Drugs Commonly Applied to Kidney Patients May Compromise Renal Tubular Uremic Toxins Excretion

 , ,
, , 
Abstract
1. Introduction
2. Results
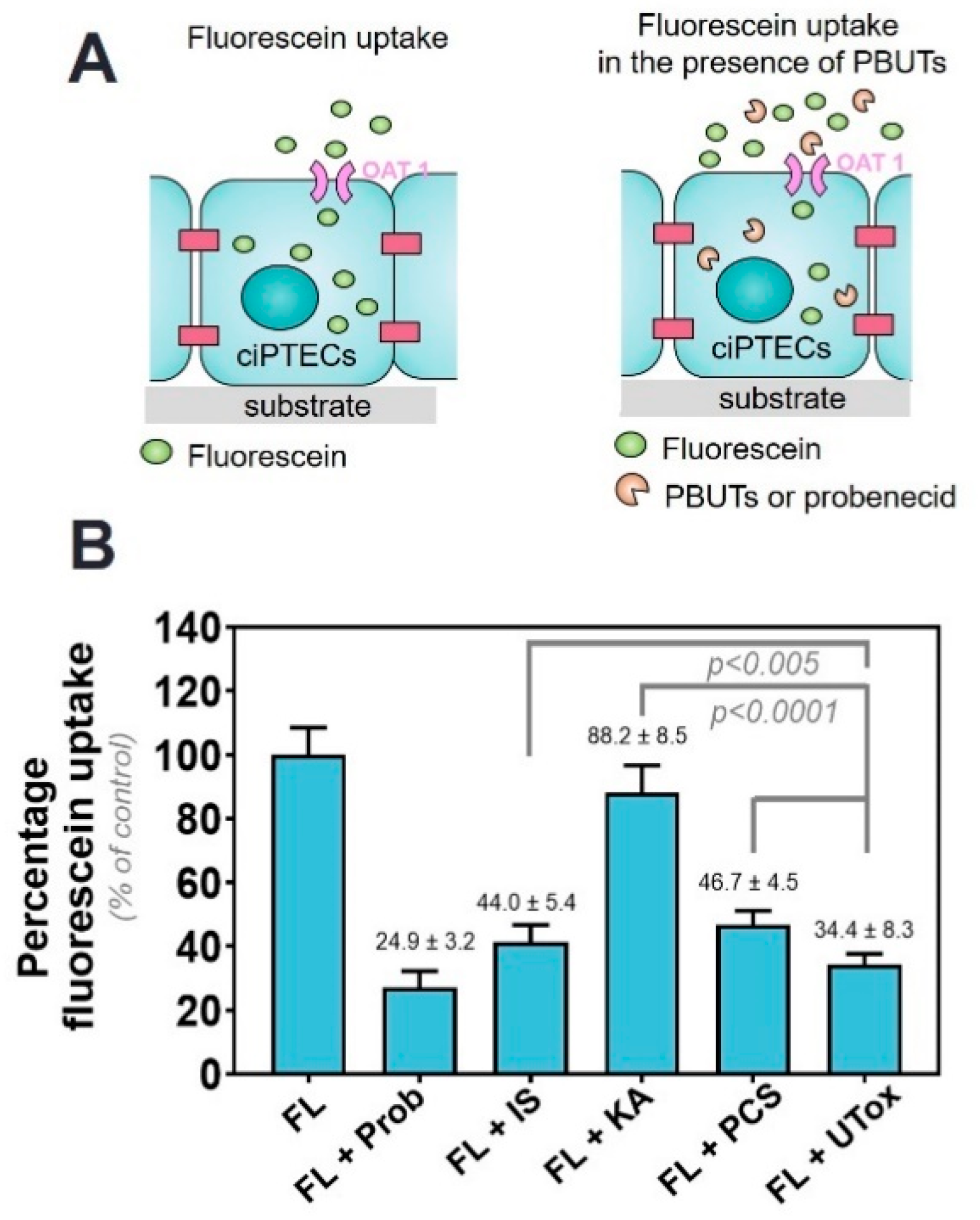
2.1. Protein-Bound Uremic Toxins Reduce OAT1-Mediated Uptake at Clinically Relevant Concentrations
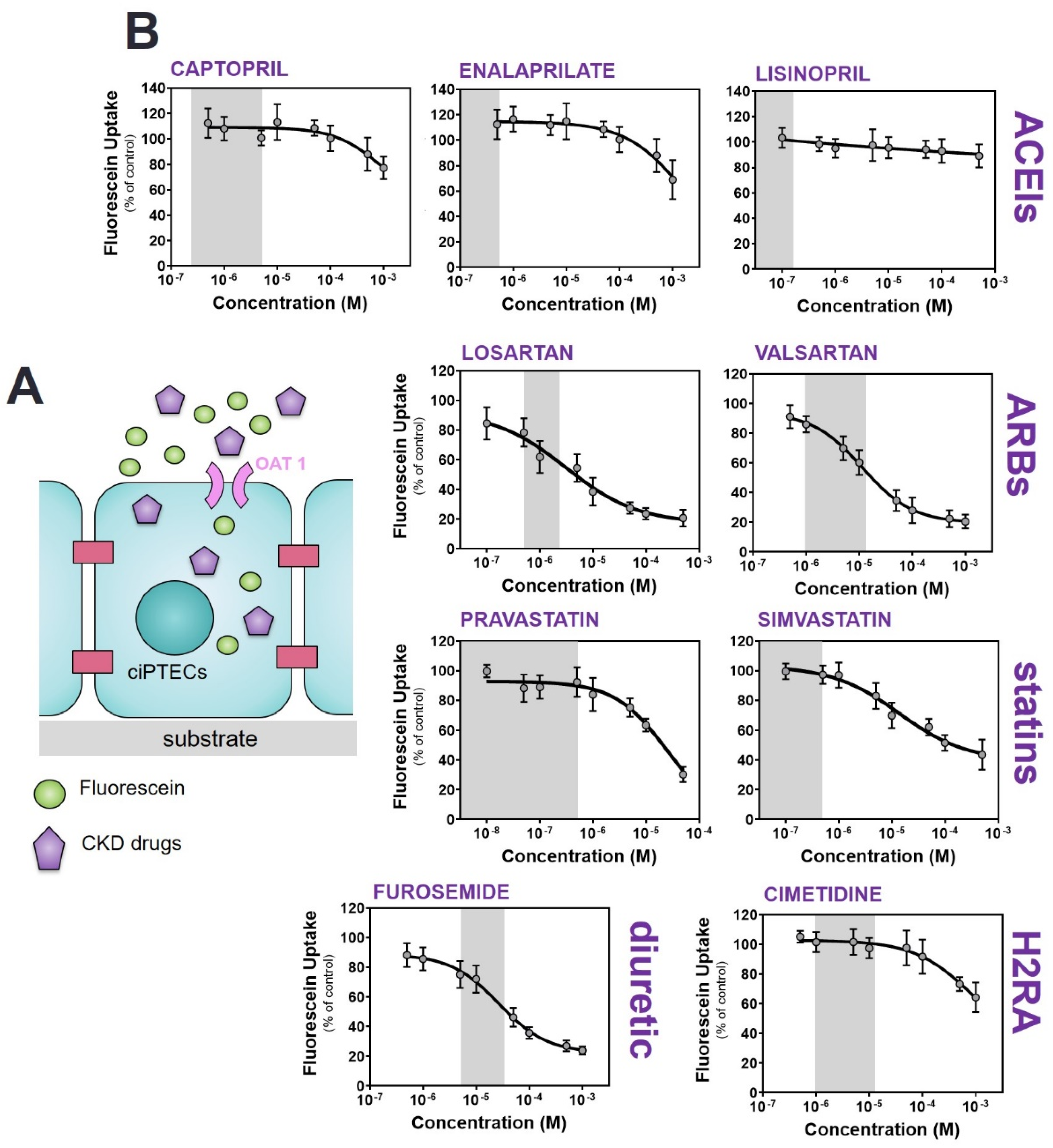
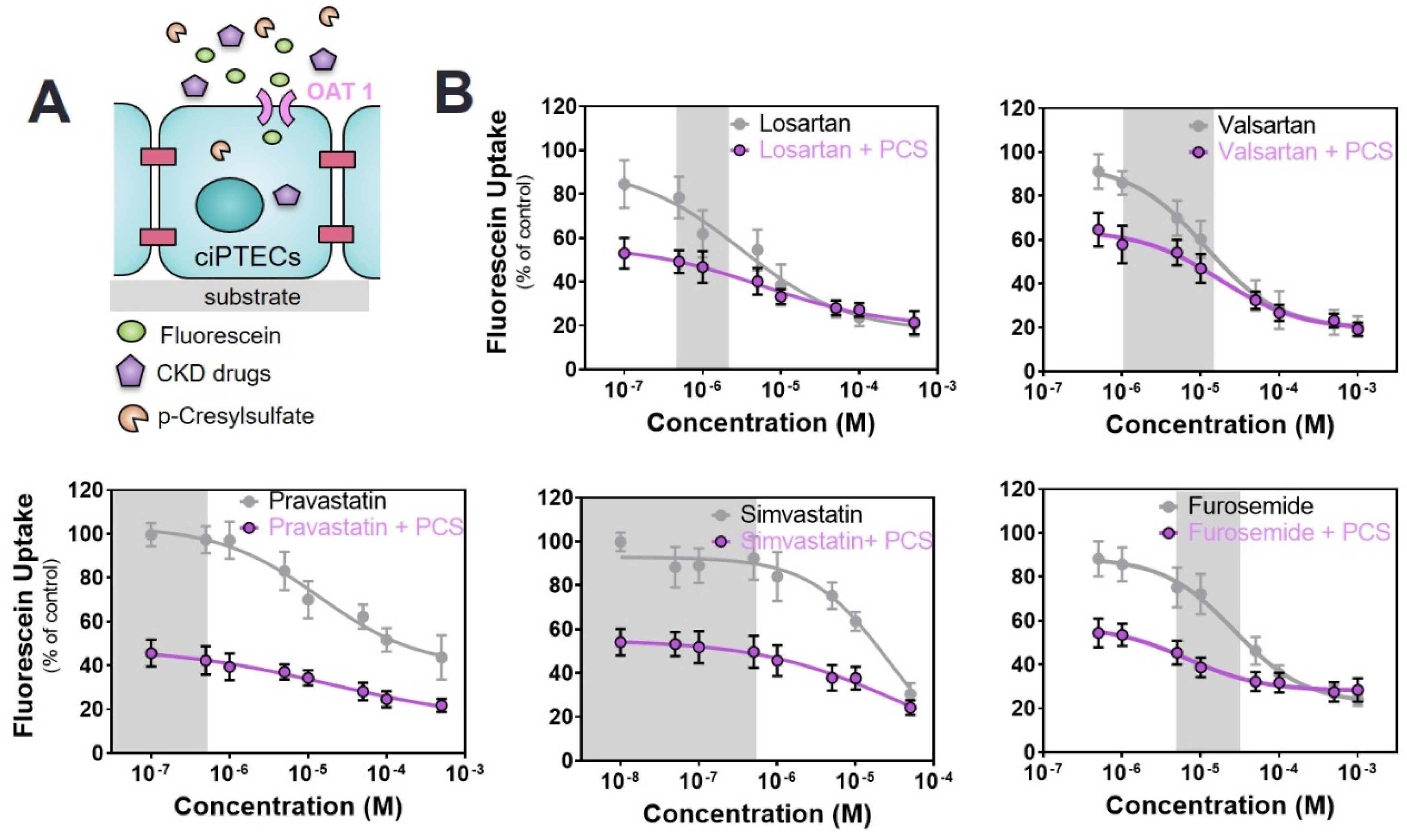
2.2. Commonly Prescribed Drugs in CKD Management Reduce OAT1-Mediated Uptake
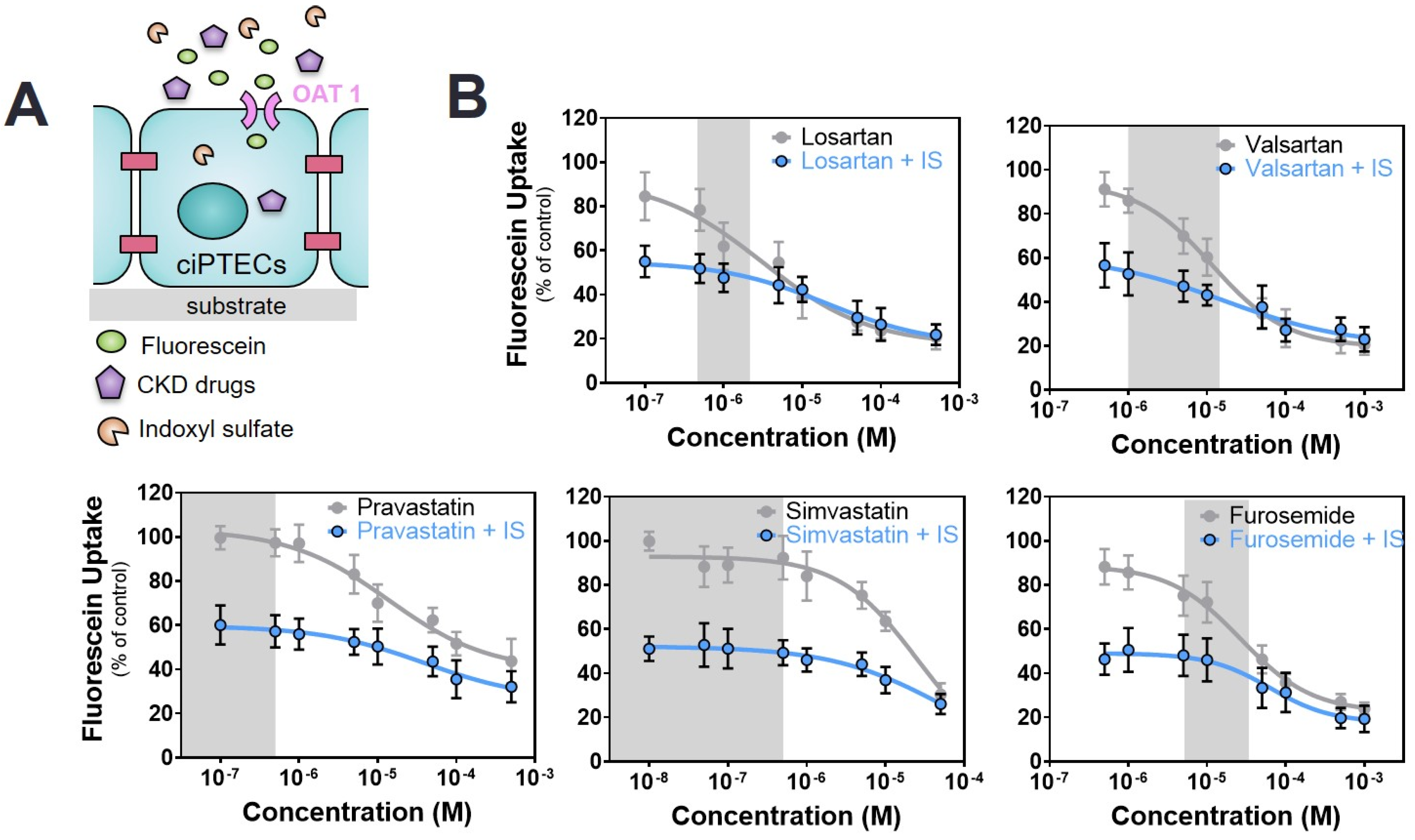
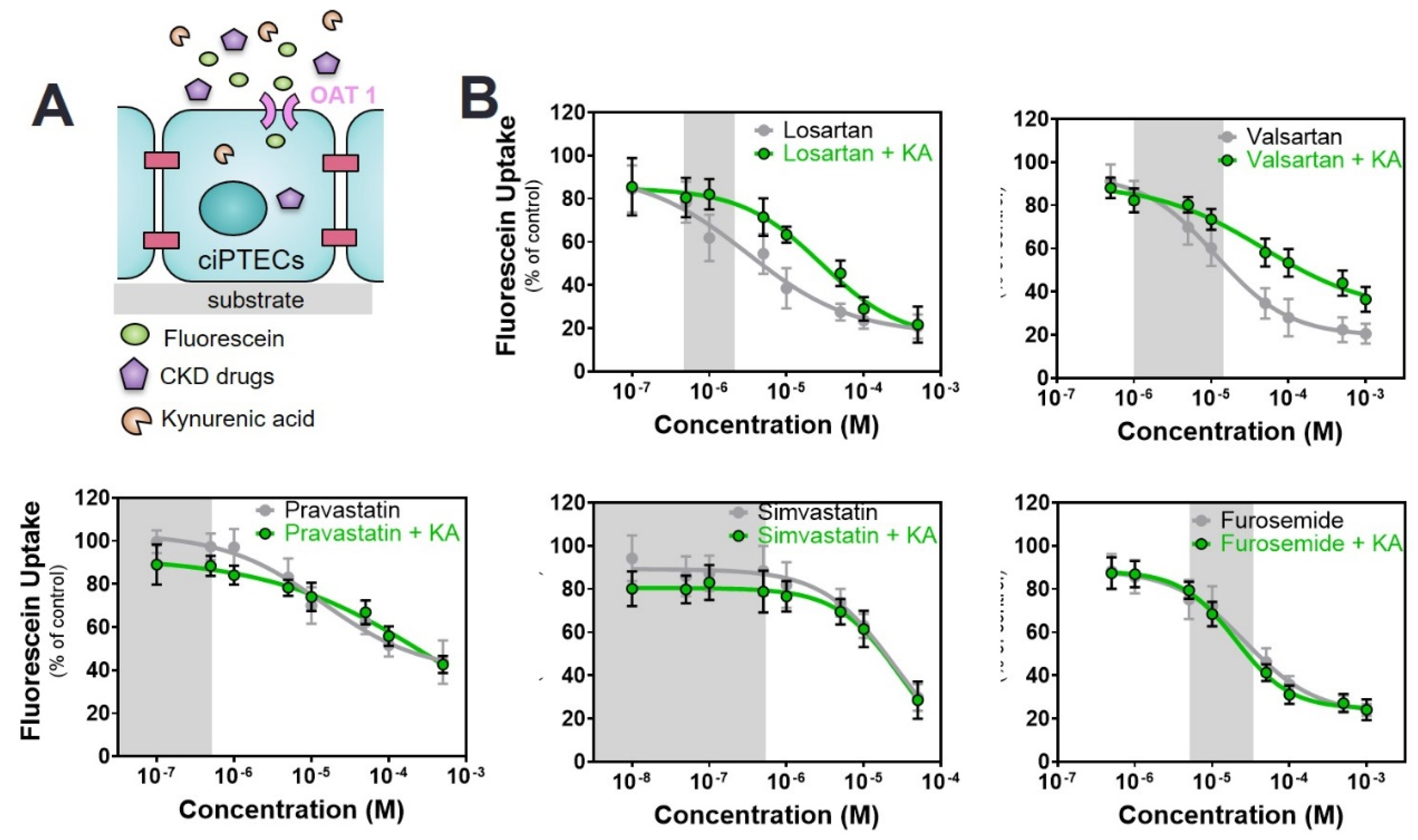
2.3. Commonly Prescribed Drugs in CKD in Combination with PBUTs Further Reduce OAT1-Mediated Uptake
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Cultures
4.3. Fluorescein Inhibition Assay
4.4. Co-Exposure of ciPTEC-OAT1 to Selected PBUTs and Drugs
4.5. Data Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- James, M.T.; Hemmelgarn, B.R.; Tonelli, M. Early recognition and prevention of chronic kidney disease. Lancet 2010, 375, 1296–1309. [Google Scholar] [CrossRef]
- Ronco, C.; Haapio, M.; House, A.A.; Anavekar, N.; Bellomo, R. Cardiorenal syndrome. J. Am. Coll. Cardiol. 2008, 52, 1527–1539. [Google Scholar] [CrossRef] [PubMed]
- Fujii, H.; Goto, S.; Fukagawa, M. Role of uremic toxins for kidney, cardiovascular, and bone dysfunction. Toxins 2018, 10, 2660. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; de Smet, R.; Lameire, N. Protein-bound uremic solutes: The forgotten toxins. Kidney Int. Suppl. 2001, 78, S266–S270. [Google Scholar] [CrossRef] [PubMed]
- Liabeuf, S.; Drueke, T.B.; Massy, Z.A. Protein-bound uremic toxins: New insight from clinical studies. Toxins 2011, 3, 911–919. [Google Scholar] [CrossRef]
- Ito, S.; Yoshida, M. Protein-bound uremic toxins: New culprits of cardiovascular events in chronic kidney disease patients. Toxins 2014, 6, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Prokopienko, A.J.; Nolin, T.D. Microbiota-derived uremic retention solutes: Perpetrators of altered nonrenal drug clearance in kidney disease. Expert Rev. Clin. Pharmacol. 2018, 11, 71–82. [Google Scholar] [CrossRef]
- Liu, W.C.; Wu, C.C.; Lim, P.S.; Chien, S.W.; Hou, Y.C.; Zheng, C.M.; Shyu, J.F.; Lin, Y.F.; Lu, K.C. Effect of uremic toxin-indoxyl sulfate on the skeletal system. Clin. Chim. Acta 2018, 484, 197–206. [Google Scholar] [CrossRef]
