Chronic Kidney Disease-Associated Immune Dysfunctions: Impact of Protein-Bound Uremic Retention Solutes on Immune Cells


Abstract
1. Introduction
2. Protein-Bound Uremic Retention Solutes
2.1. Classification of Uremic Retention Solutes (URS)
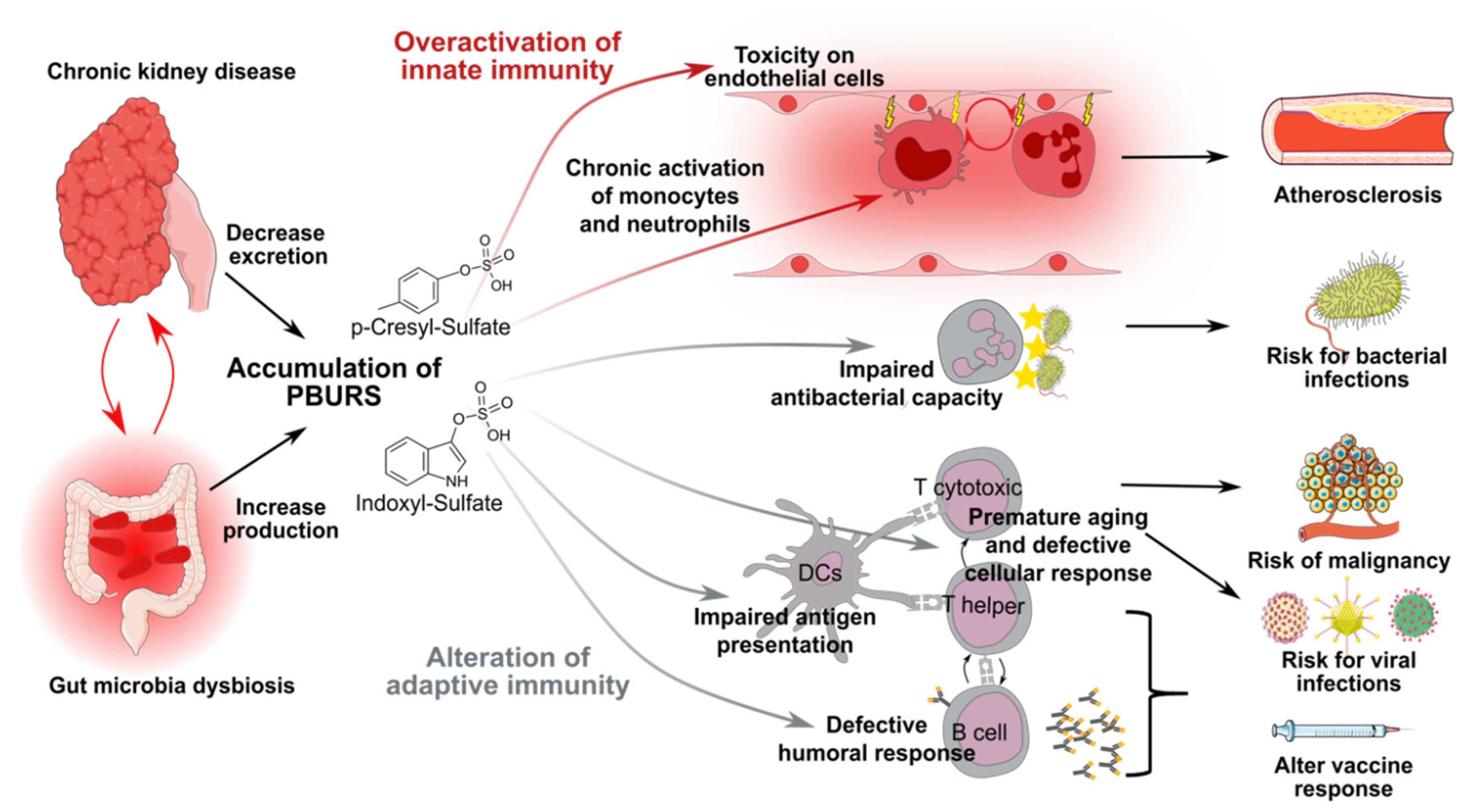
2.2. CKD-Associated Dysbiosis and PBURS
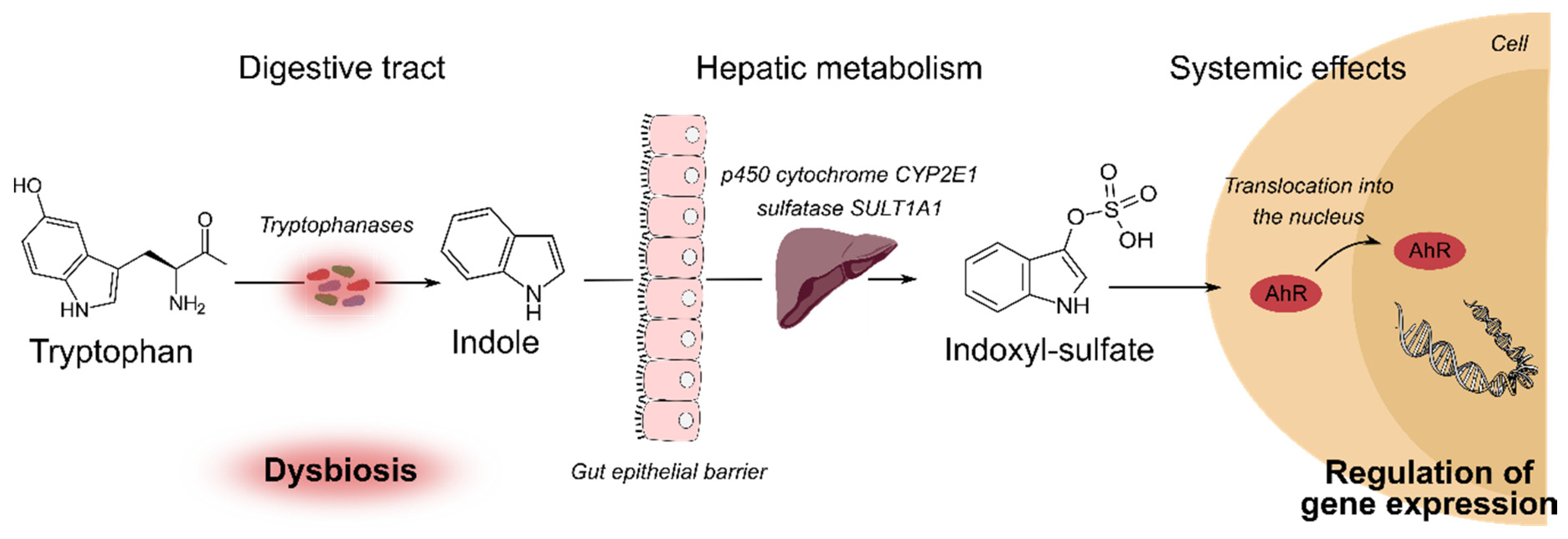
2.3. Tryptophan Catabolites
2.4. Phenols Derivates
2.5. Role of PBURS in CKD-Associated Complications
3. CKD Induces Chronic Activation of Innate Effectors and Endothelial Damages
4. Neutrophils Responses Against Extracellular Bacteria are Impaired During CKD
5. Adaptive T-Cell Responses are Impaired in CKD Patients
6. CKD Induces Defective Humoral Responses
7. Conclusions and Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Webster, A.C.; Nagler, E.V.; Morton, R.L.; Masson, P. Chronic Kidney Disease. Lancet 2017, 389, 1238–1252. [Google Scholar] [CrossRef]
- Eckardt, K.U.; Coresh, J.; Devuyst, O.; Johnson, R.J.; Köttgen, A.; Levey, A.S.; Levin, A. Evolving importance of kidney disease: From subspecialty to global health burden. Lancet 2013, 382, 158–169. [Google Scholar] [CrossRef]
- Vanholder, R.; Pletinck, A.; Schepers, E.; Glorieux, G. Biochemical and Clinical Impact of Organic Uremic Retention Solutes: A Comprehensive Update. Toxins 2018, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Mathew, R.O.; Bangalore, S.; Lavelle, M.P.; Pellikka, P.A.; Sidhu, M.S.; Boden, W.E.; Asif, A. Diagnosis and management of atherosclerotic cardiovascular disease in chronic kidney disease: A review. Kidney Int. 2017, 91, 797–807. [Google Scholar] [CrossRef]
- Xu, H.; Matsushita, K.; Su, G.; Trevisan, M.; Ärnlöv, J.; Barany, P.; Lindholm, B.; Elinder, C.G.; Lambe, M.; Carrero, J.J. Estimated Glomerular Filtration Rate and the Risk of Cancer. Clin. J. Am. Soc. Nephrol. 2019, 14, 530–539. [Google Scholar] [CrossRef]
- Sarnak, M.; Jaber, B. Mortality caused by sepsis in patients with end-stage renal disease compared with the general population. Kidney Int. 2000, 58, 1758–1764. [Google Scholar] [CrossRef]
- Ishigami, J.; Grams, M.E.; Chang, A.R.; Carrero, J.J.; Coresh, J.; Matsushita, K. CKD and Risk for Hospitalization with Infection: The Atherosclerosis Risk in Communities (ARIC) Study. Am. J. Kidney Dis. 2017, 69, 752–761. [Google Scholar] [CrossRef]
- Thompson, S.; James, M.; Wiebe, N.; Hemmelgarn, B.; Manns, B.; Klarenbach, S.; Tonelli, M. Cause of Death in Patients with Reduced Kidney Function. J. Am. Soc. Nephrol. 2015, 26, 2504–2511. [Google Scholar] [CrossRef]
- Ma, Y.; Diao, B.; Lv, X.; Zhu, J.; Liang, W.; Liu, L.; Bu, W.; Cheng, H.; Zhang, S.; Yang, L.; et al. 2019 novel coronavirus disease in hemodialysis (HD) patients: Report from one HD center in Wuhan, China. medRxiv 2020. [Google Scholar] [CrossRef]
- Vanholder, R.; Meert, N.; Schepers, E.; Glorieux, G.; Argiles, A.; Brunet, P.; Cohen, G.; Drüeke, T.; Mischak, H.; Spasovski, G.; et al. Review on uraemic solutes II—Variability in reported concentrations: Causes and consequences. Nephrol. Dial. Transplant. 2007, 22, 3115–3121. [Google Scholar] [CrossRef]
- Koppe, L.; Nyam, E.; Vivot, K.; Fox, J.E.M.; Dai, X.Q.; Nguyen, B.N.; Trudel, D.; Attané, C.; Moullé, V.S.; MacDonald, P.E.; et al. Urea impairs β cell glycolysis and insulin secretion in chronic kidney disease. J. Clin. Investig. 2016, 126, 3598–3612. [Google Scholar] [CrossRef] [PubMed]
- Vervloet, M.G.; Sezer, S.; Massy, Z.A.; Johansson, L.; Cozzolino, M.; Fouque, D. ERA–EDTA Working Group on Chronic Kidney Disease–Mineral and Bone Disorders and the European Renal Nutrition Working Group. The role of phosphate in kidney disease. Nat. Rev. Nephrol. 2017, 13, 27–38. [Google Scholar] [CrossRef]
- Duranton, F.; Cohen, G.; De Smet, R.; Rodriguez, M.; Jankowski, J.; Vanholder, R.; Argiles, A. Normal and Pathologic Concentrations of Uremic Toxins. J. Am. Soc. Nephrol. 2012, 23, 1258–1270. [Google Scholar] [CrossRef] [PubMed]
- Gryp, T.; Vanholder, R.; Vaneechoutte, M.; Glorieux, G. p-Cresyl Sulfate. Toxins 2017, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; Schepers, E.; Pletinck, A.; Nagler, E.V.; Glorieux, G. The uremic toxicity of indoxyl sulfate and p-cresyl sulfate: A systematic review. J. Am. Soc. Nephrol. 2014, 25, 1897–1907. [Google Scholar] [CrossRef] [PubMed]
- Tremaroli, V.; Bäckhed, F. Functional interactions between the gut microbiota and host metabolism. Nature 2012, 489, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Wong, J.; Pahl, M.; Piceno, Y.M.; Yuan, J.; DeSantis, T.Z.; Ni, Z.; Nguyen, T.H.; Andersen, G.L. Chronic kidney disease alters intestinal microbial flora. Kidney Int. 2013, 83, 308–315. [Google Scholar] [CrossRef]
