Immunoaffinity Extraction and Alternative Approaches for the Analysis of Toxins in Environmental, Food or Biological Matrices



Abstract
:1. Introduction
2. Immunoaffinity Sorbents
2.1. Antibody Production and Development of Immunosorbents
2.2. Immunoextraction Procedure on IS Cartridges
2.3. Immunoextraction Using Other Formats
2.4. Potential of Immunosorbents for the Reliable Quantification in Real Samples
3. Molecularly Imprinted Polymers
4. Oligosorbents
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pichon, V. Immunoaffinity Extraction. In Reference Module in Chemistry, Molecular Sciences and Chemical Engineering; Elsevier: Amsterdam, The Netherlands, 2016; ISBN 978-0-12-409547-2. [Google Scholar]
- Pichon, V. 6-Aptamer-based and immunosorbents. In Solid-Phase Extraction; Poole, C.F., Ed.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 151–183. ISBN 978-0-12-816906-3. [Google Scholar]
- Pichon, V.; Combès, A.; Delaunay, N. Immunosorbents in microextraction. TrAC Trends Anal. Chem. 2019, 113, 246–255. [Google Scholar] [CrossRef]
- Tsikas, D. Quantitative analysis of biomarkers, drugs and toxins in biological samples by immunoaffinity chromatography coupled to mass spectrometry or tandem mass spectrometry: A focused review of recent applications. J. Chromatogr. B 2010, 878, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Şenyuva, H.Z.; Gilbert, J. Immunoaffinity column clean-up techniques in food analysis: A review. J. Chromatogr. B 2010, 878, 115–132. [Google Scholar] [CrossRef] [PubMed]
- Pichon, V.; Delaunay, N.; Combès, A. Sample Preparation Using Molecularly Imprinted Polymers. Anal. Chem. 2020, 92, 16–33. [Google Scholar] [CrossRef] [PubMed]
- Malik, M.I.; Shaikh, H.; Mustafa, G.; Bhanger, M.I. Recent Applications of Molecularly Imprinted Polymers in Analytical Chemistry. Sep. Purif. Rev. 2019, 48, 179–219. [Google Scholar] [CrossRef]
- Ye, W.; Liu, T.; Zhang, W.; Zhu, M.; Liu, Z.; Kong, Y.; Liu, S. Marine Toxins Detection by Biosensors Based on Aptamers. Toxins 2020, 12, 1. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Huang, Y.; Dong, Y.; Han, X.; Wang, S.; Liang, X. Aptamers and Aptasensors for Highly Specific Recognition and Sensitive Detection of Marine Biotoxins: Recent Advances and Perspectives. Toxins 2018, 10, 427. [Google Scholar] [CrossRef] [Green Version]
- Pichon, V.; Delaunay-Bertoncini, N.; Hennion, M.-C. Immunosorbents in sample preparation. Compr. Anal. Chem. 2002, 37, 1081–1100. [Google Scholar]
- Marco, M.-P.; Gee, S.; Hammock, B.D. Immunochemical techniques for environmental analysis I. Immunosensors. TrAC 1995, 14, 341–350. [Google Scholar] [CrossRef]
- Bao, L.; Liang, C.; Trucksess, M.W.; Xu, Y.; Lv, N.; Wu, Z.; Jing, P.; Fry, F.S. Determination of Aflatoxins B-1, B-2, G(1), and G(2) in Olive Oil, Peanut Oil, and Sesame Oil Using Immunoaffinity Column Cleanup, Postcolumn Derivatization, and Liquid Chromatography/Fluorescence Detection: Collaborative Study. J. AOAC Int. 2012, 95, 1689–1700. [Google Scholar] [CrossRef]
- AlFaris, N.A.; Altamimi, J.Z.; Alothman, Z.A.; Al Qahtani, S.F.; Wabaidur, S.M.; Ghfar, A.A.; Aldayel, T.S. Saleh Analysis of aflatoxins in foods retailed in Saudi Arabia using immunoaffinity column cleanup and high-performance liquid chromatography-fluorescence detection. J. King Saud Univ. Sci. 2020, 32, 1437–1443. [Google Scholar] [CrossRef]
- AlFaris, N.A.; Wabaidur, S.M.; Alothman, Z.A.; Altamimi, J.Z.; Aldayel, T.S. Fast and efficient immunoaffinity column cleanup and liquid chromatography–tandem mass spectrometry method for the quantitative analysis of aflatoxins in baby food and feeds. J. Sep. Sci. 2020, 43, 2079–2087. [Google Scholar] [CrossRef] [PubMed]
- Edinboro, L.E.; Karnes, H.T. Determination of aflatoxin B1 in sidestream cigarette smoke by immunoaffinity column extraction coupled with liquid chromatography/mass spectrometry. J. Chromatogr. A 2005, 1083, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Tosun, H.; Arslan, R. Determination of Aflatoxin B1 Levels in Organic Spices and Herbs. Sci. World J. 2013, 874093. [Google Scholar] [CrossRef] [Green Version]
- Chiavaro, E.; Cacchioli, C.; Berni, E.; Spotti, E. Immunoaffinity clean-up and direct fluorescence measurement of aflatoxins B-1 and M-1 in pig liver: Comparison with high-performance liquid chromatography determination. Food Addit. Contam. PART Chem. Anal. Control Expo. Risk Assess. 2005, 22, 1154–1161. [Google Scholar] [CrossRef]
- Cahill, L.M.; Kruger, S.C.; McAlice, B.T.; Ramsey, C.S.; Prioli, R.; Kohn, B. Quantification of deoxynivalenol in wheat using an immunoaffinity column and liquid chromatography. J. Chromatogr. A 1999, 859, 23–28. [Google Scholar] [CrossRef]
- Ok, H.E.; Lee, S.Y.; Chun, H.S. Occurrence and simultaneous determination of nivalenol and deoxynivalenol in rice and bran by HPLC-UV detection and immunoaffinity cleanup. Food Control 2018, 87, 53–59. [Google Scholar] [CrossRef]
- Solfrizzo, M.; De Girolamo, A.; Visconti, A. Determination of fumonisins B-1 and B-2 in cornflakes by high performance liquid chromatography and immunoaffinity clean-up. Food Addit. Contam. 2001, 18, 227–235. [Google Scholar] [CrossRef]
- Sharman, M.; MacDonald, S.; Gilbert, J. Automated liquid chromatographic determination of ochratoxin A in cereals and animal products using immunoaffinity column clean-up. J. Chromatogr. A 1992, 603, 285–289. [Google Scholar] [CrossRef]
- Castellari, M.; Fabbri, S.; Fabiani, A.; Amati, A.; Galassi, S. Comparison of different immunoaffinity clean-up procedures for high-performance liquid chromatographic analysis of ochratoxin A in wines. J. Chromatogr. A 2000, 888, 129–136. [Google Scholar] [CrossRef]
- Visconti, A.; Pascale, M.; Centonze, G. Determination of ochratoxin a in domestic and imported beers in Italy by immunoaffinity clean-up and liquid chromatography. J. Chromatogr. A 2000, 888, 321–326. [Google Scholar] [CrossRef]
- Pascale, M.; Visconti, A. Rapid method for the determination of ochratoxin A in urine by immunoaffinity column clean-up and high-performance liquid chromatography. Mycopathologia 2001, 152, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Bascarán, V.; De Rojas, A.H.; Chouciño, P.; Delgado, T. Analysis of ochratoxin A in milk after direct immunoaffinity column clean-up by high-performance liquid chromatography with fluorescence detection. J. Chromatogr. A 2007, 1167, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Solfrizzo, M.; Panzarini, G.; Visconti, A. Determination of Ochratoxin A in Grapes, Dried Vine Fruits, and Winery Byproducts by High-Performance Liquid Chromatography with Fluorometric Detection (HPLC-FLD) and Immunoaffinity Cleanup. J. Agric. Food Chem. 2008, 56, 11081–11086. [Google Scholar] [CrossRef]
- Noba, S.; Uyama, A.; Mochizuki, N. Determination of Ochratoxin A in Ready-To-Drink Coffee by Immunoaffinity Cleanup and Liquid Chromatography—Tandem Mass Spectrometry. J. Agric. Food Chem. 2009, 57, 6036–6040. [Google Scholar] [CrossRef]
- Longobardi, F.; Iacovelli, V.; Catucci, L.; Panzarini, G.; Pascale, M.; Visconti, A.; Agostiano, A. Determination of Ochratoxin A in Wine by Means of Immunoaffinity and Aminopropyl Solid-Phase Column Cleanup and Fluorometric Detection. J. Agric. Food Chem. 2013, 61, 1604–1608. [Google Scholar] [CrossRef]
- Liu, X.; Liu, X.; Huang, P.; Wei, F.; Ying, G.; Lu, J.; Zhou, L.; Kong, W. Regeneration and Reuse of Immunoaffinity Column for Highly Efficient Clean-Up and Economic Detection of Ochratoxin A in Malt and Ginger. Toxins 2018, 10, 462. [Google Scholar] [CrossRef] [Green Version]
- Boudra, H.; Morgavi, D.P. Development and validation of a HPLC method for the quantitation of ochratoxins in plasma and raw milk. J. Chromatogr. B 2006, 843, 295–301. [Google Scholar] [CrossRef]
- Marley, E.; Brown, P.; Mackie, J.; Donnelly, C.; Wilcox, J.; Pietri, A.; Macdonald, S. Analysis of sterigmatocystin in cereals, animal feed, seeds, beer and cheese by immunoaffinity column clean-up and HPLC and LC-MS/MS quantification. Food Addit. Contam. Part A 2015, 32, 2131–2137. [Google Scholar] [CrossRef]
- Pascale, M.; Haidukowski, M.; Visconti, A. Determination of T-2 toxin in cereal grains by liquid chromatography with fluorescence detection after immunoaffinity column clean-up and derivatization with 1-anthroylnitrile. J. Chromatogr. A 2003, 989, 257–264. [Google Scholar] [CrossRef]
