Basel) 2018, 10, 410. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Uremic Toxins | Effect on Vascular Reactivity |
|---|---|
| Phosphate | Vasoconstriction and decrease of vasorelaxation, decrease in NO production, stimulation of ROS production, induction of endothelial cells apoptosis. |
| p-cresyl sulfate | Vasoconstriction, stimulation of ROS production, increase in EMP release, vascular remodeling. |
| Indoxyl sulfate | Decrease of endothelium dependent vasorelaxation, decrease of NO production, stimulation of ROS production, reduction of endothelial cells viability. |
| Klotho deficiency | Arterial stiffness, decrease of eNOS expression, stimulation of ROS production, decrease of vasorelaxation. |
| FGF23 | Stimulation of ROS production, increase in vasoconstriction or decrease of vasorelaxation, reduction of NO production. |
| ADMA, SDMA | Inhibition of NO synthase, stimulation of ROS production. |
| AGE | Stimulation of ROS production, inhibition of NO synthase activity, induction of ET-1 expression. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Six, I.; Flissi, N.; Lenglet, G.; Louvet, L.; Kamel, S.; Gallet, M.; Massy, Z.A.; Liabeuf, S. Uremic Toxins and Vascular Dysfunction. Toxins 2020, 12, 404. https://doi.org/10.3390/toxins12060404
Six I, Flissi N, Lenglet G, Louvet L, Kamel S, Gallet M, Massy ZA, Liabeuf S. Uremic Toxins and Vascular Dysfunction. Toxins. 2020; 12(6):404. https://doi.org/10.3390/toxins12060404
Chicago/Turabian StyleSix, Isabelle, Nadia Flissi, Gaëlle Lenglet, Loïc Louvet, Said Kamel, Marlène Gallet, Ziad A. Massy, and Sophie Liabeuf. 2020. "Uremic Toxins and Vascular Dysfunction" Toxins 12, no. 6: 404. https://doi.org/10.3390/toxins12060404
APA StyleSix, I., Flissi, N., Lenglet, G., Louvet, L., Kamel, S., Gallet, M., Massy, Z. A., & Liabeuf, S. (2020). Uremic Toxins and Vascular Dysfunction. Toxins, 12(6), 404. https://doi.org/10.3390/toxins12060404