- Koppe, L.; Mafra, D.; Fouque, D. Probiotics and chronic kidney disease. Kidney Int. 2015, 88, 958–966. [Google Scholar] [CrossRef]
- Vaziri, N.D.; Yuan, J.; Nazertehrani, S.; Ni, Z.; Liu, S. Chronic Kidney Disease Causes Disruption of Gastric and Small Intestinal Epithelial Tight Junction. Am. J. Nephrol. 2013, 38, 99–103. [Google Scholar] [CrossRef]
- Mishima, E.; Fukuda, S.; Mukawa, C.; Yuri, A.; Kanemitsu, Y.; Matsumoto, Y.; Akiyama, Y.; Fukuda, N.N.; Tsukamoto, H.; Asaji, K.; et al. Evaluation of the impact of gut microbiota on uremic solute accumulation by a CE-TOFMS-based metabolomics approach. Kidney Int. 2017, 92, 634–645. [Google Scholar] [CrossRef]
- Aronov, P.A.; Luo, F.J.G.; Plummer, N.S.; Quan, Z.; Holmes, S.; Hostetter, T.H.; Meyer, T.W. Colonic contribution to uremic solutes. J. Am. Soc. Nephrol. 2011, 22, 1769–1776. [Google Scholar] [CrossRef]
- Roager, H.M.; Licht, T.R. Microbial tryptophan catabolites in health and disease. Nat. Commun. 2018, 9, 3294. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.A.; Macfarlane, G.T. Enumeration of human colonic bacteria producing phenolic and indolic compounds: Effects of pH, carbohydrate availability and retention time on dissimilatory aromatic amino acid metabolism. J. Appl. Bacteriol. 1996, 81, 288–302. [Google Scholar] [CrossRef] [PubMed]
- Elsden, S.R.; Hilton, M.G.; Waller, J.M. The end products of the metabolism of aromatic amino acids by clostridia. Arch. Microbiol. 1976, 107, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Bansal, T.; Alaniz, R.C.; Wood, T.K.; Jayaraman, A. The bacterial signal indole increases epithelial-cell tight-junction resistance and attenuates indicators of inflammation. Proc. Natl. Acad. Sci. USA 2010, 107, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Shimada, Y.; Kinoshita, M.; Harada, K.; Mizutani, M.; Masahata, K.; Kayama, H.; Takeda, K. Commensal Bacteria-Dependent Indole Production Enhances Epithelial Barrier Function in the Colon. PLoS ONE 2013, 8, e80604. [Google Scholar] [CrossRef]
- Schirmer, M.; Smeekens, S.P.; Vlamakis, H.; Jaeger, M.; Oosting, M.; Franzosa, E.A.; Horst, R.T.; Jansen, T.; Jacobs, L.; Bonder, M.J.; et al. Linking the Human Gut Microbiome to Inflammatory Cytokine Production Capacity. Cell 2016, 167, 1897. [Google Scholar] [CrossRef]
- Wlodarska, M.; Luo, C.; Kolde, R.; D’Hennezel, E.; Annand, J.W.; Heim, C.E.; Krastel, P.; Schmitt, E.K.; Omar, A.S.; Creasey, E.A.; et al. Indoleacrylic Acid Produced by Commensal Peptostreptococcus Species Suppresses Inflammation. Cell Host Microbe 2017, 22, 25–37.e6. [Google Scholar] [CrossRef]
- Hwang, I.K.; Yoo, K.Y.; Li, H.; Park, O.K.; Lee, C.H.; Choi, J.H.; Jeong, Y.G.; Lee, Y.L.; Kim, Y.M.; Kwon, Y.G.; et al. Indole-3-propionic acid attenuates neuronal damage and oxidative stress in the ischemic hippocampus. J. Neurosci. Res. 2009, 87, 2126–2137. [Google Scholar] [CrossRef]
- Sonowal, R.; Swimm, A.; Sahoo, A.; Luo, L.; Matsunaga, Y.; Wu, Z.; Bhingarde, J.A.; Ejzak, E.A.; Ranawade, A.; Qadota, H.; et al. Indoles from commensal bacteria extend healthspan. Proc. Natl. Acad. Sci. USA 2017, 114, E7506–E7515. [Google Scholar] [CrossRef]
- Hubbard, T.D.; Murray, I.A.; Bisson, W.H.; Lahoti, T.S.; Gowda, K.; Amin, S.G.; Patterson, A.D.; Perdew, G.H. Adaptation of the human aryl hydrocarbon receptor to sense microbiota-derived indoles. Sci. Rep. 2015, 5, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Rothhammer, V.; Quintana, F.J. The aryl hydrocarbon receptor: An environmental sensor integrating immune responses in health and disease. Nat. Rev. Immunol. 2019, 19, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Missailidis, C.; Hällqvist, J.; Qureshi, A.R.; Barany, P.; Heimbürger, O.; Lindholm, B.; Stenvinkel, P.; Bergman, P. Serum Trimethylamine-N-Oxide Is Strongly Related to Renal Function and Predicts Outcome in Chronic Kidney Disease. PLoS ONE 2016, 11, e0141738. [Google Scholar] [CrossRef] [PubMed]
- Koppe, L.; Fouque, D.; Soulage, C.O. The Role of Gut Microbiota and Diet on Uremic Retention Solutes Production in the Context of Chronic Kidney Disease. Toxins 2018, 10, 155. [Google Scholar] [CrossRef]
- Suchy-Dicey, A.M.; Laha, T.; Hoofnagle, A.; Newitt, R.; Sirich, T.L.; Meyer, T.W.; Thummel, K.E.; Yanez, N.D.; Himmelfarb, J.; Weiss, N.S.; et al. Tubular Secretion in CKD. J. Am. Soc. Nephrol. 2016, 27, 2148–2155. [Google Scholar] [CrossRef]