- Lin, Y.T.; Wu, P.H.; Liang, S.S.; Mubanga, M.; Yang, Y.H.; Hsu, Y.L.; Kuo, M.C.; Hwang, S.J.; Kuo, P.L. Protein-bound uremic toxins are associated with cognitive function among patients undergoing maintenance hemodialysis. Sci. Rep. 2019, 9, 20388. [Google Scholar] [CrossRef]
- Nigam, S.K.; Wu, W.; Bush, K.T.; Hoenig, M.P.; Blantz, R.C.; Bhatnagar, V. Handling of drugs, metabolites, and uremic toxins by kidney proximal tubule drug transporters. Clin. J. Am. Soc. Nephrol. 2015, 10, 2039–2049. [Google Scholar] [CrossRef]
- Jansen, J.; Jankowski, J.; Gajjala, P.R.; Wetzels, J.F.M.; Masereeuw, R. Disposition and clinical implications of protein-bound uremic toxins. Clin. Sci. 2017, 131, 1631–1647. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; Schepers, E.; Pletinck, A.; Nagler, E.V.; Glorieux, G. The uremic toxicity of indoxyl sulfate and p-cresyl sulfate: A systematic review. J. Am. Soc. Nephrol. 2014, 25, 1897–1907. [Google Scholar] [CrossRef] [PubMed]
- Murray, M.; Zhou, F. Trafficking and other regulatory mechanisms for organic anion transporting polypeptides and organic anion transporters that modulate cellular drug and xenobiotic influx and that are dysregulated in disease. Br. J. Pharmacol. 2017, 174, 1908–1924. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; You, G. Loops and layers of post-translational modifications of drug transporters. Adv. Drug Deliv. Rev. 2017, 116, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Yamaguchi, H.; Kogawa, T.; Abe, T.; Mano, N. Organic anion transporting polypeptides 1b1 and 1b3 play an important role in uremic toxin handling and drug-uremic toxin interactions in the liver. J. Pharm. Sci. 2014, 17, 475–484. [Google Scholar] [CrossRef]
- Manley, H.J.; Garvin, C.G.; Drayer, D.K.; Reid, G.M.; Bender, W.L.; Neufeld, T.K.; Hebbar, S.; Muther, R.S. Medication prescribing patterns in ambulatory haemodialysis patients: Comparisons of usrds to a large not-for-profit dialysis provider. Nephrol. Dial. Transplant. 2004, 19, 1842–1848. [Google Scholar] [CrossRef]
- Chiu, Y.W.; Teitelbaum, I.; Misra, M.; de Leon, E.M.; Adzize, T.; Mehrotra, R. Pill burden, adherence, hyperphosphatemia, and quality of life in maintenance dialysis patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 1089–1096. [Google Scholar] [CrossRef]