- Visconti, A.; Lattanzio, V.M.T.; Pascale, M.; Haidukowski, M. Analysis of T-2 and HT-2 toxins in cereal grains by immunoaffinity clean-up and liquid chromatography with fluorescence detection. J. Chromatogr. A 2005, 1075, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Trebstein, A.; Seefelder, W.; Lauber, U.; Humpf, H.-U. Determination of T-2 and HT-2 toxins in cereals including oats after immunoaffinity cleanup by liquid chromatography and fluorescence detection. J. Agric. Food Chem. 2008, 56, 4968–4975. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, C.; Pollock, A.; Heidtmann, Y.; Marley, E. Development of an Immunoaffinity Column for the Determination of T-2 and HT-2 Toxins in Cereals Using Liquid Chromatography with Fluorescence Detection. In Food Contaminants; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2008; Volume 1001, pp. 276–284. ISBN 978-0-8412-6954-5. [Google Scholar]
- Kong, W.; Zhang, X.; Shen, H.; Ou-Yang, Z.; Yang, M. Validation of a gas chromatography-electron capture detection of T-2 and HT-2 toxins in Chinese herbal medicines and related products after immunoaffinity column clean-up and pre-column derivatization. Food Chem. 2012, 132, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Pascale, M.; Panzarini, G.; Visconti, A. Determination of HT-2 and T-2 toxins in oats and wheat by ultra-performance liquid chromatography with photodiode array detection. Talanta 2012, 89, 231–236. [Google Scholar] [CrossRef]
- Di Marco Pisciottano, I.; Imperato, C.; Urbani, V.; Guadagnuolo, G.; Imbimbo, S.; De Crescenzo, M.; Soprano, V.; Esposito, M.; Gallo, P. T-2 and HT-2 toxins in feed and food from Southern Italy, determined by LC-MS/MS after immunoaffinity clean-up. Food Addit. Contam. Part B 2020, 13, 1–9. [Google Scholar] [CrossRef]
- Visconti, A.; Pascale, M. Determination of zearalenone in corn by means of immunoaffinity clean-up and high-performance liquid chromatography with fluorescence detection. J. Chromatogr. A 1998, 815, 133–140. [Google Scholar] [CrossRef]
- Trucksess, M.W.; Fu, W.-S.; Oles, C.J.; White, K.D. Determination of Zearalenone in Botanical Dietary Supplements, Soybeans, Grains, and Grain Products by Immunoaffinity Column Cleanup and Liquid Chromatography: Single-Laboratory Validation. J. AOAC Int. 2011, 94, 589–595. [Google Scholar]
- Kruger, S.; Kohn, B.; Ramsey, C.; Prioli, R. Rapid immunoaffinity-based method for determination of zearalenone in corn by fluorometry and liquid chromatography. J. AOAC Int. 1999, 82, 1364–1368. [Google Scholar] [CrossRef] [Green Version]
- Trucksess, M.W.; Weaver, C.M.; Oles, C.J.; Rump, L.V.; White, K.D.; Betz, J.M.; Rader, J.I. Use of multitoxin immunoaffinity columns for determination of aflatoxins and ochratoxin a in ginseng and ginger. J. AOAC Int. 2007, 90, 1042–1049. [Google Scholar] [CrossRef] [Green Version]
- Kabak, B. Determination of aflatoxins and ochratoxin A in retail cereal products from Turkey by high performance liquid chromatography with fluorescence detection. Food Control 2012, 28, 1–6. [Google Scholar] [CrossRef]
- Di Stefano, V.; Pitonzo, R.; Avellone, G.; Di Fiore, A.; Monte, L.; Ogorka, A. Determination of Aflatoxins and Ochratoxins in Sicilian Sweet Wines by High-Performance Liquid Chromatography with Fluorometric Detection and Immunoaffinity Cleanup. Food Anal. Methods 2015, 8, 569–577. [Google Scholar] [CrossRef]
- Abd-Elghany, S.M.; Sallam, K.I. Rapid determination of total aflatoxins and ochratoxins A in meat products by immuno-affinity fluorimetry. Food Chem. 2015, 179, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Ainiza, W.W.; Jinap, S.; Sanny, M. Simultaneous determination of aflatoxins and ochratoxin A in single and mixed spices. Food Control 2015, 50, 913–918. [Google Scholar] [CrossRef]
- Lippolis, V.; Irurhe, O.; Porricelli, A.C.R.; Cortese, M.; Schena, R.; Imafidon, T.; Oluwadun, A.; Pascale, M. Natural co-occurrence of aflatoxins and ochratoxin A in ginger (Zingiber officinale) from Nigeria. Food Control 2017, 73, 1061–1067. [Google Scholar] [CrossRef] [Green Version]
- Lattanzio, V.M.T.; Solfrizzo, M.; Powers, S.; Visconti, A. Simultaneous determination of aflatoxins, ochratoxin A and Fusarium toxins in maize by liquid chromatography/tandem mass spectrometry after multitoxin immunoaffinity cleanup. Rapid Commun. Mass Spectrom. 2007, 21, 3253–3261. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chai, T.; Lu, G.; Quan, C.; Duan, H.; Yao, M.; Zucker, B.-A.; Schlenker, G. Simultaneous detection of airborne Aflatoxin, Ochratoxin and Zearalenone in a poultry house by immunoaffinity clean-up and high-performance liquid chromatography. Environ. Res. 2008, 107, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Solfrizzo, M.; Gambacorta, L.; Bibi, R.; Ciriaci, M.; Paoloni, A.; Pecorelli, I. Multimycotoxin Analysis by LC-MS/MS in Cereal Food and Feed: Comparison of Different Approaches for Extraction, Purification, and Calibration. J. AOAC Int. 2018, 101, 647–657. [Google Scholar] [CrossRef]
- Sakin, F.; Tekeli, I.O.; Yipel, M.; Kurekci, C. Occurrence and health risk assessment of aflatoxins and ochratoxin a in Surk, a Turkish dairy food, as studied by HPLC. Food Control 2018, 90, 317–323. [Google Scholar] [CrossRef]
- Bessaire, T.; Mujahid, C.; Mottier, P.; Desmarchelier, A. Multiple Mycotoxins Determination in Food by LC-MS/MS: An International Collaborative Study. Toxins 2019, 11, 658. [Google Scholar] [CrossRef] [Green Version]
- Ren, Y.; Zhang, Y.; Shao, S.; Cai, Z.; Feng, L.; Pan, H.; Wang, Z. Simultaneous determination of multi-component mycotoxin contaminants in foods and feeds by ultra-performance liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2007, 1143, 48–64. [Google Scholar] [CrossRef]
- Lattanzio, V.M.T.; Ciasca, B.; Powers, S.; Visconti, A. Improved method for the simultaneous determination of aflatoxins, ochratoxin A and Fusarium toxins in cereals and derived products by liquid chromatography–tandem mass spectrometry after multi-toxin immunoaffinity clean up. J. Chromatogr. A 2014, 1354, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Takino, M.; Sugita-Konishi, Y.; Tanaka, T. Development of a liquid chromatography/time-of-flight mass spectrometric method for the simultaneous determination of trichothecenes, zearalenone and aflatoxins in foodstuffs. Rapid Commun. Mass Spectrom. 2006, 20, 1422–1428. [Google Scholar] [CrossRef] [PubMed]
- Desmarchelier, A.; Tessiot, S.; Bessaire, T.; Racault, L.; Fiorese, E.; Urbani, A.; Chan, W.-C.; Cheng, P.; Mottier, P. Combining the quick, easy, cheap, effective, rugged and safe approach and clean-up by immunoaffinity column for the analysis of 15 mycotoxins by isotope dilution liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2014, 1337, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Kim, D.-H.; Moon, J.-Y.; An, J.-A.; Kim, Y.-W.; Chung, S.-H.; Lee, C. Distribution Analysis of Twelve Mycotoxins in Corn and Corn-Derived Products by LC-MS/MS to Evaluate the Carry-Over Ratio during Wet-Milling. Toxins 2018, 10, 319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.-H.; Hong, S.-Y.; Kang, J.W.; Cho, S.M.; Lee, K.R.; An, T.K.; Lee, C.; Chung, S.H. Simultaneous Determination of Multi-Mycotoxins in Cereal Grains Collected from South Korea by LC/MS/MS. Toxins 2017, 9, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, H.-D.; Suh, J.H.; Feng, S.; Eom, T.; Kim, J.; Hyun, S.M.; Kim, J.; Wang, Y.; Han, S.B. Comprehensive analysis of multi-class mycotoxins in twenty different species of functional and medicinal herbs using liquid chromatography–tandem mass spectrometry. Food Control 2019, 96, 517–526. [Google Scholar] [CrossRef]
- Vaclavikova, M.; MacMahon, S.; Zhang, K.; Begley, T.H. Application of single immunoaffinity clean-up for simultaneous determination of regulated mycotoxins in cereals and nuts. Talanta 2013, 117, 345–351. [Google Scholar] [CrossRef]
- Wilcox, J.; Donnelly, C.; Leeman, D.; Marley, E. The use of immunoaffinity columns connected in tandem for selective and cost-effective mycotoxin clean-up prior to multi-mycotoxin liquid chromatographic–tandem mass spectrometric analysis in food matrices. J. Chromatogr. A 2015, 1400, 91–97. [Google Scholar] [CrossRef]
- Solfrizzo, M.; Gambacorta, L.; Lattanzio, V.M.T.; Powers, S.; Visconti, A. Simultaneous LC–MS/MS determination of aflatoxin M1, ochratoxin A, deoxynivalenol, de-epoxydeoxynivalenol, α and β-zearalenols and fumonisin B1 in urine as a multi-biomarker method to assess exposure to mycotoxins. Anal. Bioanal. Chem. 2011, 401, 2831. [Google Scholar] [CrossRef]
- Zhang, Y.; Pei, F.; Fang, Y.; Li, P.; Zhao, Y.; Shen, F.; Zou, Y.; Hu, Q. Comparison of concentration and health risks of 9 Fusarium mycotoxins in commercial whole wheat flour and refined wheat flour by multi-IAC-HPLC. Food Chem. 2019, 275, 763–769. [Google Scholar] [CrossRef]
- Versilovskis, A.; Huybrecht, B.; Tangni, E.K.; Pussemier, L.; De Saeger, S.; Callebaut, A. Cross-reactivity of some commercially available deoxynivalenol (DON) and zearalenone (ZEN) immunoaffinity columns to DON- and ZEN-conjugated forms and metabolites. Food Addit. Contam. Part Chem. Anal. Control Expo. Risk Assess. 2011, 28, 1687–1693. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Takino, M.; Sugita-Konishi, Y.; Tanaka, T.; Leeman, D.; Toriba, A.; Hayakawa, K. Determination of Fusarium mycotoxins by liquid chromatography/tandem mass spectrometry coupled with immunoaffinity extraction. Rapid Commun. Mass Spectrom. 2010, 24, 2445–2452. [Google Scholar] [CrossRef] [PubMed]