- Koppe, L.; Pillon, N.J.; Vella, R.E.; Croze, M.L.; Pelletier, C.C.; Chambert, S.; Massy, Z.; Glorieux, G.; Vanholder, R.; Dugenet, Y.; et al. p-Cresyl Sulfate Promotes Insulin Resistance Associated with CKD. J. Am. Soc. Nephrol. 2013, 24, 88–99. [Google Scholar] [CrossRef]
- Watanabe, H.; Miyamoto, Y.; Honda, D.; Tanaka, H.; Wu, Q.; Endo, M.; Noguchi, T.; Kadowaki, D.; Ishima, Y.; Kotani, S.; et al. p-Cresyl sulfate causes renal tubular cell damage by inducing oxidative stress by activation of NADPH oxidase. Kidney Int. 2013, 83, 582–592. [Google Scholar] [CrossRef]
- Poveda, J.; Sanchez-Niño, M.D.; Glorieux, G.; Sanz, A.B.; Egido, J.; Vanholder, R.; Ortiz, A. p-cresyl sulphate has pro-inflammatory and cytotoxic actions on human proximal tubular epithelial cells. Nephrol. Dial. Transplant. 2014, 29, 56–64. [Google Scholar] [CrossRef]
- Schepers, E.; Meert, N.; Glorieux, G.; Goeman, J.; Van der Eycken, J.; Vanholder, R. P-cresylsulphate, the main In Vivo metabolite of p-cresol, activates leucocyte free radical production. Nephrol. Dial. Transplant. 2007, 22, 592–596. [Google Scholar] [CrossRef]
- Lau, W.L.; Savoj, J.; Nakata, M.B.; Vaziri, N.D. Altered microbiome in chronic kidney disease: Systemic effects of gut-derived uremic toxins. Clin. Sci. 2018, 132, 509–522. [Google Scholar] [CrossRef]
- Opdebeeck, B.; Maudsley, S.; Azmi, A.; De Maré, A.; De Leger, W.; Meijers, B.; Verhulst, A.; Evenepoel, P.; D’Haese, P.C.; Neven, E. Indoxyl Sulfate and p-Cresyl Sulfate Promote Vascular Calcification and Associate with Glucose Intolerance. J. Am. Soc. Nephrol. 2019, 30, 751–766. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.S.; Chen, J.; Zou, J.Z.; Zhong, Y.H.; Teng, J.; Ji, J.; Chen, Z.W.; Liu, Z.H.; Shen, B.; Nie, Y.X.; et al. Association of Indoxyl Sulfate with Heart Failure among Patients on Hemodialysis. Clin. J. Am. Soc. Nephrol. 2015, 10, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Barreto, F.C.; Barreto, D.V.; Liabeuf, S.; Meert, N.; Glorieux, G.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A. on behalf of the European Uremic Toxin Work Group (EUTox). Serum Indoxyl Sulfate is Associated with Vascular Disease and Mortality in Chronic Kidney Disease Patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 1551–1558. [Google Scholar] [CrossRef] [PubMed]
- Chiang, C.K.; Tanaka, T.; Inagi, R.; Fujita, T.; Nangaku, M. Indoxyl sulfate, a representative uremic toxin, suppresses erythropoietin production in a HIF-dependent manner. Lab. Investig. 2011, 91, 1564–1571. [Google Scholar] [CrossRef] [PubMed]
- Hirata, J.; Hirai, K.; Asai, H.; Matsumoto, C.; Inada, M.; Miyaura, C.; Yamato, H.; Watanabe-Akanuma, M. Indoxyl sulfate exacerbates low bone turnover induced by parathyroidectomy in young adult rats. Bone 2015, 79, 252–258. [Google Scholar] [CrossRef]
- Hörl, W.H. Hemodialysis membranes: Interleukins, biocompatibility, and middle molecules. J. Am. Soc. Nephrol. 2002, 13, S62–S71. [Google Scholar]
- Paul, E.W. Fundamental Immunology; Wolters Kluwer: Alphen aan den Rijn, The Netherlands, 2012. [Google Scholar]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Grabulosa, C.C.; Manfredi, S.R.; Canziani, M.E.; Quinto, B.M.R.; Barbosa, R.B.; Rebello, J.F.; Batista, M.C.; Cendoroglo, M.; Dalboni, M.A. Chronic kidney disease induces inflammation by increasing Toll-like receptor-4, cytokine and cathelicidin expression in neutrophils and monocytes. Exp. Cell Res. 2018, 365, 157–162. [Google Scholar] [CrossRef]
- Mihai, S.; Codrici, E.; Popescu, I.D.; Enciu, A.M.; Albulescu, L.; Necula, L.G.; Mambet, C.; Anton, G.; Tanase, C. Inflammation-Related Mechanisms in Chronic Kidney Disease Prediction, Progression, and Outcome. J. Immunol Res. 2018, 2018, 2180373. [Google Scholar] [CrossRef]
- Amdur, R.L.; Feldman, H.I.; Gupta, J.; Yang, W.; Kanetsky, P.; Shlipak, M.; Rahman, M.; Lash, J.P.; Townsend, R.R.; Ojo, A.; et al. Inflammation and Progression of CKD: The CRIC Study. Clin. J. Am. Soc. Nephrol. 2016, 11, 1546–1556. [Google Scholar] [CrossRef]
- Castillo-Rodríguez, E.; Pizarro-Sánchez, S.; Sanz, A.B.; Ramos, A.M.; Sanchez-Niño, M.D.; Martin-Cleary, C.; Fernandez-Fernandez, B.; Ortiz, A. Inflammatory Cytokines as Uremic Toxins: “Ni Son Todos Los Que Estan, Ni Estan Todos Los Que Son”. Toxins 2017. [Google Scholar] [CrossRef] [PubMed]
- Sela, S.; Shurtz-Swirski, R.; Cohen-Mazor, M.; Mazor, R.; Chezar, J.; Shapiro, G.; Hassan, K.; Shkolnik, G.; Geron, R.; Kristal, B. Primed peripheral polymorphonuclear leukocyte: A culprit underlying chronic low-grade inflammation and systemic oxidative stress in chronic kidney disease. J. Am. Soc. Nephrol. 2005, 16, 2431–2438. [Google Scholar] [CrossRef] [PubMed]