- Zandi-Nejad, K.; Brenner, B.M. Strategies to retard the progression of chronic kidney disease. Med. Clin. N. Am. 2005, 89, 489–509. [Google Scholar] [CrossRef]
- Kazancioglu, R. Risk factors for chronic kidney disease: An update. Kidney Int. Suppl. 2013, 3, 368–371. [Google Scholar] [CrossRef]
- Said, S.; Hernandez, G.T. The link between chronic kidney disease and cardiovascular disease. J. Nephropathol. 2014, 3, 99–104. [Google Scholar] [CrossRef]
- Bello, A.K.; Alrukhaimi, M.; Ashuntantang, G.E.; Basnet, S.; Rotter, R.C.; Douthat, W.G.; Kazancioglu, R.; Kottgen, A.; Nangaku, M.; Powe, N.R.; et al. Complications of chronic kidney disease: Current state, knowledge gaps, and strategy for action. Kidney Int. Suppl. 2017, 7, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Go, A.S.; Chertow, G.M.; Fan, D.; McCulloch, C.E.; Hsu, C.Y. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N. Engl. J. Med. 2004, 351, 1296–1305. [Google Scholar] [CrossRef] [PubMed]
- Keith, D.S.; Nichols, G.A.; Gullion, C.M.; Brown, J.B.; Smith, D.H. Longitudinal follow-up and outcomes among a population with chronic kidney disease in a large managed care organization. Arch. Intern. Med. 2004, 164, 659–663. [Google Scholar] [CrossRef]
- Ku, E.; McCulloch, C.E.; Vittinghoff, E.; Lin, F.; Johansen, K.L. Use of antihypertensive agents and association with risk of adverse outcomes in chronic kidney disease: Focus on angiotensin-converting enzyme inhibitors and angiotensin receptor blockers. J. Am. Heart Assoc. 2018, 7, e009992. [Google Scholar] [CrossRef] [PubMed]
- Weir, M.R.; Lakkis, J.I.; Jaar, B.; Rocco, M.V.; Choi, M.J.; Kramer, H.J.; Ku, E. Use of renin-angiotensin system blockade in advanced ckd: An nkf-kdoqi controversies report. Am. J. Kidney Dis. 2018, 72, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Liu, Y.; Perkovic, V.; Li, X.; Ninomiya, T.; Hou, W.; Zhao, N.; Liu, L.; Lv, J.; Zhang, H.; et al. Renin-angiotensin system inhibitors and kidney and cardiovascular outcomes in patients with ckd: A bayesian network meta-analysis of randomized clinical trials. Am. J. Kidney Dis. 2016, 67, 728–741. [Google Scholar] [CrossRef]