- Berthiller, F.; Schuhmacher, R.; Buttinger, G.; Krska, R. Rapid simultaneous determination of major type A- and B-trichothecenes as well as zearalenone in maize by high performance liquid chromatography–tandem mass spectrometry. J. Chromatogr. A 2005, 1062, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Biselli, S.; Hummert, C. Development of a multicomponent method for Fusarium toxins using LC-MS/MS and its application during a survey for the content of T-2 toxin and deoxynivalenol in various feed and food samples. Food Addit. Contam. 2005, 22, 752–760. [Google Scholar] [CrossRef] [PubMed]
- Senyuva, H.Z.; Gilbert, J.; Türköz, G.; Leeman, D.; Donnelly, C. Analysis of Deoxynivalenol, Zearalenone, T-2, and HT-2 Toxins in Animal Feed by LC/MS/MS–A Critical Comparison of Immunoaffinity Column Cleanup with No Cleanup. J. AOAC Int. 2012, 95, 1701–1708. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yan, Z.; Wang, Y.; Jiang, T.; Wang, J.; Sun, X.; Guo, Y. Immunoaffinity Chromatography Purification and Ultrahigh Performance Liquid Chromatography Tandem Mass Spectrometry Determination of Tetrodotoxin in Marine Organisms. J. Agric. Food Chem. 2015, 63, 3129–3134. [Google Scholar] [CrossRef]
- Le, T.; Esteve-Turrillas, F.A.; Armenta, S.; de la Guardia, M.; Quiñones-Reyes, G.; Abad-Fuentes, A.; Abad-Somovilla, A. Dispersive magnetic immunoaffinity extraction. Anatoxin—A determination. J. Chromatogr. A 2017, 1529, 57–62. [Google Scholar] [CrossRef]
- Brothier, F.; Pichon, V. Immobilized antibody on a hybrid organic–inorganic monolith: Capillary immunoextraction coupled on-line to nanoLC-UV for the analysis of microcystin-LR. Anal. Chim. Acta 2013, 792, 52–58. [Google Scholar] [CrossRef]
- Wharton, R.E.; Ojeda-Torres, G.; Cunningham, B.; Feyereisen, M.C.; Hill, K.L.; Abbott, N.L.; Seymour, C.; Hill, D.; Lang, J.; Hamelin, E.I.; et al. Quantification of Microcystin-LR in Human Urine by Immunocapture Liquid Chromatography Tandem Mass Spectrometry. Chem. Res. Toxicol. 2018, 31, 898–903. [Google Scholar] [CrossRef]
- Neumann, A.-C.; Melnik, S.; Niessner, R.; Stoeger, E.; Knopp, D. Microcystin-LR Enrichment from Freshwater by a Recombinant Plant-derived Antibody Using Sol-Gel-Glass Immunoextraction. Anal. Sci. 2019, 35, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Rivasseau, C.; Hennion, M.-C. Potential of immunoextraction coupled to analytical and bioanalytical methods (liquid chromatography, ELISA kit and phosphatase inhibition test) for an improved environmental monitoring of cyanobacterial toxins. Anal. Chim. Acta 1999, 399, 75–87. [Google Scholar] [CrossRef]
- Aguete, E.C.; Gago-Martínez, A.; Leão, J.M.; Rodríguez-Vázquez, J.A.; Menàrd, C.; Lawrence, J.F. HPLC and HPCE analysis of microcystins RR, LR and YR present in cyanobacteria and water by using immunoaffinity extraction. Talanta 2003, 59, 697–705. [Google Scholar] [CrossRef]
- Lawrence, J.F.; Menard, C. Determination of microcystins in blue-green algae, fish and water using liquid chromatography with ultraviolet detection after sample clean-up employing immunoaffinity chromatography. J. Chromatogr. A 2001, 922, 111–117. [Google Scholar] [CrossRef]
- Mhadhbi, H.; Ben-Rejeb, S.; Cléroux, C.; Martel, A.; Delahaut, P. Generation and characterization of polyclonal antibodies against microcystins—Application to immunoassays and immunoaffinity sample preparation prior to analysis by liquid chromatography and UV detection. Talanta 2006, 70, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Aranda-Rodriguez, R.; Kubwabo, C.; Benoit, F.M. Extraction of 15 microcystins and nodularin using immunoaffinity columns. Toxicon 2003, 42, 587–599. [Google Scholar] [CrossRef] [PubMed]
- Wharton, R.E.; Cunningham, B.R.; Schaefer, A.M.; Guldberg, S.M.; Hamelin, E.I.; Johnson, R.C. Measurement of Microcystin and Nodularin Activity in Human Urine by Immunocapture-Protein Phosphatase 2A Assay. Toxins 2019, 11, 729. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Tan, Z.; Wu, H.; Peng, J.; Zhai, Y.; Guo, M. Selective enrichment and quantification of okadaic acid in shellfish using an immunomagnetic-bead-based liquid chromatography with tandem mass spectrometry assay. J. Sep. Sci. 2019, 42, 1423–1431. [Google Scholar] [CrossRef] [Green Version]
- Ten-Hage, L.; Delaunay, N.; Pichon, V.; Couté, A.; Puiseux-Dao, S.; Turquet, J. Okadaic acid production from the marine benthic dinoflagellate Prorocentrum arenarium Faust (Dinophyceae) isolated from Europa Island coral reef ecosystem (SW Indian Ocean). Toxicon 2000, 38, 1043–1054. [Google Scholar] [CrossRef]
- Delaunay, N.; Pichon, V.; Caer, J.-P.L.; Hennion, M.-C. Immunoaffinity extraction as a new approach for an improved liquid chromatography-mass spectrometric or fluorimetric determination of okadaic acid in shellfish and algae. Anal. Chim. Acta 2000, 407, 173–186. [Google Scholar] [CrossRef]
- Puech, L.; Dragacci, S.; Gleizes, E.; Fremy, J. Use of immunoaffinity columns for clean-up of diarrhetic toxins (okadaic acid and dinophysistoxins) extracts from shellfish prior to their analysis by HPLC fluorimetry. Food Addit. Contam. Part Chem. Anal. Control Expo. Risk Assess. 1999, 16, 239–251. [Google Scholar] [CrossRef]
- Hu, X.; Hu, R.; Zhang, Z.; Li, P.; Zhang, Q.; Wang, M. Development of a multiple immunoaffinity column for simultaneous determination of multiple mycotoxins in feeds using UPLC-MS/MS. Anal. Bioanal. Chem. 2016, 408, 6027–6036. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Hu, X.; Zhang, Q.; Li, P. Determination for multiple mycotoxins in agricultural products using HPLC–MS/MS via a multiple antibody immunoaffinity column. J. Chromatogr. B 2016, 1021, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Peng, T.; He, J.-L.; Shao, Y.; Fan, C.-L.; Chen, Y.; Jiang, W.-X.; Chen, M.; Wang, Q.; Pei, X.-Y.; et al. Preparation and characterization of an immunoaffinity column for the selective extraction of aflatoxin B1 in 13 kinds of foodstuffs. J. Chromatogr. B 2015, 998–999, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Maurer, H.H.; Schmitt, C.J.; Weber, A.A.; Kraemer, T. Validated electrospray liquid chromatographic–mass spectrometric assay for the determination of the mushroom toxins α- and β-amanitin in urine after immunoaffinity extraction. J. Chromatogr. B. Biomed. Sci. Appl. 2000, 748, 125–135. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Z.; De Saeger, S.; Shi, W.; Li, C.; Zhang, S.; Cao, X.; Shen, J. Determination of deoxynivalenol in cereals by immunoaffinity clean-up and ultra-high performance liquid chromatography tandem mass spectrometry. Methods 2012, 56, 192–197. [Google Scholar] [CrossRef]
- Brenn-Struckhofova, Z.; Cichna-Markl, M.; Bohm, C.; Razzazi-Fazeli, E. Selective Sample Cleanup by Reusable Sol-Gel Immunoaffinity Columns for Determination of Deoxynivalenol in Food and Feed Samples. Anal. Chem. 2006, 79, 710–717. [Google Scholar] [CrossRef]
- Newkirk, D.; Benson, R.; Howard, P.; Churchwell, M.; Doerge, D.; Roberts, D. On-line immunoaffinity capture, coupled with HPLC and electrospray ionization mass spectrometry, for automated determination of fumonisins. J. Agric. Food Chem. 1998, 46, 1677–1688. [Google Scholar] [CrossRef]
- Sheng, W.; Wu, H.; Ji, W.; Li, Z.; Chu, F.; Wang, S. Visual Non-Instrumental On-Site Detection of Fumonisin B-1, B-2, and B-3 in Cereal Samples Using a Clean-Up Combined with Gel-Based Immunoaffinity Test Column Assay. Toxins 2018, 10, 165. [Google Scholar] [CrossRef] [Green Version]
- Goryacheva, I.Y.; Basova, E.Y.; Van Peteghem, C.; Eremin, S.A.; Pussemier, L.; Motte, J.-C.; De Saeger, S. Novel gel-based rapid test for non-instrumental detection of ochratoxin A in beer. Anal. Bioanal. Chem. 2008, 390, 723–727. [Google Scholar] [CrossRef]
- Chamieh, J.; Faye, C.; Dugas, V.; Moreau, T.; Vandenabeele-Trambouze, O.; Demesmay, C. Preparation and full characterization of a micro-immunoaffinity monolithic column and its in-line coupling with capillary zone electrophoresis with Ochratoxin A as model solute. J. Chromatogr. A 2012, 1232, 93–100. [Google Scholar] [CrossRef]