- Koc, M.; Toprak, A.; Arikan, H.; Odabasi, Z.; Elbir, Y.; Tulunay, A.; Asicioglu, E.; Eksioglu-Demiralp, E.; Glorieux, G.; Vanholder, R.; et al. Toll-like receptor expression in monocytes in patients with chronic kidney disease and haemodialysis: Relation with inflammation. Nephrol. Dial. Transplant. 2011, 26, 955–963. [Google Scholar] [CrossRef]
- Gollapudi, P.; Yoon, J.W.; Gollapudi, S.; Pahl, M.V.; Vaziri, N.D. Leukocyte Toll-Like Receptor Expression in End-Stage Kidney Disease. Am. J. Nephrol. 2010, 31, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Gondouin, B.; Cerini, C.; Dou, L.; Sallée, M.; Duval-Sabatier, A.; Pletinck, A.; Calaf, R.; Lacroix, R.; Jourde-Chiche, N.; Poitevin, S.; et al. Indolic uremic solutes increase tissue factor production in endothelial cells by the aryl hydrocarbon receptor pathway. Kidney Int. 2013, 84, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Yoo, T.H.; Cho, J.Y.; Kim, H.C.; Lee, W.W. Indoxyl sulfate-induced TNF-α is regulated by crosstalk between the aryl hydrocarbon receptor, NF-κB, and SOCS2 in human macrophages. FASEB J. 2019, 33, 10844–10858. [Google Scholar] [CrossRef]
- Kim, H.Y.; Yoo, T.H.; Hwang, Y.; Lee, G.H.; Kim, B.; Jang, J.; Yu, H.T.; Kim, M.C.; Cho, J.Y.; Lee, C.J.; et al. Indoxyl sulfate (IS)-mediated immune dysfunction provokes endothelial damage in patients with end-stage renal disease (ESRD). Sci. Rep. 2017, 7, 3057. [Google Scholar] [CrossRef]
- Zimmermann, J.; Herrlinger, S.; Pruy, A.; Metzger, T.; Wanner, C. Inflammation enhances cardiovascular risk and mortality in hemodialysis patients. Kidney Int. 1999, 55, 648–658. [Google Scholar] [CrossRef]
- Gross, P.; Massy, Z.A.; Henaut, L.; Boudot, C.; Cagnard, J.; March, C.; Kamel, S.; Drueke, T.B.; Six, I. Para-cresyl sulfate acutely impairs vascular reactivity and induces vascular remodeling. J. Cell. Physiol. 2015, 230, 2927–2935. [Google Scholar] [CrossRef]
- Huang, M.; Wei, R.; Wang, Y.; Su, T.; Li, P.; Chen, X. The uremic toxin hippurate promotes endothelial dysfunction via the activation of Drp1-mediated mitochondrial fission. Redox Biol 2018, 16, 303–313. [Google Scholar] [CrossRef]
- Sallée, M.; Dou, L.; Cerini, C.; Poitevin, S.; Brunet, P.; Burtey, S. The aryl hydrocarbon receptor-activating effect of uremic toxins from tryptophan metabolism: A new concept to understand cardiovascular complications of chronic kidney disease. Toxins 2014, 6, 934–949. [Google Scholar] [CrossRef] [PubMed]
- Addi, T.; Dou, L.; Burtey, S. Tryptophan-Derived Uremic Toxins and Thrombosis in Chronic Kidney Disease. Toxins 2018, 10, 412. [Google Scholar] [CrossRef] [PubMed]
- Jalal, D.; Renner, B.; Laskowski, J.; Stites, E.; Cooper, J.; Valente, K.; You, Z.; Perrenoud, L.; Le Quintrec, M.; Muhamed, I.; et al. Endothelial Microparticles and Systemic Complement Activation in Patients With Chronic Kidney Disease. J. Am. Heart Assoc. 2018, 7. [Google Scholar] [CrossRef]
- Yu, M.; Kim, Y.J.; Kang, D.H. Indoxyl Sulfate-Induced Endothelial Dysfunction in Patients with Chronic Kidney Disease via an Induction of Oxidative Stress. Clin. J. Am. Soc. Nephrol. 2011, 6, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Pletinck, A.; Glorieux, G.; Schepers, E.; Cohen, G.; Gondouin, B.; Landschoot, M.V.; Eloot, S.; Rops, A.; de Voorde, J.V.; Vriese, A.D.; et al. Protein-Bound Uremic Toxins Stimulate Crosstalk between Leukocytes and Vessel Wall. J. Am. Soc. Nephrol. 2013, 24, 1981–1994. [Google Scholar] [CrossRef] [PubMed]
- Cendoroglo, M.; Jaber, B.L.; Balakrishnan, V.S.; Perianayagam, M.; King, A.J.; Pereira, B.J. Neutrophil apoptosis and dysfunction in uremia. J. Am. Soc. Nephrol. 1999, 10, 93–100. [Google Scholar] [PubMed]
- Xiang, F.; Zhu, J.; Cao, X.; Shen, B.; Zou, J.; Liu, Z.; Zhang, H.; Teng, J.; Liu, H.; Ding, X. Lymphocyte depletion and subset alteration correlate to renal function in chronic kidney disease patients. Ren. Fail. 2016, 38, 7–14. [Google Scholar] [CrossRef]
- Sarabandi, A.; Manafi Shabestari, R.; Farshi, Y.; Tabibian, S.; Dorgalaleh, A.; Esmaeili Reykande, S.; Kia, S.H.; Varmaghani, B.; Rashidpanah, J. Uremia Effect on White Blood Cell Count in Patients with Renal Failure. Int. J. Med. Lab. 2015, 2, 21–24. [Google Scholar]
- Ferrante, A.; Maxwell, G.M.; Rencis, V.O.; Allison, A.C.; Morgan, D.M.L. Inhibition of the respiratory burst of human neutrophils by the polyamine oxidase-polyamine system. Int. J. Immunopharmacol. 1986, 8, 411–417. [Google Scholar] [CrossRef]