- Navaneethan, S.D.; Pansini, F.; Strippoli, G.F. Statins in patients with chronic kidney disease: Evidence from systematic reviews and randomized clinical trials. PLoS Med. 2006, 3, e123. [Google Scholar] [CrossRef]
- Tsimihodimos, V.; Mitrogianni, Z.; Elisaf, M. Dyslipidemia associated with chronic kidney disease. Open Cardiovasc. Med. J. 2011, 5, 41–48. [Google Scholar] [CrossRef]
- Masereeuw, R.; Mutsaers, H.A.; Toyohara, T.; Abe, T.; Jhawar, S.; Sweet, D.H.; Lowenstein, J. The kidney and uremic toxin removal: Glomerulus or tubule? Semin. Nephrol. 2014, 34, 191–208. [Google Scholar] [CrossRef]
- Nigam, S.K.; Bush, K.T.; Martovetsky, G.; Ahn, S.Y.; Liu, H.C.; Richard, E.; Bhatnagar, V.; Wu, W. The organic anion transporter (oat) family: A systems biology perspective. Physiol. Rev. 2015, 95, 83–123. [Google Scholar] [CrossRef]
- Deguchi, T.; Kusuhara, H.; Takadate, A.; Endou, H.; Otagiri, M.; Sugiyama, Y. Characterization of uremic toxin transport by organic anion transporters in the kidney. Kidney Int. 2004, 65, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Bush, K.T.; Nigam, S.K. Key role for the organic anion transporters, oat1 and oat3, in the in vivo handling of uremic toxins and solutes. Sci. Rep. 2017, 7, 4939. [Google Scholar] [CrossRef] [PubMed]
- Nigam, S.K.; Bush, K.T. Uraemic syndrome of chronic kidney disease: Altered remote sensing and signalling. Nat. Rev. Nephrol. 2019, 15, 301–316. [Google Scholar] [CrossRef] [PubMed]
- Jansen, J.; Schophuizen, C.M.; Wilmer, M.J.; Lahham, S.H.; Mutsaers, H.A.; Wetzels, J.F.; Bank, R.A.; van den Heuvel, L.P.; Hoenderop, J.G.; Masereeuw, R. A morphological and functional comparison of proximal tubule cell lines established from human urine and kidney tissue. Exp Cell Res. 2014, 323, 87–99. [Google Scholar] [CrossRef]
- Wilmer, M.J.; Saleem, M.A.; Masereeuw, R.; Ni, L.; van der Velden, T.J.; Russel, F.G.; Mathieson, P.W.; Monnens, L.A.; van den Heuvel, L.P.; Levtchenko, E.N. Novel conditionally immortalized human proximal tubule cell line expressing functional influx and efflux transporters. Cell Tissue Res. 2010, 339, 449–457. [Google Scholar] [CrossRef]