- Li, Y.; Lin, S.; Wang, Y.; Mao, X.; Wu, Y.; Liu, Y.; Chen, D. Broad-specific monoclonal antibody based IACs purification coupled UPLC-MS/MS method for T-2 and HT-2 toxin determination in maize and cherry samples. Food Agric. Immunol. 2020, 31, 291–302. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, H.; Wang, J.; Li, X.; Jiang, H.; Shen, J. Multiresidue determination of zeranol and related compounds in bovine muscle by gas chromatography/mass spectrometry with immunoaffinity cleanup. J. AOAC Int. 2006, 89, 1677–1681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.J.; Zhang, J.N.; Ding, K.; Chen, N.; Han, T. The development and characterisation of an immunoaffinity column used for the simultaneous selective extraction of Fusarium toxins from grain products. Qual. Assur. Saf. Crop. Foods 2019, 11, 325–331. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, J.; Ding, K.; Chen, X.; Han, T. Preparation of multitarget Fusarium toxin (zearalenone, deoxynivalenol, T-2, and HT-2) immunoaffinity columns using poly(glycidyl methacrylate–divinylbenzene) as a matrix. Polym. Bull. 2020, 77, 4507–4522. [Google Scholar] [CrossRef]
- Basova, E.Y.; Goryacheva, I.Y.; Rusanova, T.Y.; Burmistrova, N.A.; Dietrich, R.; Maertlbauer, E.; Detavernier, C.; Van Peteghem, C.; De Saeger, S. An immunochemical test for rapid screening of zearalenone and T-2 toxin. Anal. Bioanal. Chem. 2010, 397, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Bragg, W.A.; Garrett, A.; Hamelin, E.I.; Coleman, R.M.; Campbell, K.; Elliott, C.T.; Johnson, R.C. Quantitation of saxitoxin in human urine using immunocapture extraction and LC–MS. Bioanalysis 2018, 10, 229–239. [Google Scholar] [CrossRef]
- Devlin, R.; Campbell, K.; Kawatsu, K.; Elliott, C. Studies in the Use of Magnetic Microspheres for Immunoaffinity Extraction of Paralytic Shellfish Poisoning Toxins from Shellfish. Toxins 2011, 3, 1–16. [Google Scholar] [CrossRef]
- Devlin, R.A.; Campbell, K.; Kawatsu, K.; Elliott, C.T. Physical and immunoaffinity extraction of paralytic shellfish poisoning toxins from cultures of the dinoflagellate Alexandrium tamarense. Harmful Algae 2011, 10, 542–548. [Google Scholar] [CrossRef]
- Hansbauer, E.-M.; Worbs, S.; Volland, H.; Simon, S.; Junot, C.; Fenaille, F.; Dorner, B.G.; Becher, F. Rapid Detection of Abrin Toxin and Its Isoforms in Complex Matrices by Immuno-Extraction and Quantitative High Resolution Targeted Mass Spectrometry. Anal. Chem. 2017, 89, 11719–11727. [Google Scholar] [CrossRef]
- De Dianous, S.; Kopeyan, C.; Bahraoui, E.; Rochat, H. Purification of contracture-inducing insect toxins from Buthinae scorpion venoms by immunoaffinity and high pressure liquid chromatography. Toxicon 1987, 25, 731–741. [Google Scholar] [CrossRef]
- Morineaux, V.; Mazuet, C.; Hilaire, D.; Enche, J.; Popoff, M.R. Characterization of botulinum neurotoxin type A subtypes by immunocapture enrichment and liquid chromatography-tandem mass spectrometry. Anal. Bioanal. Chem. 2015, 407, 5559–5570. [Google Scholar] [CrossRef] [PubMed]
- Seyer, A.; Fenaille, F.; Féraudet-Tarisse, C.; Volland, H.; Popoff, M.R.; Tabet, J.-C.; Junot, C.; Becher, F. Rapid Quantification of Clostridial Epsilon Toxin in Complex Food and Biological Matrixes by Immunopurification and Ultraperformance Liquid Chromatography-Tandem Mass Spectrometry. Anal. Chem. 2012, 84, 5103–5109. [Google Scholar] [CrossRef] [PubMed]
- Becher, F.; Duriez, E.; Volland, H.; Tabet, J.C.; Ezan, E. Detection of Functional Ricin by Immunoaffinity and Liquid Chromatography−Tandem Mass Spectrometry. Anal. Chem. 2007, 79, 659–665. [Google Scholar] [CrossRef] [PubMed]
- Duriez, E.; Fenaille, F.; Tabet, J.-C.; Lamourette, P.; Hilaire, D.; Becher, F.; Ezan, E. Detection of Ricin in Complex Samples by Immunocapture and Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry. J. Proteome Res. 2008, 7, 4154–4163. [Google Scholar] [CrossRef] [PubMed]
- Kalb, S.R.; Barr, J.R. Mass Spectrometric Detection of Ricin and its Activity in Food and Clinical Samples. Anal. Chem. 2009, 81, 2037–2042. [Google Scholar] [CrossRef]
- Ma, X.; Tang, J.; Li, C.; Liu, Q.; Chen, J.; Li, H.; Guo, L.; Xie, J. Identification and quantification of ricin in biomedical samples by magnetic immunocapture enrichment and liquid chromatography electrospray ionization tandem mass spectrometry. Anal. Bioanal. Chem. 2014, 406, 5147–5155. [Google Scholar] [CrossRef]
- Kull, S.; Pauly, D.; Störmann, B.; Kirchner, S.; Stämmler, M.; Dorner, M.B.; Lasch, P.; Naumann, D.; Dorner, B.G. Multiplex Detection of Microbial and Plant Toxins by Immunoaffinity Enrichment and Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry. Anal. Chem. 2010, 82, 2916–2924. [Google Scholar] [CrossRef]
- Dupre, M.; Gilquin, B.; Fenaille, F.; Feraudet-Tarisse, C.; Dano, J.; Ferro, M.; Simon, S.; Junot, C.; Brun, V.; Becher, F. Multiplex Quantification of Protein Toxins in Human Biofluids and Food Matrices Using Immunoextraction and High-Resolution Targeted Mass Spectrometry. Anal. Chem. 2015, 87, 8473–8480. [Google Scholar] [CrossRef]
- Kongmuang, U.; Honda, T.; Miwatani, T. A simple method for purification of Shiga or Shiga-like toxin from Shigella-Dysenteria-Coli O157-H7 by immunoaffinity column chromatography. FEMS Microbiol. Lett. 1987, 48, 379–383. [Google Scholar] [CrossRef]
- Shinagawa, K.; Mitsumori, M.; Matsusaka, N.; Suggi, S. Purification of staphylococcal enterotoxins A and E by immunoaffinity chromatography using a murine monoclonal antibody with dual specificity for both of these toxins. J. Immunol. Methods 1991, 139, 49–53. [Google Scholar] [CrossRef]
- Yue, Y.; Zhu, B.; Lun, L.; Xu, N. Quantifications of saxitoxin concentrations in bivalves by high performance liquid chromatography-tandem mass spectrometry with the purification of immunoaffinity column. J. Chromatogr. B 2020, 1147, 122133. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.W.C.; Wu, A.H.T. Development and validation of an analytical method for the analysis of Sterigmatocystin in roasted coffee beans and black pepper using liquid chromatography-tandem mass spectrometry. Food Addit. Contam. Part Chem. Anal. Control Expo. Risk Assess. 2020, 37, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; He, J.; Li, Y.; Chen, N.; Huang, Z.; You, L.; He, L.; Zhang, S. Solid-phase extraction of aflatoxins using a nanosorbent consisting of a magnetized nanoporous carbon core coated with a molecularly imprinted polymer. Microchim. Acta 2018, 185, 515. [Google Scholar] [CrossRef] [PubMed]
- Rui, C.; He, J.; Li, Y.; Liang, Y.; You, L.; He, L.; Li, K.; Zhang, S. Selective extraction and enrichment of aflatoxins from food samples by mesoporous silica FDU-12 supported aflatoxins imprinted polymers based on surface molecularly imprinting technique. Talanta 2019, 201, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Jayasinghe, G.D.T.M.; Domínguez-González, R.; Bermejo-Barrera, P.; Moreda-Piñeiro, A. Miniaturized vortex assisted-dispersive molecularly imprinted polymer micro-solid phase extraction and HPLC-MS/MS for assessing trace aflatoxins in cultured fish. Anal. Methods 2020, 12, 4351–4362. [Google Scholar] [CrossRef] [PubMed]
- Rico-Yuste, A.; Walravens, J.; Urraca, J.L.; Abou-Hany, R.A.G.; Descalzo, A.B.; Orellana, G.; Rychlik, M.; De Saeger, S.; Moreno-Bondi, M.C. Analysis of alternariol and alternariol monomethyl ether in foodstuffs by molecularly imprinted solid-phase extraction and ultra-high-performance liquid chromatography tandem mass spectrometry. Food Chem. 2018, 243, 357–364. [Google Scholar] [CrossRef]
- Tan, L.; He, R.; Li, Y.; Liang, Y.; Li, H.; Tang, Y. Fabrication of a biomimetic adsorbent imprinted with a common specificity determinant for the removal of α- and β-amanitin from plasma. J. Chromatogr. A 2016, 1459, 1–8. [Google Scholar] [CrossRef]
- Svoboda, P.; Combes, A.; Petit, J.; Nováková, L.; Pichon, V.; BMAALS Group. Synthesis of a molecularly imprinted sorbent for selective solid-phase extraction of β-N-methylamino-l-alanine. Talanta 2015, 144, 1021–1029. [Google Scholar] [CrossRef]
- Appell, M.; Jackson, M.A.; Wang, L.C.; Bosma, W.B. Determination of Citrinin Using Molecularly Imprinted Solid Phase Extraction Purification, HPLC Separation, and Fluorescence Detection. J. Liq. Chromatogr. Relat. Technol. 2015, 38, 1815–1819. [Google Scholar] [CrossRef]
- Urraca, J.L.; Huertas-Pérez, J.F.; Cazorla, G.A.; Gracia-Mora, J.; García-Campaña, A.M.; Moreno-Bondi, M.C. Development of magnetic molecularly imprinted polymers for selective extraction: Determination of citrinin in rice samples by liquid chromatography with UV diode array detection. Anal. Bioanal. Chem. 2016, 408, 3033–3042. [Google Scholar] [CrossRef]
- Lhotská, I.; Kholová, A.; Machyňáková, A.; Hroboňová, K.; Solich, P.; Švec, F.; Šatínský, D. Preparation of citrinin-selective molecularly imprinted polymer and its use for on-line solid-phase extraction coupled to liquid chromatography. Anal. Bioanal. Chem. 2019, 411, 2395–2404. [Google Scholar] [CrossRef]