- Ottonello, L.; Gnerre, P.; Bertolotto, M.; Mancini, M.; Dapino, P.; Russo, R.; Garibotto, G.; Barreca, T.; Dallegri, F. Leptin as a Uremic Toxin Interferes with Neutrophil Chemotaxis. J. Am. Soc. Nephrol. 2004, 15, 2366–2372. [Google Scholar] [CrossRef]
- Mahajan, S.; Kalra, O.P.; Asit, K.T.; Ahuja, G.; Kalra, V. Phagocytic polymorphonuclear function in patients with progressive uremia and the effect of acute hemodialysis. Ren. Fail. 2005, 27, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Schulz, A.M.; Terne, C.; Jankowski, V.; Cohen, G.; Schaefer, M.; Boehringer, F.; Tepel, M.; Kunkel, D.; Zidek, W.; Jankowski, J. Modulation of NADPH oxidase activity by known uraemic retention solutes. Eur. J. Clin. Investig. 2014, 44, 802–811. [Google Scholar] [CrossRef]
- Hirayama, A.; Noronha-Dutra, A.A.; Gordge, M.P.; Neild, G.H.; Hothersall, J.S. Inhibition of Neutrophil Superoxide Production by Uremic Concentrations of Guanidino Compounds. J. Am. Soc. Nephrol. 2000, 11, 684–689. [Google Scholar] [PubMed]
- Vanholder, R.; De Smet, R.; Waterloos, M.A.; Van Landschoot, N.; Vogeleere, P.; Hoste, E.; Ringoir, S. Mechanisms of uremic inhibition of phagocyte reactive species production: Characterization of the role of p-cresol. Kidney Int. 1995, 47, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; Ringoir, S.; Dhondt, A.; Hakim, R. Phagocytosis in uremic and hemodialysis patients: A prospective and cross-sectional study. Kidney Int. 1991, 39, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Lim, W.H.; Kireta, S.; Leedham, E.; Russ, G.R.; Coates, P.T. Uremia impairs monocyte and monocyte-derived dendritic cell function in hemodialysis patients. Kidney Int. 2007, 72, 1138–1148. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, M.L.V.; Bonan, N.B.; Dias, G.; Brehm, F.; Steiner, T.M.; Souza, W.M.; Stinghen, A.E.M.; Barreto, F.C.; Elifio-Esposito, S.; Pecoits-Filho, R.; et al. p-Cresyl sulfate affects the oxidative burst, phagocytosis process, and antigen presentation of monocyte-derived macrophages. Toxicol. Lett. 2016, 263, 1–5. [Google Scholar] [CrossRef]
- Christensson, A.; Savage, C.; Sjoberg, D.D.; Cronin, A.M.; O’Brien, M.F.; Lowrance, W.; Nilsson, P.M.; Vickers, A.J.; Russo, P.; Lilja, H. Association of cancer with moderately impaired renal function at baseline in a large, representative, population-based cohort followed for up to 30 years. Int. J. Cancer 2013, 133, 1452–1458. [Google Scholar] [CrossRef]
- Kim, J.U.; Kim, M.; Kim, S.; Nguyen, T.T.; Kim, E.; Lee, S.; Kim, S.; Kim, H. Dendritic Cell Dysfunction in Patients with End-stage Renal Disease. Immune Netw. 2017, 17, 152–162. [Google Scholar] [CrossRef]
- Hesselink, D.A.; Betjes, M.G.H.; Verkade, M.A.; Athanassopoulos, P.; Baan, C.C.; Weimar, W. The effects of chronic kidney disease and renal replacement therapy on circulating dendritic cells. Nephrol. Dial. Transplant. 2005, 20, 1868–1873. [Google Scholar] [CrossRef]
- Paul, K.; Kretzschmar, D.; Yilmaz, A.; Bärthlein, B.; Titze, S.; Wolf, G.; Busch, M. Circulating dendritic cell precursors in chronic kidney disease: A cross-sectional study. BMC Nephrol. 2013, 14, 274. [Google Scholar] [CrossRef] [PubMed]
- Verkade, M.A.; van Druningen, C.J.; Op de Hoek, C.T.; Weimar, W.; Betjes, M.G.H. Decreased antigen-specific T-cell proliferation by moDC among hepatitis B vaccine non-responders on haemodialysis. Clin. Exp. Med. 2007, 7, 65–71. [Google Scholar] [CrossRef]
- Lim, W.H.; Kireta, S.; Thomson, A.W.; Russ, G.R.; Coates, P.T.H. Renal transplantation reverses functional deficiencies in circulating dendritic cell subsets in chronic renal failure patients. Transplantation 2006, 81, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Shiba, T.; Makino, I.; Kawakami, K.; Kato, I.; Kobayashi, T.; Kaneko, K. p-Cresyl sulfate suppresses lipopolysaccharide-induced anti-bacterial immune responses in murine macrophages In Vitro. Toxicol. Lett. 2016, 245, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Ghimire, S.; Matos, C.; Caioni, M.; Weber, D.; Peter, K.; Holler, E.; Kreutz, M.; Renner, K. Indoxyl 3-sulfate inhibits maturation and activation of human monocyte-derived dendritic cells. Immunobiology 2018, 223, 239–245. [Google Scholar] [CrossRef]
- Kurz, P.; Köhler, H.; Meuer, S.; Hütteroth, T.; Meyer zum Büschenfelde, K.H. Impaired cellular immune responses in chronic renal failure: Evidence for a T cell defect. Kidney Int. 1986, 29, 1209–1214. [Google Scholar] [CrossRef]
- Meier, P.; Dayer, E.; Blanc, E.; Wauters, J.P. Early T Cell Activation Correlates with Expression of Apoptosis Markers in Patients with End-Stage Renal Disease. J. Am. Soc. Nephrol. 2002, 13, 204–212. [Google Scholar]