- Nieskens, T.T.; Peters, J.G.; Schreurs, M.J.; Smits, N.; Woestenenk, R.; Jansen, K.; van der Made, T.K.; Roring, M.; Hilgendorf, C.; Wilmer, M.J.; et al. A human renal proximal tubule cell line with stable organic anion transporter 1 and 3 expression predictive for antiviral-induced toxicity. AAPS J. 2016, 18, 465–475. [Google Scholar] [CrossRef]
- Jansen, J.; Fedecostante, M.; Wilmer, M.J.; Peters, J.G.; Kreuser, U.M.; van den Broek, P.H.; Mensink, R.A.; Boltje, T.J.; Stamatialis, D.; Wetzels, J.F.; et al. Bioengineered kidney tubules efficiently excrete uremic toxins. Sci. Rep. 2016, 6, 26715. [Google Scholar] [CrossRef]
- Truong, D.M.; Kaler, G.; Khandelwal, A.; Swaan, P.W.; Nigam, S.K. Multi-level analysis of organic anion transporters 1, 3, and 6 reveals major differences in structural determinants of antiviral discrimination. J. Biol. Chem. 2008, 283, 8654–8663. [Google Scholar] [CrossRef]
- Lacy, S.A.; Hitchcock, M.J.; Lee, W.A.; Tellier, P.; Cundy, K.C. Effect of oral probenecid coadministration on the chronic toxicity and pharmacokinetics of intravenous cidofovir in cynomolgus monkeys. Toxicol. Sci. 1998, 44, 97–106. [Google Scholar] [CrossRef]
- Tune, B.M.; Wu, K.Y.; Kempson, R.L. Inhibition of transport and prevention of toxicity of cephaloridine in the kidney. Dose-responsiveness of the rabbit and the guinea pig to probenecid. J. Pharmacol. Exp. Ther. 1977, 202, 466–471. [Google Scholar]
- Vanholder, R.; de Smet, R.; Glorieux, G.; Argiles, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; de Deyn, P.P.; Deppisch, R.; et al. Review on uremic toxins: Classification, concentration, and interindividual variability. Kidney Int. 2003, 63, 1934–1943. [Google Scholar] [CrossRef] [PubMed]
- Duranton, F.; Cohen, G.; de Smet, R.; Rodriguez, M.; Jankowski, J.; Vanholder, R.; Argiles, A.; European Uremic Toxin Work. Normal and pathologic concentrations of uremic toxins. J. Am. Soc. Nephrol. 2012, 23, 1258–1270. [Google Scholar] [CrossRef] [PubMed]
- Ehrsson, H.; Wallin, I. Cimetidine as an organic cation transporter antagonist. Am. J. Pathol. 2010, 177, 1573–1574, author reply 74. [Google Scholar] [CrossRef] [PubMed]
- Schulz, M.; Iwersen-Bergmann, S.; Andresen, H.; Schmoldt, A. Therapeutic and toxic blood concentrations of nearly 1,000 drugs and other xenobiotics. Crit. Care 2012, 16, R136. [Google Scholar] [CrossRef] [PubMed]