- Kubo, T.; Nomachi, M.; Nemoto, K.; Sano, T.; Hosoya, K.; Tanaka, N.; Kaya, K. Chromatographic separation for domoic acid using a fragment imprinted polymer. Anal. Chim. Acta 2006, 577, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.-H.; Guo, X.-C.; Zhao, H.-Q.; Wu, S.-X.; Yang, H.-H.; Wang, X.-R. Molecularly imprinted polymer for selective extraction of domoic acid from seafood coupled with high-performance liquid chromatographic determination. Talanta 2011, 84, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Wang, D.; Peng, A.; Huang, Z.; Lin, Y. Determination of domoic acid in shellfish extracted by molecularly imprinted polymers. J. Sep. Sci. 2016, 39, 3254–3259. [Google Scholar] [CrossRef] [PubMed]
- Ao, J.; Gu, J.; Yuan, T.; Li, D.; Ma, Y.; Shen, Z. Applying molecular modelling and experimental studies to develop molecularly imprinted polymer for domoic acid enrichment from both seawater and shellfish. Chemosphere 2018, 199, 98–106. [Google Scholar] [CrossRef]
- Pascale, M.; De Girolamo, A.; Visconti, A.; Magan, N.; Chianella, I.; Piletska, E.V.; Piletsky, S.A. Use of itaconic acid-based polymers for solid-phase extraction of deoxynivalenol and application to pasta analysis. Anal. Chim. Acta 2008, 609, 131–138. [Google Scholar] [CrossRef]
- Pan, S.-D.; Ye, M.-J.; Gao, G.-S.; He, Q.; Wang, L.; Chen, X.-H.; Qiu, Q.-L.; Jin, M.-C. Synthesis of a monodisperse well-defined core–shell magnetic molecularly-imprinted polymer prior to LC-MS/MS for fast and sensitive determination of mycotoxin residues in rice. Anal Methods 2017, 9, 5281–5292. [Google Scholar] [CrossRef]
- Mei, X.-Q.; He, X.-P.; Wang, J.-T. Molecularly imprinted polymer as efficient sorbent of solid-phase extraction for determination of gonyautoxin 1,4 in seawater followed by high-performance liquid chromatography-fluorescence detection. Anal. Bioanal. Chem. 2016, 408, 5737–5743. [Google Scholar] [CrossRef]
- He, X.; Wang, J.; Mei, X.-Q. Dummy Fragment Template Molecularly Imprinted Polymers for the Selective Solid-phase Extraction of Gonyautoxins from Seawater. Anal. Lett. 2017, 50, 1877–1886. [Google Scholar] [CrossRef]
- Lian, Z.; Wang, J. Selective isolation of gonyautoxins 1,4 from the dinoflagellate Alexandrium minutum based on molecularly imprinted solid-phase extraction. Mar. Pollut. Bull. 2017, 122, 500–504. [Google Scholar] [CrossRef]
- Lian, Z.-R.; Wang, J.-T. Study of molecularly imprinted solid-phase extraction of gonyautoxins 2,3 in the cultured dinoflagellate Alexandrium tamarense by high-performance liquid chromatography with fluorescence detection. Environ. Pollut. 2013, 182, 385–391. [Google Scholar] [CrossRef]
- Zhang, Y.; Qu, J.; Du, W.; Wu, M.; Liu, L. Molecularly imprinted polymer solid phase extraction coupled with liquid chromatography-high resolution mass spectrometry for the detection of gonyautoxins 2&3 in seawater. Mar. Pollut. Bull. 2020, 157, 111333. [Google Scholar] [CrossRef]
- Chianella, I.; Piletsky, S.A.; Tothill, I.E.; Chen, B.; Turner, A.P.F. MIP-based solid phase extraction cartridges combined with MIP-based sensors for the detection of microcystin-LR. Biosens. Bioelectron. 2003, 18, 119–127. [Google Scholar] [CrossRef]
- Tian, X.; She, C.; Qi, Z.; Xu, X. Magnetic-graphene oxide based molecularly imprinted polymers for selective extraction of microsystin-LR prior to the determination by HPLC. Microchem. J. 2019, 146, 1126–1133. [Google Scholar] [CrossRef]
- Wu, Z.; He, D.; Cui, B.; Jin, Z. Ultrasensitive detection of microcystin-LR with gold immunochromatographic assay assisted by a molecular imprinting technique. Food Chem. 2019, 283, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Maier, N.M.; Buttinger, G.; Welhartizki, S.; Gavioli, E.; Lindner, W. Molecularly imprinted polymer-assisted sample clean-up of ochratoxin A from red wine: Merits and limitations. J. Chromatogr. B 2004, 804, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Krushkova, S.; Lai, E.P.; Dabek-Zlotorzynska, E. Molecularly-imprinted polypyrrole-modified stainless steel frits for selective solid phase preconcentration of ochratoxin A. Anal. Bioanal. Chem. 2005, 382, 1534–1540. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.C.C.; Lai, E.P.C. Determination of ochratoxin A in red wines by multiple pulsed elutions from molecularly imprinted polypyrrole. Food Chem. 2007, 105, 301–310. [Google Scholar] [CrossRef]
- Vidal, J.C.; Duato, P.; Bonel, L.; Castillo, J.R. Molecularly Imprinted On-Line Solid-Phase Extraction Coupled with Fluorescence Detection for the Determination of Ochratoxin A in Wheat Samples. Anal. Lett. 2012, 45, 51–62. [Google Scholar] [CrossRef]
- Lee, T.P.; Saad, B.; Khayoon, W.S.; Salleh, B. Molecularly imprinted polymer as sorbent in micro-solid phase extraction of ochratoxin A in coffee, grape juice and urine. Talanta 2012, 88, 129–135. [Google Scholar] [CrossRef]
- Cao, J.; Kong, W.; Zhou, S.; Yin, L.; Wan, L.; Yang, M. Molecularly imprinted polymer-based solid phase clean-up for analysis of ochratoxin A in beer, red wine, and grape juice. J. Sep. Sci. 2013, 36, 1291–1297. [Google Scholar] [CrossRef] [PubMed]
- Ali, W.H.; Derrien, D.; Alix, F.; Pérollier, C.; Lépine, O.; Bayoudh, S.; Chapuis-Hugon, F.; Pichon, V. Solid-phase extraction using molecularly imprinted polymers for selective extraction of a mycotoxin in cereals. J. Chromatogr. A 2010, 1217, 6668–6673. [Google Scholar] [CrossRef] [PubMed]
- Giovannoli, C.; Passini, C.; Di Nardo, F.; Anfossi, L.; Baggiani, C. Determination of Ochratoxin A in Italian Red Wines by Molecularly Imprinted Solid Phase Extraction and HPLC Analysis. J. Agric. Food Chem. 2014, 62, 5220–5225. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Huang, P.; Suo, L.; Wu, F. Polydopamine-based molecularly imprinting polymers on magnetic nanoparticles for recognition and enrichment of ochratoxins prior to their determination by HPLC. Microchim. Acta 2018, 185, 300. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Jia, J.; Yu, X.; Sun, X. Preparation and characterization of a molecularly imprinted polymer by grafting on silica supports: A selective sorbent for patulin toxin. Anal. Bioanal. Chem. 2011, 401, 2259. [Google Scholar] [CrossRef] [PubMed]
- Anene, A.; Hosni, K.; Chevalier, Y.; Kalfat, R.; Hbaieb, S. Molecularly imprinted polymer for extraction of patulin in apple juice samples. Food Control 2016, 70, 90–95. [Google Scholar] [CrossRef]
- Lhotská, I.; Holznerova, A.; Solich, P.; Šatínský, D. Critical comparison of the on-line and off-line molecularly imprinted solid-phase extraction of patulin coupled with liquid chromatography. J. Sep. Sci. 2017, 40, 4599–4609. [Google Scholar] [CrossRef]
- Yang, Y.; Li, Q.; Fang, G.; Wang, S. Preparation and evaluation of novel surface molecularly imprinted polymers by sol–gel process for online solid-phase extraction coupled with high performance liquid chromatography to detect trace patulin in fruit derived products. RSC Adv. 2016, 6, 54510–54517. [Google Scholar] [CrossRef]
- Zhao, M.; Shao, H.; He, Y.; Li, H.; Yan, M.; Jiang, Z.; Wang, J.; Abd El-Aty, A.M.; Hacımüftüoğlu, A.; Yan, F.; et al. The determination of patulin from food samples using dual-dummy molecularly imprinted solid-phase extraction coupled with LC-MS/MS. J. Chromatogr. B 2019, 1125, 121714. [Google Scholar] [CrossRef]
- Luo, Z.; Chen, G.; Li, X.; Wang, L.; Shu, H.; Cui, X.; Chang, C.; Zeng, A.; Fu, Q. Molecularly imprinted polymer solid -phase microextraction coupled with ultra high performance liquid chromatography and tandem mass spectrometry for rapid analysis of pyrrolizidine alkaloids in herbal medicine. J. Sep. Sci. 2019, 42, 3352–3362. [Google Scholar] [CrossRef]
- De Smet, D.; Monbaliu, S.; Dubruel, P.; Van Peteghem, C.; Schacht, E.; De Saeger, S. Synthesis and application of a T-2 toxin imprinted polymer. J. Chromatogr. A 2010, 1217, 2879–2886. [Google Scholar] [CrossRef] [PubMed]
- Lhotská, I.; Gajdošová, B.; Solich, P.; Šatínský, D. Molecularly imprinted vs. reversed-phase extraction for the determination of zearalenone: A method development and critical comparison of sample clean-up efficiency achieved in an on-line coupled SPE chromatography system. Anal. Bioanal. Chem. 2018, 410, 3265–3273. [Google Scholar] [CrossRef] [PubMed]
- Cavaliere, C.; Antonelli, M.; Cerrato, A.; La Barbera, G.; Laganà, A.; Laus, M.; Piovesana, S.; Capriotti, A.L. A Novel Magnetic Molecular Imprinted Polymer for Selective Extraction of Zearalenone from Cereal Flours before Liquid Chromatography-Tandem Mass Spectrometry Determination. Toxins 2019, 11, 493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.; He, J.; Li, Y.; Wu, C.; You, L.; Wei, H.; Li, K.; Zhang, S. Preparation of dummy molecularly imprinted polymers for extraction of Zearalenone in grain samples. J. Chromatogr. A 2019, 1602, 11–18. [Google Scholar] [CrossRef]
- Mausia, T.; De Smet, D.; Guorun, Q.; Van Peteghem, C.; Zhang, D.; Wu, A.; De Saeger, S. Molecularly Imprinted Polymers as Specific Adsorbents for Zearalenone Produced by Precipitation Polymerization and Applied to Mycotoxin Production. Anal. Lett. 2011, 44, 2633–2643. [Google Scholar] [CrossRef]