- Yoon, J.W.; Gollapudi, S.; Pahl, M.V.; Vaziri, N.D. Naïve and central memory T-cell lymphopenia in end-stage renal disease. Kidney Int. 2006, 70, 371–376. [Google Scholar] [CrossRef]
- Huang, L.; Betjes, M.G.H.; Klepper, M.; Langerak, A.W.; Baan, C.C.; Litjens, N.H.R. End-Stage Renal Disease Causes Skewing in the TCR Vβ-Repertoire Primarily within CD8+ T Cell Subsets. Front. Immunol. 2017, 8, 1826. [Google Scholar] [CrossRef]
- Betjes, M.G.H.; Langerak, A.W.; van der Spek, A.; de Wit, E.A.; Litjens, N.H.R. Premature aging of circulating T cells in patients with end-stage renal disease. Kidney Int. 2011, 80, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Yadav, A.K.; Jha, V. CD4+CD28null cells are expanded and exhibit a cytolytic profile in end-stage renal disease patients on peritoneal dialysis. Nephrol. Dial. Transplant. 2011, 26, 1689–1694. [Google Scholar] [CrossRef][Green Version]
- Betjes, M.G.H.; Huisman, M.; Weimar, W.; Litjens, N.H.R. Expansion of cytolytic CD4+CD28- T cells in end-stage renal disease. Kidney Int. 2008, 74, 760–767. [Google Scholar] [CrossRef][Green Version]
- Litjens, N.H.R.; van Druningen, C.J.; Betjes, M.G.H. Progressive loss of renal function is associated with activation and depletion of naive T lymphocytes. Clin. Immunol. 2006, 118, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Cano-Romero, F.L.; Laguna Goya, R.; Utrero-Rico, A.; Gómez-Massa, E.; Arroyo-Sánchez, D.; Suárez-Fernández, P.; Lora, D.; Andrés, A.; Castro-Panete, M.J.; Paz-Artal, E. Longitudinal profile of circulating T follicular helper lymphocytes parallels anti-HLA sensitization in renal transplant recipients. Am. J. Transplant. 2019, 19, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Fresnedo, G.; Ramos, M.A.; González-Pardo, M.C.; de Francisco, A.L.; López-Hoyos, M.; Arias, M. B lymphopenia in uremia is related to an accelerated In Vitro apoptosis and dysregulation of Bcl-2. Nephrol. Dial. Transplant. 2000, 15, 502–510. [Google Scholar] [CrossRef] [PubMed]
- Jacque, E.; Schweighoffer, E.; Tybulewicz, V.L.J.; Ley, S.C. BAFF activation of the ERK5 MAP kinase pathway regulates B cell survival. J. Exp. Med. 2015, 212, 883–892. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vanholder, R.; Ringoir, S. Infectious morbidity and defects of phagocytic function in end-stage renal disease: A review. J. Am. Soc. Nephrol. 1993, 3, 1541–1554. [Google Scholar] [PubMed]
- Dammin, G.J.; Couch, N.P.; Murray, J.E. Prolonged survival of skin homografts in uremic patients. Ann. N. Y. Acad. Sci. 1957, 64, 967–976. [Google Scholar] [CrossRef]
- Chen, C.C.; Pouliquen, E.; Broisat, A.; Andreata, F.; Racapé, M.; Bruneval, P.; Kessler, L.; Ahmadi, M.; Bacot, S.; Saison-Delaplace, C.; et al. Endothelial chimerism and vascular sequestration protect pancreatic islet grafts from antibody-mediated rejection. J. Clin. Investig. 2018, 128, 219–232. [Google Scholar] [CrossRef]
- Thommen, D.S.; Schumacher, T.N. T Cell Dysfunction in Cancer. Cancer Cell 2018, 33, 547–562. [Google Scholar] [CrossRef]
- Lowrance, W.T.; Ordoñez, J.; Udaltsova, N.; Russo, P.; Go, A.S. CKD and the risk of incident cancer. J. Am. Soc. Nephrol. 2014, 25, 2327–2334. [Google Scholar] [CrossRef] [PubMed]
- Verkade, M.A.; van Druningen, C.J.; Vaessen, L.M.B.; Hesselink, D.A.; Weimar, W.; Betjes, M.G.H. Functional impairment of monocyte-derived dendritic cells in patients with severe chronic kidney disease. Nephrol. Dial. Transplant. 2007, 22, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Ye, Z.; Kijlstra, A.; Zhou, Y.; Yang, P. Activation of the aryl hydrocarbon receptor affects activation and function of human monocyte-derived dendritic cells. Clin. Exp. Immunol. 2014, 177, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Platzer, B.; Richter, S.; Kneidinger, D.; Waltenberger, D.; Woisetschläger, M.; Strobl, H. Aryl hydrocarbon receptor activation inhibits In Vitro differentiation of human monocytes and Langerhans dendritic cells. J. Immunol. 2009, 183, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, B.P.; Denison, M.S.; Novak, H.; Vorderstrasse, B.A.; Harrer, N.; Neruda, W.; Reichel, C.; Woisetschläger, M. Activation of the aryl hydrocarbon receptor is essential for mediating the anti-inflammatory effects of a novel low-molecular-weight compound. Blood 2008, 112, 1158–1165. [Google Scholar] [CrossRef] [PubMed]
- Hauben, E.; Gregori, S.; Draghici, E.; Migliavacca, B.; Olivieri, S.; Woisetschläger, M.; Roncarolo, M.G. Activation of the aryl hydrocarbon receptor promotes allograft-specific tolerance through direct and dendritic cell-mediated effects on regulatory T cells. Blood 2008, 112, 1214–1222. [Google Scholar] [CrossRef] [PubMed]