- Siekmeier, R.; Gross, W.; März, W. Determination of pravastatin by high performance liquid chromatography. Int. J. Clin. Pharmacol. Ther. 2000, 38, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Patrick, K.S. Goodman and Gilman’s the Pharmacological Basis of Therapeutics, 10th ed.; Hardman, J.G., Limbird, L.E., Gilman, A.G., Eds.; Mcgraw Hill: New York, NY, USA, 2001. [Google Scholar]
- Takeda, M.; Noshiro, R.; Onozato, M.L.; Tojo, A.; Hasannejad, H.; Huang, X.L.; Narikawa, S.; Endou, H. Evidence for a role of human organic anion transporters in the muscular side effects of hmg-coa reductase inhibitors. Eur. J. Pharmacol. 2004, 483, 133–138. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Watanabe, H.; Noguchi, T.; Kotani, S.; Nakajima, M.; Kadowaki, D.; Otagiri, M.; Maruyama, T. Organic anion transporters play an important role in the uptake of p-cresyl sulfate, a uremic toxin, in the kidney. Nephrol. Dial. Transplant. 2011, 26, 2498–2502. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.L.; Yoshida, K.; Zhao, P.; Zhang, L.; Nolin, T.D.; Piquette-Miller, M.; Galetin, A.; Huang, S.M. Effect of chronic kidney disease on nonrenal elimination pathways: A systematic assessment of cyp1a2, cyp2c8, cyp2c9, cyp2c19, and oatp. Clin. Pharmacol. Ther. 2018, 103, 854–867. [Google Scholar] [CrossRef]
- Saleem, A.; Masood, I.; Khan, T.M. Clinical relevancy and determinants of potential drug-drug interactions in chronic kidney disease patients: Results from a retrospective analysis. Integr. Pharm. Res. Pract. 2017, 6, 71–77. [Google Scholar] [CrossRef]
- Rowland Yeo, K.; Aarabi, M.; Jamei, M.; Rostami-Hodjegan, A. Modeling and predicting drug pharmacokinetics in patients with renal impairment. Expert Rev. Clin. Pharmacol. 2011, 4, 261–274. [Google Scholar] [CrossRef]
- Verbeeck, R.K.; Musuamba, F.T. Pharmacokinetics and dosage adjustment in patients with renal dysfunction. Eur. J. Clin. Pharmacol. 2009, 65, 757–773. [Google Scholar] [CrossRef] [PubMed]
- U.S. Department of Health and Human Services, F., Center for Drug Evaluation and Research (CDER). Clinical Drug Interaction Studies—Study Design, Data Analysis, and Clinical Implications Guidance for Industry. Available online: http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/default.htm (accessed on 10 May 2020).