- Urraca, J.L.; Marazuela, M.D.; Moreno-Bondi, M.C. Molecularly imprinted polymers applied to the clean-up of zearalenone and α-zearalenol from cereal and swine feed sample extracts. Anal. Bioanal. Chem. 2006, 385, 1155–1161. [Google Scholar] [CrossRef]
- Szumski, M.; Grzywiński, D.; Prus, W.; Buszewski, B. Monolithic molecularly imprinted polymeric capillary columns for isolation of aflatoxins. J. Chromatogr. A 2014, 1364, 163–170. [Google Scholar] [CrossRef]
- Guo, X.; Wen, F.; Zheng, N.; Saive, M.; Fauconnier, M.-L.; Wang, J. Aptamer-Based Biosensor for Detection of Mycotoxins. Front. Chem. 2020, 8, 195. [Google Scholar] [CrossRef] [Green Version]
- Kudlak, B.; Wieczerzak, M. Aptamer based tools for environmental and therapeutic monitoring: A review of developments, applications, future perspectives. Crit. Rev. Environ. Sci. Technol. 2019, 50, 816–867. [Google Scholar] [CrossRef]
- ZOU, X.-M.; ZHOU, J.-W.; SONG, S.-H.; CHEN, G.-H. Screening of Oligonucleotide Aptamers and Application in Detection of Pesticide and Veterinary Drug Residues. Chin. J. Anal. Chem. 2019, 47, 488–499. [Google Scholar] [CrossRef]
- Pichon, V.; Chapuis-Hugon, F.; Hennion, M.C. 2.19-Bioaffinity Sorbents. In Comprehensive Sampling and Sample Preparation; Pawliszyn, J., Ed.; Academic Press: Oxford, UK, 2012; Volume 2, pp. 359–388. ISBN 978-0-12-381374-9. [Google Scholar]
- Stoltenburg, R.; Reinemann, C.; Strehlitz, B. SELEX—A (r)evolutionary method to generate high-affinity nucleic acid ligands. Biomol. Eng. 2007, 24, 381–403. [Google Scholar] [CrossRef] [PubMed]
- Peyrin, E. Nucleic acid aptamer molecular recognition principles and application in liquid chromatography and capillary electrophoresis. J. Sep. Sci. 2009, 32, 1531–1536. [Google Scholar] [CrossRef] [PubMed]
- Peltomaa, R.; Benito-Peña, E.; Moreno-Bondi, M.C. Bioinspired recognition elements for mycotoxin sensors. Anal. Bioanal. Chem. 2018, 410, 747–771. [Google Scholar] [CrossRef] [PubMed]
- Pichon, V.; Brothier, F.; Combès, A. Aptamer-based-sorbents for sample treatment—A review. Anal. Bioanal. Chem. 2015, 407, 681–698. [Google Scholar] [CrossRef]
- Cruz-Aguado, J.A.; Penner, G. Fluorescence Polarization Based Displacement Assay for the Determination of Small Molecules with Aptamers. Anal. Chem. 2008, 80, 8853–8855. [Google Scholar] [CrossRef]
- Setlem, K.; Monde, B.; Ramlal, S.; Kingston, J. Immuno Affinity SELEX for Simple, Rapid, and Cost-Effective Aptamer Enrichment and Identification against Aflatoxin B1. Front. Microbiol. 2016, 7, 1909. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Wang, W.; Chen, X.; Xia, Y.; Wu, S.; Duan, N.; Wang, Z. Selection, identification, and application of Aflatoxin B1 aptamer. Eur. Food Res. Technol. 2014, 238, 919–925. [Google Scholar] [CrossRef]
- Ma, X.; Wang, W.; Chen, X.; Xia, Y.; Duan, N.; Wu, S.; Wang, Z. Selection, characterization and application of aptamers targeted to Aflatoxin B2. Food Control 2015, 47, 545–551. [Google Scholar] [CrossRef]
- Liu, H.; Luan, Y.; Lu, A.; Li, B.; Yang, M.; Wang, J. An oligosorbent-based aptamer affinity column for selective extraction of aflatoxin B2 prior to HPLC with fluorometric detection. Microchim. Acta 2018, 185, 71. [Google Scholar] [CrossRef]
- Liu, H.; Zhao, Y.; Lu, A.; Ye, J.; Wang, J.; Wang, S.; Luan, Y. An aptamer affinity column for purification and enrichment of aflatoxin B1 and aflatoxin B2 in agro-products. Anal. Bioanal. Chem. 2020, 412, 895–904. [Google Scholar] [CrossRef]
- Liu, H.; Lu, A.; Fu, H.; Li, B.; Yang, M.; Wang, J.; Luan, Y. Affinity capture of aflatoxin B1 and B2 by aptamer-functionalized magnetic agarose microspheres prior to their determination by HPLC. Microchim. Acta 2018, 185, 326. [Google Scholar] [CrossRef] [PubMed]
- Khodadadi, M.; Malekpour, A.; Mehrgardi, M.A. Aptamer functionalized magnetic nanoparticles for effective extraction of ultratrace amounts of aflatoxin M1 prior its determination by HPLC. J. Chromatogr. A 2018, 1564, 85–93. [Google Scholar] [CrossRef] [PubMed]
- De Girolamo, A.; McKeague, M.; Miller, J.D.; DeRosa, M.C.; Visconti, A. Determination of ochratoxin A in wheat after clean-up through a DNA aptamer-based solid phase extraction column. Food Chem. 2011, 127, 1378–1384. [Google Scholar] [CrossRef] [PubMed]
- Chapuis-Hugon, F.; du Boisbaudry, A.; Madru, B.; Pichon, V. New extraction sorbent based on aptamers for the determination of ochratoxin A in red wine. Anal. Bioanal. Chem. 2011, 400, 1199–1207. [Google Scholar] [CrossRef]
- Ali, W.H.; Pichon, V. Characterization of oligosorbents and application to the purification of ochratoxin A from wheat extracts. Anal. Bioanal. Chem. 2014, 406, 1233–1240. [Google Scholar]
- Yang, X.; Kong, W.; Hu, Y.; Yang, M.; Huang, L.; Zhao, M.; Ouyang, Z. Aptamer-affinity column clean-up coupled with ultra high performance liquid chromatography and fluorescence detection for the rapid determination of ochratoxin A in ginger powder. J. Sep. Sci. 2014, 37, 853–860. [Google Scholar] [CrossRef]
- Yang, X.; Hu, Y.; Kong, W.; Chu, X.; Yang, M.; Zhao, M.; Ouyang, Z. Ultra-fast liquid chromatography with tandem mass spectrometry determination of ochratoxin A in traditional Chinese medicines based on vortex-assisted solid–liquid microextraction and aptamer-affinity column clean-up. J. Sep. Sci. 2014, 37, 3052–3059. [Google Scholar] [CrossRef]
- Yu, X.; Song, H.; Huang, J.; Chen, Y.; Dai, M.; Lin, X.; Xie, Z. An aptamer@AuNP-modified POSS–polyethylenimine hybrid affinity monolith with a high aptamer coverage density for sensitive and selective recognition of ochratoxin A. J. Mater. Chem. B 2018, 6, 1965–1972. [Google Scholar] [CrossRef]
- Wu, X.; Hu, J.; Zhu, B.; Lu, L.; Huang, X.; Pang, D. Aptamer-targeted magnetic nanospheres as a solid-phase extraction sorbent for determination of ochratoxin A in food samples. J. Chromatogr. A 2011, 1218, 7341–7346. [Google Scholar] [CrossRef]
- Zhang, Q.; Yang, Y.; Zhi, Y.; Wang, X.; Wu, Y.; Zheng, Y. Aptamer-modified magnetic metal-organic framework MIL-101 for highly efficient and selective enrichment of ochratoxin A. J. Sep. Sci. 2019, 42, 716–724. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, M.; Chi, J.; Yu, X.; Chen, Y.; Lin, X.; Xie, Z. Aptamer-based polyhedral oligomeric silsesquioxane (POSS)-containing hybrid affinity monolith prepared via a “one-pot” process for selective extraction of ochratoxin A. J. Chromatogr. A 2018, 1563, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Chi, J.; Chen, M.; Deng, L.; Lin, X.; Xie, Z. A facile AuNPs@aptamer-modified mercaptosiloxane-based hybrid affinity monolith with an unusually high coverage density of aptamer for on-column selective extraction of ochratoxin A. Analyst 2018, 143, 5210–5217. [Google Scholar] [CrossRef] [PubMed]
- Brothier, F.; Pichon, V. Miniaturized DNA aptamer-based monolithic sorbent for selective extraction of a target analyte coupled on-line to nanoLC. Anal. Bioanal. Chem. 2014, 406, 7875–7886. [Google Scholar] [CrossRef] [PubMed]
- Marechal, A.; Jarrosson, F.; Randon, J.; Dugas, V.; Demesmay, C. In-line coupling of an aptamer based miniaturized monolithic affinity preconcentration unit with capillary electrophoresis and Laser Induced Fluorescence detection. J. Chromatogr. A 2015, 1406, 109–117. [Google Scholar] [CrossRef]
- Chen, X.; Huang, Y.; Duan, N.; Wu, S.; Ma, X.; Xia, Y.; Zhu, C.; Jiang, Y.; Wang, Z. Selection and identification of ssDNA aptamers recognizing zearalenone. Anal. Bioanal. Chem. 2013, 405, 6573–6581. [Google Scholar] [CrossRef]
- Pichon, V. Selective sample treatment using molecularly imprinted polymers. J. Chromatogr. A 2007, 1152, 41–53. [Google Scholar] [CrossRef]
- Cruz-Aguado, J.A.; Penner, G. Determination of Ochratoxin A with a DNA Aptamer. J. Agric. Food Chem. 2008, 56, 10456–10461. [Google Scholar] [CrossRef]
- Tang, J.; Xie, J.; Shao, N.; Yan, Y. The DNA aptamers that specifically recognize ricin toxin are selected by two in vitro selection methods. Electrophoresis 2006, 27, 1303–1311. [Google Scholar] [CrossRef]
- Ng, A.; Chinnappan, R.; Eissa, S.; Liu, H.; Tlili, C.; Zourob, M. Selection, Characterization, and Biosensing Application of High Affinity Congener-Specific Microcystin-Targeting Aptamers. Environ. Sci. Technol. 2012, 46, 10697–10703. [Google Scholar] [CrossRef]
- McKeague, M.; Bradley, C.R.; De Girolamo, A.; Visconti, A.; Miller, J.D.; DeRosa, M.C. Screening and Initial Binding Assessment of Fumonisin B-1 Aptamers. Int. J. Mol. Sci. 2010, 11, 4864–4881. [Google Scholar] [CrossRef] [Green Version]