- Shiba, T.; Kawakami, K.; Sasaki, T.; Makino, I.; Kato, I.; Kobayashi, T.; Uchida, K.; Kaneko, K. Effects of intestinal bacteria-derived p-cresyl sulfate on Th1-type immune response In Vivo and In Vitro. Toxicol. Appl. Pharmacol. 2014, 274, 191–199. [Google Scholar] [CrossRef]
- Gandhi, R.; Kumar, D.; Burns, E.J.; Nadeau, M.; Dake, B.; Laroni, A.; Kozoriz, D.; Weiner, H.L.; Quintana, F.J. Activation of the aryl hydrocarbon receptor induces human type 1 regulatory T cell–like and Foxp3+ regulatory T cells. Nat. Immunol. 2010, 11, 846–853. [Google Scholar] [CrossRef]
- Funatake, C.J.; Marshall, N.B.; Steppan, L.B.; Mourich, D.V.; Kerkvliet, N.I. Cutting edge: Activation of the aryl hydrocarbon receptor by 2,3,7,8-tetrachlorodibenzo-p-dioxin generates a population of CD4+CD25+ cells with characteristics of regulatory T cells. J. Immunol. 2005, 175, 4184–4188. [Google Scholar] [CrossRef]
- Cai, L.J.; Yu, D.W.; Gao, Y.; Yang, C.; Zhou, H.M.; Chen, Z.H.K. Activation of aryl hydrocarbon receptor prolongs survival of fully mismatched cardiac allografts. J. Huazhong Univ. Sci. Technol. Med. Sci. 2013, 33, 199–204. [Google Scholar] [CrossRef]
- Ehrlich, A.K.; Pennington, J.M.; Wang, X.; Rohlman, D.; Punj, S.; Löhr, C.V.; Newman, M.T.; Kolluri, S.K.; Kerkvliet, N.I. Activation of the Aryl Hydrocarbon Receptor by 10-Cl-BBQ Prevents Insulitis and Effector T Cell Development Independently of Foxp3+ Regulatory T Cells in Nonobese Diabetic Mice. J. Immunol. 2016, 196, 264–273. [Google Scholar] [CrossRef]
- Quintana, F.J.; Basso, A.S.; Iglesias, A.H.; Korn, T.; Farez, M.F.; Bettelli, E.; Caccamo, M.; Oukka, M.; Weiner, H.L. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature 2008, 453, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Crosnier, J.; Jungers, P.; Couroucé, A.M.; Laplanche, A.; Benhamou, E.; Degos, F.; Lacour, B.; Prunet, P.; Cerisier, Y.; Guesry, P. Randomised placebo-controlled trial of hepatitis B surface antigen vaccine in french haemodialysis units: II, Haemodialysis patients. Lancet 1981, 1, 797–800. [Google Scholar] [CrossRef]
- da Silva, E.N.; Baker, A.; Alshekaili, J.; Karpe, K.; Cook, M.C. A randomized trial of serological and cellular responses to hepatitis B vaccination in chronic kidney disease. PLoS ONE 2018, 13, e0204477. [Google Scholar] [CrossRef] [PubMed]
- Udomkarnjananun, S.; Takkavatakarn, K.; Praditpornsilpa, K.; Nader, C.; Eiam-Ong, S.; Jaber, B.L.; Susantitaphong, P. Hepatitis B virus vaccine immune response and mortality in dialysis patients: A meta-analysis. J. Nephrol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Litjens, N.H.R.; Huisman, M.; van den Dorpel, M.; Betjes, M.G.H. Impaired Immune Responses and Antigen-Specific Memory CD4+ T Cells in Hemodialysis Patients. J. Am. Soc. Nephrol. 2008, 19, 1483–1490. [Google Scholar] [CrossRef] [PubMed]
- Pahl, M.V.; Gollapudi, S.; Sepassi, L.; Gollapudi, P.; Elahimehr, R.; Vaziri, N.D. Effect of end-stage renal disease on B-lymphocyte subpopulations, IL-7, BAFF and BAFF receptor expression. Nephrol. Dial. Transplant. 2010, 25, 205–212. [Google Scholar] [CrossRef]
- Shiba, T.; Makino, I.; Sasaki, T.; Fukuhara, Y.; Kawakami, K.; Kato, I.; Kobayashi, T. p-Cresyl sulfate decreases peripheral B cells in mice with adenine-induced renal dysfunction. Toxicol. Appl. Pharmacol. 2018, 342, 50–59. [Google Scholar] [CrossRef]
- Li, J.; Bhattacharya, S.; Zhou, J.; Phadnis-Moghe, A.S.; Crawford, R.B.; Kaminski, N.E. Aryl Hydrocarbon Receptor Activation Suppresses EBF1 and PAX5 and Impairs Human B Lymphopoiesis. J. Immunol. 2017, 199, 3504–3515. [Google Scholar] [CrossRef]
- Dooley, R.K.; Holsapple, M.P. Elucidation of cellular targets responsible for tetrachlorodibenzo-p-dioxin (TCDD)-induced suppression of antibody responses: I. The role of the B lymphocyte. Immunopharmacology 1988, 16, 167–180. [Google Scholar] [CrossRef]
- Villa, M.; Gialitakis, M.; Tolaini, M.; Ahlfors, H.; Henderson, C.J.; Wolf, C.R.; Brink, R.; Stockinger, B. Aryl hydrocarbon receptor is required for optimal B-cell proliferation. EMBO J. 2017, 36, 116–128. [Google Scholar] [CrossRef] [PubMed]
- Vaidyanathan, B.; Chaudhry, A.; Yewdell, W.T.; Angeletti, D.; Yen, W.F.; Wheatley, A.K.; Bradfield, C.A.; McDermott, A.B.; Yewdell, J.W.; Rudensky, A.Y.; et al. The aryl hydrocarbon receptor controls cell-fate decisions in B cells. J. Exp. Med. 2017, 214, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Maciejczyk, M.; Żukowski, P.; Zalewska, A. Salivary Biomarkers in Kidney Diseases. In Saliva in Health and Disease: The Present and Future of a Unique Sample for Diagnosis; Tvarijonaviciute, A., Martínez-Subiela, S., López-Jornet, P., Lamy, E., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 193–219. ISBN 978-3-030-37681-9. [Google Scholar]