- Giacomini, K.M.; Huang, S.M. Transporters in drug development and clinical pharmacology. Clin. Pharmacol. Ther. 2013, 94, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Soo, J.Y.; Jansen, J.; Masereeuw, R.; Little, M.H. Advances in predictive in vitro models of drug-induced nephrotoxicity. Nat. Rev. Nephrol. 2018, 14, 378–393. [Google Scholar] [CrossRef] [PubMed]
- Burckhardt, G. Drug transport by organic anion transporters (oats). Pharmacol. Ther. 2012, 136, 106–130. [Google Scholar] [CrossRef] [PubMed]
- Sica, D.A. Diuretic use in renal disease. Nat. Rev. Nephrol 2011, 8, 100–109. [Google Scholar] [CrossRef]
- Sato, M.; Iwanaga, T.; Mamada, H.; Ogihara, T.; Yabuuchi, H.; Maeda, T.; Tamai, I. Involvement of uric acid transporters in alteration of serum uric acid level by angiotensin ii receptor blockers. Pharm. Res. 2008, 25, 639–646. [Google Scholar] [CrossRef]
- Kuze, K.; Graves, P.; Leahy, A.; Wilson, P.; Stuhlmann, H.; You, G. Heterologous expression and functional characterization of a mouse renal organic anion transporter in mammalian cells. J. Biol. Chem. 1999, 274, 1519–1524. [Google Scholar] [CrossRef]
- Sugawara, M.; Mochizuki, T.; Takekuma, Y.; Miyazaki, K. Structure-affinity relationship in the interactions of human organic anion transporter 1 with caffeine, theophylline, theobromine and their metabolites. Biochim. Biophys. Acta 2005, 1714, 85–92. [Google Scholar] [CrossRef]
- Reyes, M.; Benet, L.Z. Effects of uremic toxins on transport and metabolism of different biopharmaceutics drug disposition classification system xenobiotics. J. Pharm. Sci. 2011, 100, 3831–3842. [Google Scholar] [CrossRef]
- Mutsaers, H.A.; Wilmer, M.J.; Reijnders, D.; Jansen, J.; van den Broek, P.H.; Forkink, M.; Schepers, E.; Glorieux, G.; Vanholder, R.; van den Heuvel, L.P.; et al. Uremic toxins inhibit renal metabolic capacity through interference with glucuronidation and mitochondrial respiration. Biochim. Biophys. Acta 2013, 1832, 142–150. [Google Scholar] [CrossRef]
- Sun, H.; Frassetto, L.; Benet, L.Z. Effects of renal failure on drug transport and metabolism. Pharmacol. Ther. 2006, 109, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, A.; Takeda, M.; Taki, K.; Takayama, F.; Noshiro, R.; Niwa, T.; Endou, H. Interactions of human organic anion as well as cation transporters with indoxyl sulfate. Eur. J. Pharmacol. 2003, 466, 13–20. [Google Scholar] [CrossRef]
- Uwai, Y.; Honjo, H.; Iwamoto, K. Interaction and transport of kynurenic acid via human organic anion transporters hoat1 and hoat3. Pharmacol. Res. 2012, 65, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Eloot, S.; van Biesen, W.; Roels, S.; Delrue, W.; Schepers, E.; Dhondt, A.; Vanholder, R.; Glorieux, G. Spontaneous variability of pre-dialysis concentrations of uremic toxins over time in stable hemodialysis patients. PLoS ONE 2017, 12, e0186010. [Google Scholar] [CrossRef]
- Hilmas, C.; Pereira, E.F.; Alkondon, M.; Rassoulpour, A.; Schwarcz, R.; Albuquerque, E.X. The brain metabolite kynurenic acid inhibits alpha7 nicotinic receptor activity and increases non-alpha7 nicotinic receptor expression: Physiopathological implications. J. Neurosci. 2001, 21, 7463–7473. [Google Scholar] [CrossRef]
- Viaene, L.; Annaert, P.; de Loor, H.; Poesen, R.; Evenepoel, P.; Meijers, B. Albumin is the main plasma binding protein for indoxyl sulfate and p-cresyl sulfate. Biopharm. Drug Dispos. 2013, 34, 165–175. [Google Scholar] [CrossRef]
- Fabresse, N.; Uteem, I.; Lamy, E.; Massy, Z.; Larabi, I.A.; Alvarez, J.C. Quantification of free and protein bound uremic toxins in human serum by lc-ms/ms: Comparison of rapid equilibrium dialysis and ultrafiltration. Clin. Chim. Acta 2020, 507, 228–235. [Google Scholar] [CrossRef]
- Van der Made, T.K.; Fedecostante, M.; Scotcher, D.; Rostami-Hodjegan, A.; Torano, J.S.; Middel, I.; Koster, A.S.; Gerritsen, K.G.; Jankowski, V.; Jankowski, J.; et al. Quantitative translation of microfluidic transporter in vitro data to in vivo reveals impaired albumin-facilitated indoxyl sulfate secretion in chronic kidney disease. Mol. Pharm. 2019, 16, 4551–4562. [Google Scholar] [CrossRef]