- Frohnmeyer, E.; Frisch, F.; Falke, S.; Betzel, C.; Fischer, M. Highly affine and selective aptamers against cholera toxin as capture elements in magnetic bead-based sandwich ELAA. J. Biotechnol. 2018, 269, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Hu, B.; Zheng, X.; Cao, Y.; Liu, D.; Sun, M.; Jiao, B.; Wang, L. Gonyautoxin 1/4 aptamers with high-affinity and high-specificity: From efficient selection to aptasensor application. Biosens. Bioelectron. 2016, 79, 938–944. [Google Scholar] [CrossRef] [PubMed]
- Eissa, S.; Ng, A.; Siaj, M.; Tavares, A.C.; Zourob, M. Selection and Identification of DNA Aptamers against Okadaic Acid for Biosensing Application. Anal. Chem. 2013, 85, 11794–11801. [Google Scholar] [CrossRef] [PubMed]
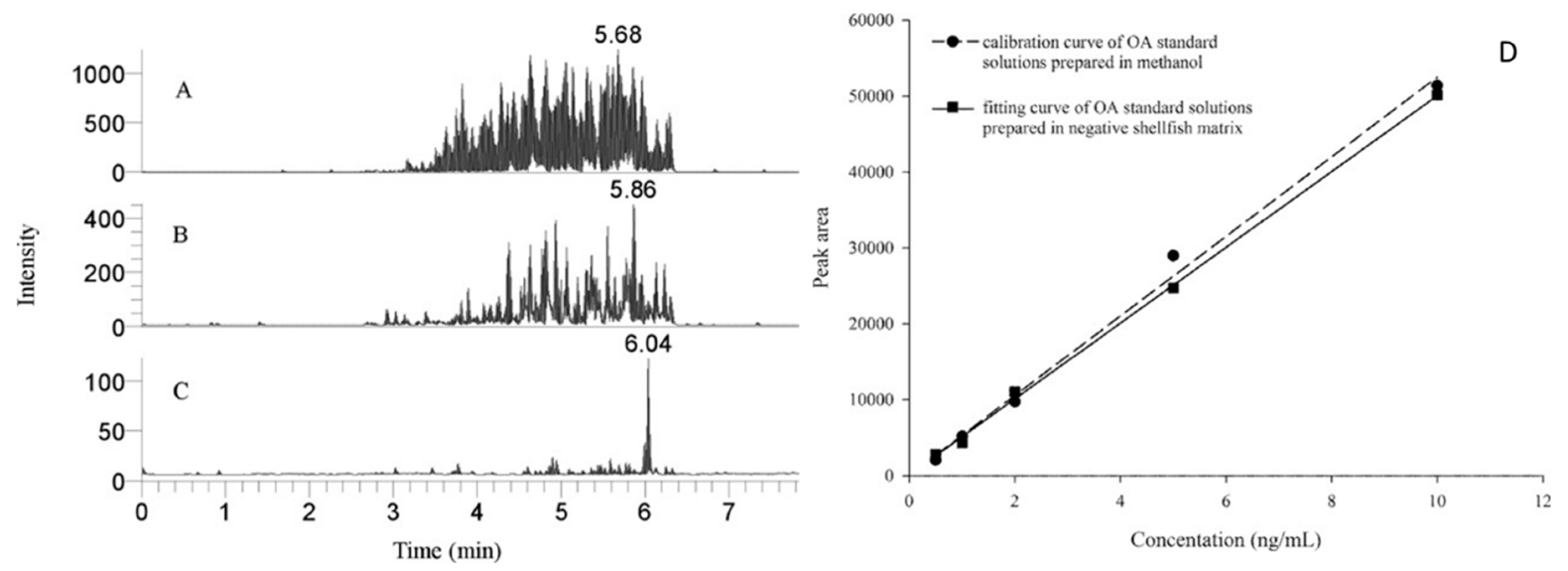


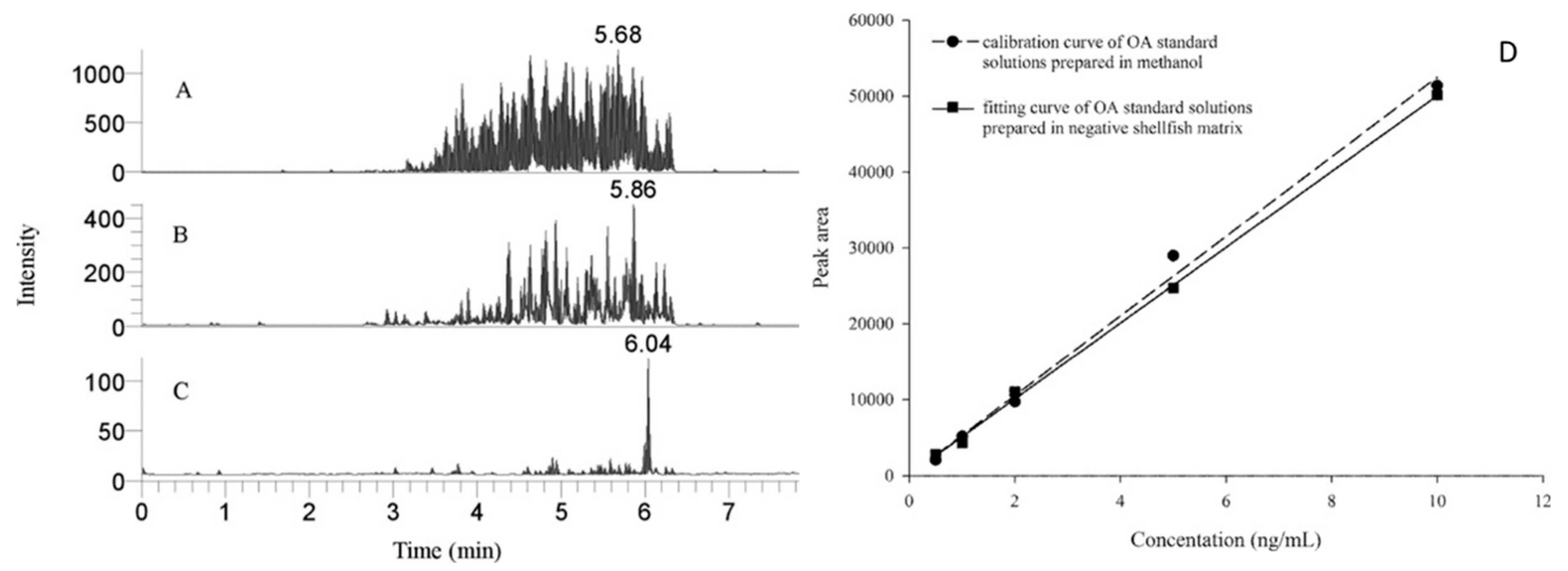{kind=link}
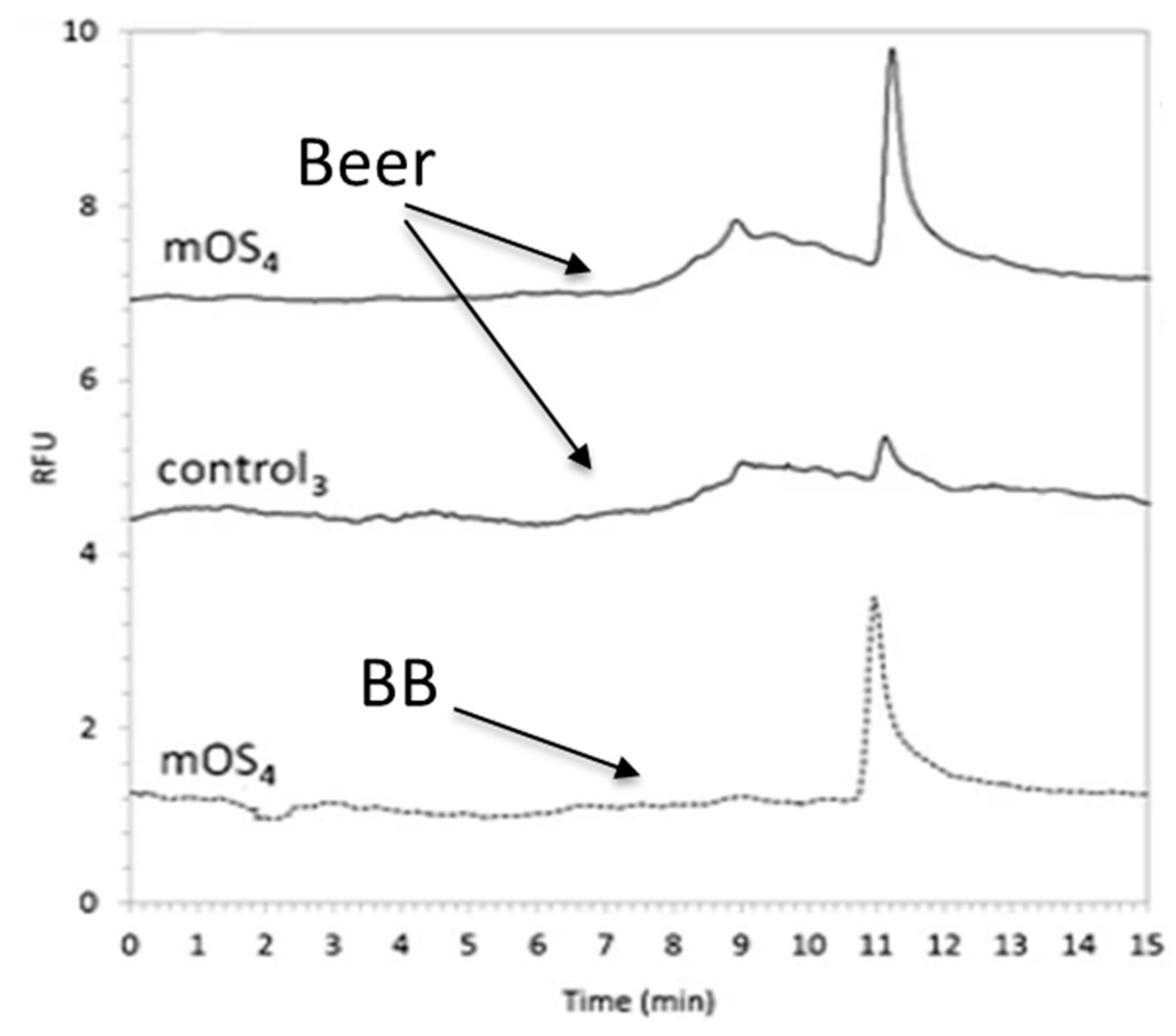
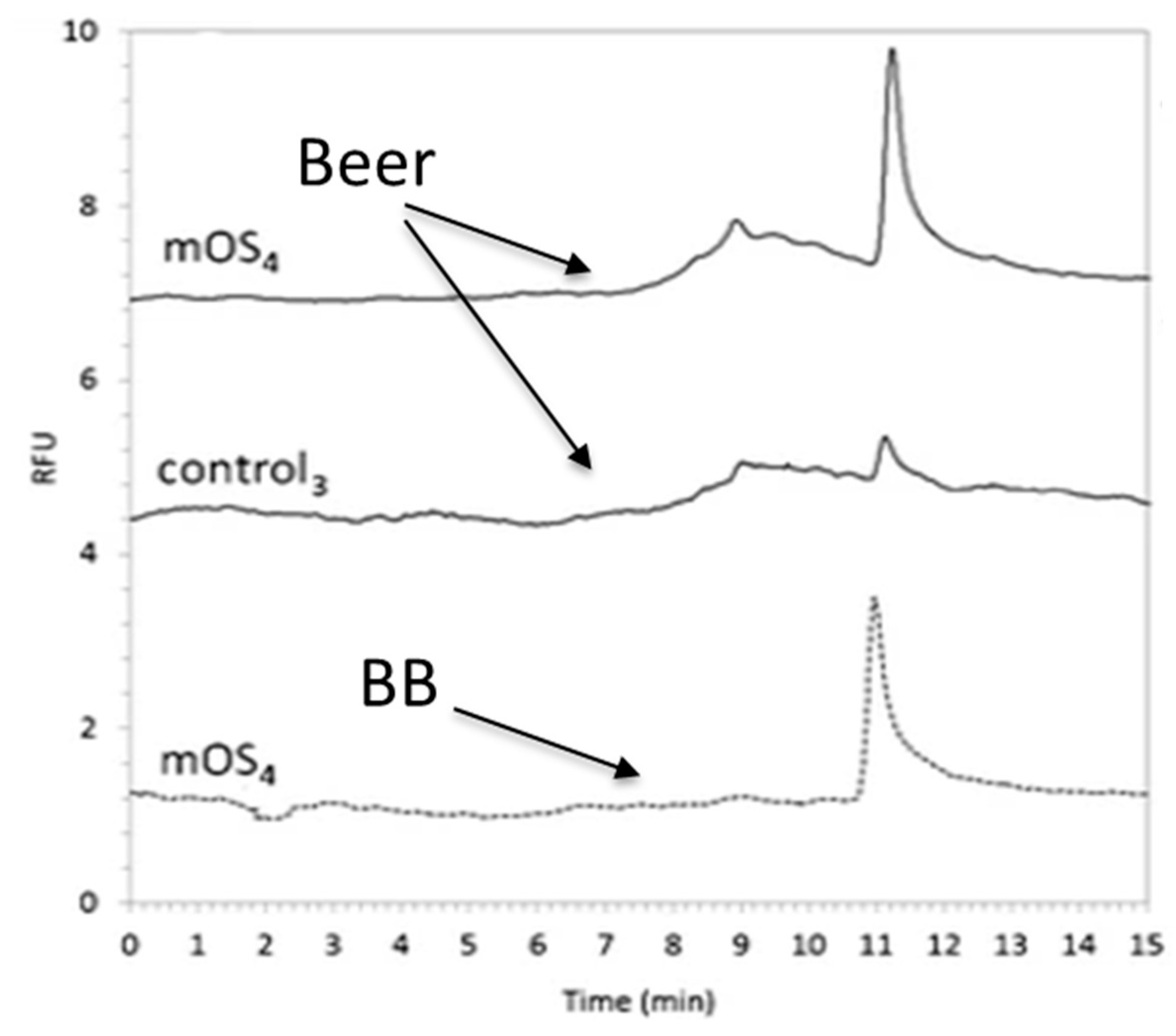
{kind=link}
{kind=link}
| Toxin(s) | Matrix | Marketed ISs (Company) | Extraction Solvent; Factor and Solvent of Dilution | Vsample (eq. of Solid Sample) | Washing | Elution | Analysis | Ref. |
|---|---|---|---|---|---|---|---|---|
| Single analyte and analogs/metabolites | ||||||||
| AFs (B1, B2, G1, G2) | Olive, peanut and sesame oils | Aflatest WB (Vicam) | MeOH/water 45/55; -, water | - | - | MeOH | LC/Fluo | [12] |
| Nuts and based-nut products | Alfaprep (R-biopharm) | MeOH/water 7/3, NaCl; ×3, water | 15 mL (eq. 1 g) | Water | [13] | |||
| Baby food and feed | AlfaOchra HPLCTM (Vicam) | ACN/water 78/22 (solid), ACN (milk); dried extract dil. in water | 10 mL | Water | MeOH | LC/MS–MS | [14] | |
| AF B1 | Sidestream cigarette smoke | Aflatest P aflatoxin (Vicam) | -; ×4, water | 20 mL | Water | ACN | LC/MS | [15] |
| Organic spices and herbs | RIDA Aflatoxin column (R-Biopharm) | MeOH/water 7/3; ×4, water | 1 mL (eq. 0.25 g) | MeOH | ELISA | [16] | ||
| AF B1 and AF M1 | Pig liver | AflaTM wide bore for M1 Aflatest-P for AFB1 (Vicam) | MeOH/water, NaCl; ×5, PBS, Tween-20 2% | 20 mL (eq. 1 g) | PBS, Tween-20 2% | MeOH | Fluo, LC/Fluo | [17] |
| DON | Wheat | DON-Test HPLC (Vicam) | Water; -, - | 1 mL (eq. 0.25 g) | - | LC/UV | [18] | |
| DON, NIV | Rice, bran | DON NIV WB (Vicam) | Water, NaCl; ×5, PBS | 10 mL (eq. 0.4 g) | PBS + water | MeOH + ACN | LC/UV, LC/MS | [19] |
| FUMs (B1, B2) | Cornflakes | Fumoni Test™ (Vicam) | ACN/MeOH/water 25/25/50; ×5, PBS | 10 mL (eq. 0.4 g) | PBS | MeOH | LC/Fluo | [20] |
| OTA | Cereals | Easi-extract (Biocode) | MeOH/water 1/1; ×3, PBS | 50 ml | Water | [21] | ||
| Wine | Ochraprep (OP, Rhone Diagnostic Technologies) et Ochratest (OT, Vicam) | pH adjusted | 10 mL + 10 mL PBS (OT) or 4 mL + 10 mL PBS (OP) | PBS + water (OP) | [22] | |||
| Beer | Ochratest (Vicam) | Degassed; ×2, PEG - NaHCO3 | 10 mL | NaCl 2.5%, NaHCO3 0.5% + water | [23] | |||
| Urine | ×2 (human) or ×3.4 (rat), NaHCO3 + filtration | Water | [24] | |||||
| Milk | Ochraprep (R-Biopharm) et Ochratest (Vicam) | - | 50 mL | [25] | ||||
| Grapes, dried wine fruit, winery products | Ochratest (Vicam) | -, ACN/water or ACN/MeOH/water | - | Water, NaCl 2.5% + NaHCO3 0.5%, PEG 1% + NaHCO3 5% | [26] | |||
| Ready-to-drink coffee | -; ×8, PB | 5 mL | NaCl + NaHCO3 0.5% + water + NH4CH3CO2 | MeOH + AA 2% | LC/MS–MS | [27] | ||
| Wine | -; ×2 PEG 8000 (1%), NaHCO3 (5%) | 10 mL | NaCl, NaHCO3 + water | MeOH | Fluo, LC/Fluo, | [28] | ||
| Cereals, spices | MeOH/water 7/3; ×1.8, water | 40 mL | Water + PBS, Tween 20 | LC/Fluo | [29] | |||
| OTA, OTB, α-OTA | Milk | Ochraprep (R-Biopharm) | LLE with CHCl3; back extraction with PBS | PBS extract | Drying | [30] | ||
| SMC | Cereal, cheese, beer | Easi-extract SMC (R-Biopharm) | ACN/water 8/2, NaCl; ×15, PBS | 10–30 mL (eq. 0.25–0.5 g) | PBS + water | ACN | LC/MS–MS | [31] |
| T-2 toxin | Cereals | T2 TAG (Vicam) | MeOH/water 8/2; ×5, water | 10 mL (eq. 1 g) | Water | MeOH | LC/Fluo | [32] |
| T-2 & HT-2 toxins | T-2 test (Vicam) | MeOH/water 9/1, NaCl; ×5, water | - | [33] | ||||
| Easi-extract T2 (R-Biopharm), T-2 Test HPLC (Vicam) | MeOH/water 9/1; -, NaCl 4% | - | MeOH (x 3, backflush) | [34] | ||||
| Easi-extract T2 (R-Biopharm) | MeOH/water 9/1 + 2% NaCl (oats) | - | - | - | [35] | |||
| Chinese herbal medicines and related products | HT-2 HPLC (Vicam) | MeOH/water 9/1, NaCl; ×5, water | 10 mL (eq. 0.5 g) | Water | MeOH | GC/ECD, GC/MS | [36] | |
| Oats | Easi-extract T-2 and HT-2 (R-Biopharm) | MeOH/water 9/1, NaCl; ×5, NaCl 4% | 5–25 mL (eq. 1.5–0.3 g) | Tween 20 0.01% + water | LC/UV | [37] | ||
| Food, Feed | MeOH/water 9/1, NaCl; ×5, water | 25 mL (eq. 1 g) | Water | LC/MS–MS | [38] | |||
| ZON | Corn | ZearalaTest (Vicam) | ACN/water 9/1; ×10, water | 10 mL | LC/Fluo | [39] | ||
| Botanical root products, soybeans, grains, grain products extracted | MeOH/water 75/25; ×10, PBS, Tween 20 (0.5%) | 50 mL (eq. 1 g) | MeOH/PBS, 15/85 + Tween 20 (0.5%) + water | LC/Fluo, LC/MS | [40] | |||
| ZON and metabolites (5) | Maize | ACN/water 9/1, NaCl; ×5, PBS, Tween 20 (0.1%) | 10 mL | Water | Fluo or LC/Fluo | [41] | ||
| Multi-analytes | ||||||||
| AFs (B1, B2, G1, G2), OTA | Ginseng, ginger | AflaOchraTest (Vicam) | MeOH/water 7/3, 0.5%NaHCO3; ×5, PBS, Tween 20 (1%) | - | PBS + water + water/MeOH 85/15 | MeOH | LC/Fluo, LC/MS | [42] |
| Cereals | Aflatest and Ochratest (Vicam) | ACN/water 6/4 (OTA); MeOH/water 8/2 (AFs); ×5, PBS | 50 mL (eq. 0.5 g) | Water | MeOH | LC/Fluo | [43] | |
| Sicilian sweet wines | Ochraprep, Easi-extract for AFs (R-Biopharm) | -; ×2 PEG 6000 (1%), NaHCO3 (5%) | 20 mL | NaCl 2.5% + NaHCO3 0.5% + water | MeOH/AA 2% (OTA), ACN (AFs) | LC/Fluo | [44] | |
| Meat products | Aflatest and Ochratest (Vicam) | MeOH/water 6/4, NaCl; × 2, water (AF) MeOH/water, NaHCO3 1% 7/3; ×5, water (OTA); | 10 mL (eq. 1 g) | Water (AFs); Tween 20/PBS (OTA) | MeOH (AFs) | LC/Fluo | [45] | |
| Spices and spices mixtures | AflaOchra HPLC (Vicam) | MeOH/water 8/2, NaCl; ×10, PBS, Tween 20 | 20 mL | Tween 20 0.01%, PBS + water | MeOH | LC/Fluo | [46] | |
| Ginger | MeOH/water 7/3, NaHCO3 0.5%; ×4, PBS, Tween 20 (1%) | 25 mL (eq. 0.3 g) | PBS + water | LC/Fluo | [47] | |||
| AFs (B1, B2, G1, G2), OTA, FUMs (B1, B2), DON, ZON, T-2 and HT-2 | Maize | AOFZDT2TM (Vicam) | Water (A) and then water /MeOH 3/7; PBS | Percolation of B and then of A | PBS (B), water (A) | LC/MS | [48] | |
| AFs (B1, B2, G1, G2), OTA, ZON | Airborne from poultry house | AOZ (Vicam) | aqueous extract + NaCl; -, - | - | - | LC/Fluo | [49] | |
| AFs (B1, B2, G1, G2), OTA, ZON, FUM (B1, B2, B3), T-2 and HT-2 | Cereals | AOF-MS-Prep and DZT-MS-Prep used in tandem | MeOH/water 7/3, NaCl; -, - | - | - | - | LC/MS–MS | [50] |
| AFs (B1, B2, G1, G2, M1), OTA | Turkish dairy food | 3 ISs (no supplier mentioned) | MeOH/water 8/2, NaCl (AF B and G), 7/3 (OTA) and CHCl3, NaCl (AF M1); ×7, PBS (AF B/G, OTA), dried residue diluted in MeOH/PBS 2/98 (AF M1) | - | PBS (AF M1) | MeOH/water 1.25/1.75 (AFs B/G); MeOH/ACN 2/3 + water (AF M1); MeOH + water (OTA) | LC/Fluo | [51] |
| AFs (B1, B2, G1, G2, M1), OTA, DON, ZON, FUM (2), T-2 and HT-2 | Food | AflaOchra Prep (R-Biopharm) | QuEChERSs method including LLE (hexane) to purify ACN extract; ×12.5, PBS | - | Water | MeOH | LC/MS–MS | [52] |
| AFs (B1, B2, G1, G2, M1), OTA, DON, ZON, NIV, FUS-X, VCG; T-2 and HT-2; CTN, 3-ADON,15-ADON, SMC | Food and feed extracts (84% ACN) | Mycosep 226 Aflazon + (COCMY2226, Romer labs) | -; ×2, ACN | - | - | - | LC/MS–MS | [53] |
| AFs (B1, B2, G1, G2), OTA, DON, ZON, FUM (3), T-2 and HT-2, NIV, 3-ADON, 5-ADON | Cereals | Myco6in 1 (Vicam) | Water + MeOH; ×3.5, PBS after partial evaporation | 7 mL (eq. 0.5 g) | Water | MeOH + water | LC/MS–MS | [54] |
| AFs (B1, B2, G1, G2), DON, ZON, NIV, FUS-X, T-2 and HT-2, 3-ADON, 15-ADON, DAS | Corn, wheat, biscuit, cornflakes | Multisep 226 (Romer Labs) | ACN/water 85/15; none | 10 mL | - | - | LC/MS | [55] |
| AFs (B1, B2, G1, G2), OTA, DON, ZON, FUMs (B1, B2), T-2 and HT-2, NIV | Spices, infant formula, coffee, nuts | AflaOchra Prep (R-Biopharm) | Water/ACN/AA 10/89.75/0.25 + salt (MgSO4/NaCl) + LLE (Hexane); ×25, PBS | 50 mL | Water | MeOH | LC/MS–MS | [56] |
| Corn and corn-derived products | Myco6in1 (Vicam) | MeOH/water 7/3; ×10, PBS | 20 mL (eq. 0.5 g) | PBS + water | [57] | |||
| AFs (B1, B2, G1, G2), OTA, DON, ZON, FUMs (B1, B2, ), T-2 and HT-2, NIV, 3-ADON | Cereal grains | ACN/water/AA 79.5/20/0.5; ×16, PBS | - | MeOH/water, 8/2, AA 0.5% | [58] | |||
| AFs (B1, B2, G1, G2), OTA, DON, ZON, FUM (B1, B2, B3), T-2 | Herbs | MeOH/PBS 7/3 + LLE (hexane); ×26, PBS | - | NH4HCO2, FA (0.1%) | MeOH | LC/MS–MS, LC/HRMS | [59] | |
| AFs (B1, B2, G1, G2), OTA, DON, ZON, FUMs (B1, B2, B3), T-2 and HT-2 | Cereals, nuts | ACN/water/AA 79.5/20/0.5 + evaporation; -, PBS | 10 mL (eq. 2.5 g) | Water | LC/MS–MS | [60] | ||
| AFs (B1, B2, G1, G2), OTA, DON, ZON, FUM, T-2 and HT-2 | Cereals | AOF MS PREP, DZT MS-PREP (R-Biopharm) | MeOH/water 7/3, NaCl; ×13, PBS | 20 mL (eq. 0.38 g) | [61] | |||