- Prendergast, G.C.; Malachowski, W.P.; DuHadaway, J.B.; Muller, A.J. Discovery of IDO1 Inhibitors: From Bench to Bedside. Cancer Res. 2017, 77, 6795–6811. [Google Scholar] [CrossRef] [PubMed]
- Piper, C.J.M.; Rosser, E.C.; Oleinika, K.; Nistala, K.; Krausgruber, T.; Rendeiro, A.F.; Banos, A.; Drozdov, I.; Villa, M.; Thomson, S.; et al. Aryl Hydrocarbon Receptor Contributes to the Transcriptional Program of IL-10-Producing Regulatory B Cells. Cell Rep. 2019, 29, 1878–1892.e7. [Google Scholar] [CrossRef] [PubMed]
- Niwa, T. Mass spectrometry in the search for uremic toxins. Mass Spectrom Rev. 1997, 16, 307–332. [Google Scholar] [CrossRef]
- Korytowska, N.; Sankowski, B.; Wyczałkowska-Tomasik, A.; Pączek, L.; Wroczyński, P.; Giebułtowicz, J. The utility of saliva testing in the estimation of uremic toxin levels in serum. Clin. Chem. Lab. Med. 2018, 57, 230–237. [Google Scholar] [CrossRef]
- Schulman, G.; Berl, T.; Beck, G.J.; Remuzzi, G.; Ritz, E.; Arita, K.; Kato, A.; Shimizu, M. Randomized Placebo-Controlled EPPIC Trials of AST-120 in CKD. J. Am. Soc. Nephrol. 2015, 26, 1732–1746. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Low Molecular Weight Molecules (<500 kDa) | Middle Molecules (500–60,000 kDa) | Protein-Bound Uremic Retention Solutes | |
|---|---|---|---|
| Selection of clinically relevant molecules | -Urea -Phosphate -Uric acid -Creatinine -Carbamylated compounds -Trimethylamine-N-oxide * | -B2 microglobuline -Parathyroid hormone -Fibroblast-growth-factor 23 -Atrial natriuretic peptide -Interleukin 6, 8, 10 -TNFα | -Indoxyl sulfate * -P-cresyl sulfate * -Indole-3- acetic acid * -Kynurenic acid * -hippuric acid * -homocysteine -Carboxymethyllysine (AGEs) -3-Carboxy-4-methyl-5-propyl-2-furan-propanoic acid -spermine |
| Cell Subset | CKD-Associated Phenotype | Impact of PBURS | |
|---|---|---|---|
| p-Cresyl Sulfate | Indoxyl Sulfate | ||
| Innate Immune Cells | |||
| Neutrophils |  response to stimulation [53] expression of TLR 2 and 4 [49,54] apoptosis [67] response to stimulation [53] expression of TLR 2 and 4 [49,54] apoptosis [67] phagocytic functions [72,76,77] phagocytic functions [72,76,77] | adhesion to endothelial cells and extravasation [66] NADPH oxidase activity [73,74,75] phagocytic functions [39] | adhesion to endothelial cells and extravasation [66] NADPH oxidase activity [73] |
| Monocytes and macrophages | expression of TLR2 and 4 [49,54] phagocytic functions [75,77] | phagocytic functions [78,79] | secretion of pro-inflammatory cytokines [57,58] |
| Dendritic cells | number [80,81,82] expression of costimulatory molecules [83,84] capacity to activate T cells [83,85] | phagocytic function and presentation of antigen [78,79] | proliferationand expression of costimulatory molecules [86,87] |
| Adaptive immune cells | |||
| Naïve T cells | apoptosis [88] number [89,90,91] thymic output [90] | Unknown | Unknown |
| Differentiated T cells | number of terminally differentiated [92,93] TCR repertoire diversity [94] | production of INFγ (Th1 cells) [95] | Unknown |
| B cells | number of naïve and memory B cells [68,96,97] apoptosis [68,96,97] by decreased prosurvival signals [68,96,97] | number of B cells [98] | Unknown |
: increase; : decrease.© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Espi, M.; Koppe, L.; Fouque, D.; Thaunat, O. Chronic Kidney Disease-Associated Immune Dysfunctions: Impact of Protein-Bound Uremic Retention Solutes on Immune Cells. Toxins 2020, 12, 300. https://doi.org/10.3390/toxins12050300
Espi M, Koppe L, Fouque D, Thaunat O. Chronic Kidney Disease-Associated Immune Dysfunctions: Impact of Protein-Bound Uremic Retention Solutes on Immune Cells. Toxins. 2020; 12(5):300. https://doi.org/10.3390/toxins12050300
Chicago/Turabian StyleEspi, Maxime, Laetitia Koppe, Denis Fouque, and Olivier Thaunat. 2020. "Chronic Kidney Disease-Associated Immune Dysfunctions: Impact of Protein-Bound Uremic Retention Solutes on Immune Cells" Toxins 12, no. 5: 300. https://doi.org/10.3390/toxins12050300
APA StyleEspi, M., Koppe, L., Fouque, D., & Thaunat, O. (2020). Chronic Kidney Disease-Associated Immune Dysfunctions: Impact of Protein-Bound Uremic Retention Solutes on Immune Cells. Toxins, 12(5), 300. https://doi.org/10.3390/toxins12050300