- Deltombe, O.; van Biesen, W.; Glorieux, G.; Massy, Z.; Dhondt, A.; Eloot, S. Exploring protein binding of uremic toxins in patients with different stages of chronic kidney disease and during hemodialysis. Toxins 2015, 7, 3933–3946. [Google Scholar] [CrossRef]
- Rueth, M.; Lemke, H.D.; Preisinger, C.; Krieter, D.; Theelen, W.; Gajjala, P.; Devine, E.; Zidek, W.; Jankowski, J.; Jankowski, V. Guanidinylations of albumin decreased binding capacity of hydrophobic metabolites. Acta Physiol. 2015, 215, 13–23. [Google Scholar] [CrossRef]
- Takamura, N.; Maruyama, T.; Otagiri, M. Effects of uremic toxins and fatty acids on serum protein binding of furosemide: Possible mechanism of the binding defect in uremia. Clin. Chem. 1997, 43, 2274–2280. [Google Scholar] [CrossRef] [PubMed]
- Mingrone, G.; de Smet, R.; Greco, A.V.; Bertuzzi, A.; Gandolfi, A.; Ringoir, S.; Vanholder, R. Serum uremic toxins from patients with chronic renal failure displace the binding of l-tryptophan to human serum albumin. Clin. Chim. Acta 1997, 260, 27–34. [Google Scholar] [CrossRef]
- Florens, N.; Yi, D.; Juillard, L.; Soulage, C.O. Using binding competitors of albumin to promote the removal of protein-bound uremic toxins in hemodialysis: Hope or pipe dream? Biochimie 2018, 144, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Santana Machado, T.; Cerini, C.; Burtey, S. Emerging roles of aryl hydrocarbon receptors in the altered clearance of drugs during chronic kidney disease. Toxins 2019, 11, 209. [Google Scholar] [CrossRef]
- Varma, M.V.; Feng, B.; Obach, R.S.; Troutman, M.D.; Chupka, J.; Miller, H.R.; El-Kattan, A. Physicochemical determinants of human renal clearance. J. Med. Chem 2009, 52, 4844–4852. [Google Scholar] [CrossRef]
- Weigand, K.M.; Schirris, T.J.J.; Houweling, M.; van den Heuvel, J.; Koenderink, J.B.; Dankers, A.C.A.; Russel, F.G.M.; Greupink, R. Uremic solutes modulate hepatic bile acid handling and induce mitochondrial toxicity. Toxicol. In Vitro 2019, 56, 52–61. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | IC50 (µM) a | R Square b | Therapeutic Concentrations (µM) [44] |
|---|---|---|---|
| ACEIs | |||
| Captopril | 2022 ± 465 | 0.5557 | 0.2–5 |
| Enalaprilate | 1853 ± 370 | 0.6753 | 0.04–0.4 |
| Lisinopril | not available | 0.1671 | 0.01–0.16 |
| ARBs | |||
| Losartan | 3.1 ± 0.7 | 0.8713 | 0.5–1.5 |
| Valsartan | 11.5 ± 3.5 | 0.9374 | 2–14 |
| Diuretics | |||
| Furosemide | 28.1 ± 9.1 | 0.9301 | 6–30 |
| Statins | |||
| Pravastatin | 13.8 ± 8.5 | 0.8757 | 0.08–0.3 [45]; 0.43 [46,47] |
| Simvastatin | 21.3 ± 3.8 | 0.8613 | 0.006–0.014 [44]; 0.55 [46,47] |
| H2RA | |||
| Cimetidine | 887.6 ± 198.4 | 0.7441 | 1–16 |
| Drug/Toxin | − | +IS 110 µM | +KA 1 µM | +PCS 125 µM |
|---|---|---|---|---|
| ARBs | ||||
| Losartan | 8.6 ± 2.5 | 13.9 ± 5.9 b | 28.2 ± 2.7 a | 15.97 ± 3.9 a |
| Valsartan | 11.5 ± 3.5 | 16.1 ± 3.6 b | 46.9 ± 4.6 a | 17.9 ± 3.8 b |
| Statins | ||||
| Pravastatin | 13.8 ± 8.35 | 40.9 ± 9.2 a | 243.0 ± 45.8 a | 19.1± 3.2 a |
| Simvastatin | 21.3 ± 3.8 | 71.8 ± 27.3 a | 28.4 ± 10.1 b | 32.8 ± 7.6 a |
| Diuretics | ||||
| Furosemide | 28.1 ± 9.1 | 44.7 ± 12.4 a | 24.8 ± 6.2 b | 60.2 ± 1.0 a |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mihaila, S.M.; Faria, J.; Stefens, M.F.J.; Stamatialis, D.; Verhaar, M.C.; Gerritsen, K.G.F.; Masereeuw, R. Drugs Commonly Applied to Kidney Patients May Compromise Renal Tubular Uremic Toxins Excretion. Toxins 2020, 12, 391. https://doi.org/10.3390/toxins12060391
Mihaila SM, Faria J, Stefens MFJ, Stamatialis D, Verhaar MC, Gerritsen KGF, Masereeuw R. Drugs Commonly Applied to Kidney Patients May Compromise Renal Tubular Uremic Toxins Excretion. Toxins. 2020; 12(6):391. https://doi.org/10.3390/toxins12060391
Chicago/Turabian StyleMihaila, Silvia M., João Faria, Maurice F. J. Stefens, Dimitrios Stamatialis, Marianne C. Verhaar, Karin G. F. Gerritsen, and Rosalinde Masereeuw. 2020. "Drugs Commonly Applied to Kidney Patients May Compromise Renal Tubular Uremic Toxins Excretion" Toxins 12, no. 6: 391. https://doi.org/10.3390/toxins12060391
APA StyleMihaila, S. M., Faria, J., Stefens, M. F. J., Stamatialis, D., Verhaar, M. C., Gerritsen, K. G. F., & Masereeuw, R. (2020). Drugs Commonly Applied to Kidney Patients May Compromise Renal Tubular Uremic Toxins Excretion. Toxins, 12(6), 391. https://doi.org/10.3390/toxins12060391