| AF M1, OTA, DON, DON analog, ZON (α,β), FUM B1 | Urine | Myco6in1 (Vicam) | Oasis HLB SPE cartridge connected to the top of the IS; ×2, water | 12 mL | MeOH + water | [62] | ||
| DON, ZON and 5 derivatives, 3-ADON, 15-ADON | Flour | Multi-IACs (Magnech Bio-Tech) | ACN/water 8/2; ×8, PBS | 20 mL (eq. 0.25 g) | Tween 20 (1%) + Water | MeOH, AA 2% | LC/DAD | [63] |
| DON, ZON (+conjugated and metabolites) | Calf serum | DON Prep and DZT MS-Prep (R-Biopharm), NeoColumns for DON and for ZEN (Neogen), AokinImmunoClean C for DON and for ZEN (Aokin), Easi-extract ZEA (R-Biopharm) | Protein precipitation + drying; PBS, 5% MeOH | 10 mL | Water | MeOH | LC/MS–MS | [64] |
| DON, ZON, HT-2 andT-2 | Wheat, biscuit | DZT MS-PREP (R-Biopharm); MultiSep 226 (Romer Labs) | MeOH/water, 75/25; ×4, PBS, MeOH (15%) | 5 mL (eq. 0.25 g) | [65] | |||
| DON, ZON, NIV, FUS-X, 3-ADON, T-2 and HT-2 | Maize | Mycosep 226 and 227 (Coring systems Diagnostix) | ACN/water, 84/16; - | 8 mL (eq. 2 g) | - | - | LC/MS | [66] |
| DON, ZON, T-2 and HT-2 | Cereal and cereal-based samples | ACN/water, 85/15; - | 5 mL (eq. 1 g) | - | - | LC/MS–MS | [67] | |
| DON, ZON, T-2 and HT-2 | Wheat, Maize | DZT MS-PREP (R-Biopharm) | ACN/water 8/2; ×40, PBS | 8 mL | Water | MeOH | [68] | |
| Target Toxin(s) | Matrix | Extraction Solvent; Dilution Factor and Solvent | Sorbent, Amount of Abs | Grafting Yield or Density; Capacity | Extraction Mode | Vsample (eq. Sample Amount)/ Amount of Sorbent | Washing | Elution | Analysis | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| Toxins with MW < 1500 | ||||||||||
| Bacterial toxin | ||||||||||
| TTX | Marine organisms | MeOH, 1% AA; PBS (20% MeOH) | CNBr-Sepharose (0.5 g); mAbs (6 mg) | 1106 ng/mL; - | Off-line SPE | 25 mL (eq. 1 g)/0.5 g | Water | MeOH, AA (1%) | LC/MS–MS | [69] |
| Phycotoxins–Cyanotoxins | ||||||||||
| Anatoxin-a | Pure water | - | NHS-Sepharose beads (27 µm, 10 µL); mAbs (100 µg) | -; 20 ng | dSPE | 20 mL/ 10 µL | - | 2-propanol | IMS | [70] |
| MC-LR | Algae extracts | - | Poly(APTES-co-TEOS) monolith; pAbs | -; 0.38 pmol (2.1 µg/g sorbent) | On-line SPE | 150 nL/ 45 × 0.1 mm i.d. capillary | PBS | ACN/water (LC mobile phase) | Nano-LC/UV | [71] |
| Urine | - | streptavidin-magnetic beads; Biotin-Abs | dSPE | 100 µL | - | Water/ACN 7/3, FA (0.5%) | LC/MS–MS | [72] | ||
| Pure water | - | Sol-gel entrapment (TEOS) | -; 4.28 µg | Off-line SPE | 1 L (eq 2.5 g)/ 0.5 g | - | ACN/water 7/3 | ELISA, LC/MS | [73] | |
| MC-LR, MC-RR, MC-YR | Real waters | - | Glutaraldehyde-silica; purified pAbs | -; 1.8 µg/g IS | 20 mL (0.5% MeOH)/ 0.25 g | Water + water/MeOH 8/2 | MeOH/water 8/2 | ELISA, PP2A, LC/MS | [74] | |
| Cyanobacteria, real waters | MeOH/water, 75/25; x0.75, PBS | pAbs | - | 100 µL | PBS + water + MeOH/water 25/75 | MeOH | LC/DAD, CE (MECK) | [75] | ||
| MC-LR, MC-RR, MC-YR, MC-LA | Algae and fish extracts, real waters | -; <15% MeOH | CNBr-Sepharose and silica; - | - | 5–15 mL/ 0.1–0.2 g | PBS + water + MeOH/water 25/75 | MeOH/water 8/2, AA (4%) | LC/UV | [76] | |
| Real waters and blue green algae extracts | - | Sepharose CL-4B; pAbs (1 mg/mg sorbent) | -; 0.2 µg | 10 mL/ 2 mg | PBS + water + water/MeOH 85/15 | MeOH/water 80/20, AA (2%) | ELISA, LC/UV | [77] | ||
| MC-RR, MC-YR, MC-LR, MC-AR, MC-FR, MC-WR, MC-LA, MC-LF, MCYST-LW and other MC variants | Real waters | Concentrated, filtered | CNBr-Sepharose and silica; pAbs | -; 200 ng/IS (Sepharose); 135 ng/IS (silica) | - | Water/MeOH 75/25 | MeOH/water (+AA) 8/2 | LC/DAD, LC/MS | [78] | |
| Urine | - | Streptavidin-beads (2.5 µL); biotinylated Abs (0.5 µg) | - | dSPE | 500 µL/ 2.5 µL | - | Water/ACN 7/3, FA (0.5%) | PP2A (inhibition assay) | [79] | |
| Phycotoxins—Diarrheic shellfish poisoning (DSP) toxins | ||||||||||
| OA | Shellfish | MeOH, NaOH; dried extract in PBS | Protein G-magnetic beads; mAbs (1 mg/mg sorbent) | - | dSPE | 1 mL/ 1 mg | PBS | MeOH | LC/MS–MS | [80] |
| Algae extract | -; PBS/ACN 8/2 | Silica; pAbs | - | Off-line SPE | -/125 mg | MeOH/water 3/7 | MeOH-water 8/2 | LC/Fluo, LC/MS | [81] | |
| OA and derived form | Shellfish (hepatopancreas) | LLE; water/ACN 8/2 | Glutaraldehyde-silica; pAbs | -; 16 µg/g IS | 2 mL/ 125 mg | MeOH/water 7/3 | PBS/ACN 7/3 | [82] | ||
| OA, DTX-1 and DTX 2 | Shellfish | -; Anti-AO mAbs | - | - | - | - | LC/Fluo | [83] | ||
| Mycotoxins | ||||||||||
| AFs (B1, B2, G1, G2), OTA, ZON, SMC, T-2 | Feed samples | ACN/water/AA 80/18/2; x3, PBS | CNBr-Crystarose; 4 mAbs (5 mg each/g sorbent) | -; 0.13 µg/mg Abs (sum of toxin) | Off-line SPE | 10 mL (eq 0.6 g)/ 0.3 ml | PBS | MeOH | LC/MS–MS | [84] |
| AFs (B1, B2, G1, G2), OTA, ZON, T-2 | Peanuts, corn, wheat | ACN/water/AA 80/19/1; x3, PBS (≤ 20% ACN) | CNBr-Sepharose (1.3 g); mAbs (20 mg each) | -; 9 µg/mL IS (sum of toxin) | 10 mL/ 0.1 mL | [85] | ||||
| AF B1 | Cereals, peanuts, vegetable oils, Chinese traditional food | MeOH/water 6/4; x6, water (<10% MeOH) | CNBr-Sepharose (1 g, 3.5 mL); mAbs (9.92 g) | 99.8%; 260 ng/mL | 30 mL (eq. 1 g)/ 1 mL | Water | LC/MS | [86] | ||
| α- and β-Amanitins | Urine | Filtration; x1.8, PB | CNBr-Sepharose 2 mL; pAbs (6.4 mg) | 9 mL/ 2 mL | PB + water + acetone/water 95/5 | Acetone/ MeOH 1/1 | [87] | |||
| DON | Cereals | MeOH/water 8/2; x2, PBS (<10% MeOH) | CNBr-Sepharose (1 g, 3.5 mL); mAbs (30 mg) | 95%; 9.67 nmol/mL | 10 mL (eq. 0.5 g)/ 1 mL | Water + MeOH/water 1/9 | MeOH | LC/MS–MS | [88] | |
| DON, 3-ADON, 15-ADON, deepoxy-DON | Foods, feeds (aqueous extracts) | - | Abs entrapped in silica gel (TMOS); mAbs | -; 1 µg/mg immob. Abs | /1 g | 1% MeOH | ACN/water 4/6 | LC/UV | [89] | |
| FUMs (B1, B2) B3, OH-B1) | Dried feed samples | -; 100 µL for 10 mg, buffer | Protein A/PS-DVB (POROS); serum/mL sorbent | - | 100 µL/ 30 × 2.1 mm column | - | Water/MeOH 7/3, FA (2%) | LC/MS | [90] | |
| FUMs (B1, B2, B3) | Cereals | - | CNBr-Sepharose 4B (0.5 g); pAbs (1.27 mg/mL, 400 µL) | - | - | - | - | bioassay on cartridge | [91] | |
| OTA | Beer | Degassed; x2, PBS, 1% PEG 6000 | anti-IgG + CNBr-Sepharose (non-covalent bonding) | - | - | PBS, 0.05%Tween | OTA-HRP: competition | [92] | ||
| pure media | None | polyGMA-co-EGDMA monolith in a capillary; - | 260 ng Ab/cm; 1.2 pmol OTA/cm | In-line SPE | 10 µL/ 8.5 cm × 75 µm i.d | PBS+ borate buffer | MeOH | CE/LIF | [93] | |
| T-2, HT-2 | Maize, cherry | MeOH/water 6/4; x6, water, ≤ 10% MeOH | CNBr-Sepharose (1 g); mAbs (30 mg) | -; 3 µg/mL IS (for each toxin) | Off-line SPE | 30 mL (eq 0.5 g)/1 mL, 10 × 0.8 mm column | - | MeOH | LC/MS–MS | [94] |
| ZER + 3 analogs | Bovine muscle | MeOH; x5, PBS | CNBr-Sepharose 2 g; mAbs (50 mg) | 96.3%; 2.7 µg/mL gel | 25 mL (eq 2.5 g)/1 mL, 10 × 0.8 mm column | PBS + water + water/MeOH 7/3 | MeOH | GC/MS | [95] | |
| ZON, DON, T-2, HT-2 | Grain products | - | CNBr-Sepharose (0.2 g); DON Abs (1.25 mg), H-2/HT-2 Abs (0.2 mg), ZON Abs (0.3 mg) | 100%; 198–281 ng (for each compound) | - | Water or PBS | MeOH | LC/MS–MS | [96] | |
| Flour | - | Activated poly(GMA-co-DVB) µSpheres (0.3 g, 1 mL); DON Abs (1.25 mg), H-2/HT-2 Abs (0.2 mg), ZON Abs (0.3 mg) | -; 210–294 ng (for each compound) | /300 mg, 1 mL | - | - | [97] | |||
| ZON, T-2, HT-2 | Feed samples | ACN/H2O, 8/2; x3, PBS | Anti-IgG-Sepharose (0.5 g, 1.8 mL); mAbs (ZON) and pAbs (T-2) | - | /0.2 g | - | - | bioassay, LC/MS–MS | [98] | |
| Phycotoxins—Paralytic shellfish poisoning toxins | ||||||||||
| STX | Human urine | - | Protein G-magnetic beads (30 mg/mL); mAbs, (1 mg/mL) | 15 µg/mg (theory); - | dSPE | 100 µL/1.5 mg | PBS + water | ACN/water 1/1, FA (2.5%) | LC/MS–MS | [99] |
| STX, NEO | Shellfish | - | NH2-coated hollow glass magnetic µSpheres; mAbs | 5.8 mg/g; - | 1 mL/25–100 mg | PBS | Glycine/HCl buffer | LC/Fluo | [100] | |
| PSP toxins | Algae culture | PBS | 5.5 mg/g; - | [101] | ||||||
| Protein toxins | ||||||||||
| Abrin | Milk | - | Tosyl-activated magnetic beads (14.8 mg); mAbs against 4 epitopes (140 µg) | - | dSPE | 500 µL/0.2 mg | PBS + Tween 0.05% + PBS + water | Trypsin digestion | LC/HRMS | [102] |
| Androctonus australis Hector | Venom | CNBr-Sepharose 2 g; purified pAbs (0.15 µmole) | Off-line SPE | /20 x1 cm column, 2 g | Tris HCl, NaCl | FA (pH 2.5), NaCl | UV | [103] | ||
| BoNT type A | Crude culture supernatant, food, environmental samples | Protein G-magnetic beads (3 µm), pAbs (BoNT A) and mAbs (ricin) | dSPE | 500 µL/10–100 µL | HEPES | Trypsin digestion | LC/MS–MS | [104] | ||
| ETX | Milk, serum | [105] | ||||||||
| Ricin | Pure media (buffer + BSA) | 500µL/100 µL | Ammonium acetate (pH 4) | RNA incubation | LC/MS on adenine | [106] | ||||
| Milk | 500µL/5 µL | Buffer | 5% FA or 0.1% TFA in water | Tryptic digestion + MALDI-MS or/and LC/MS | [107] | |||||
| Milk, apple juice, human serum, saliva | 500µL/20 µL | PBS, Tween + water | ACN, TFA | [108] | ||||||
| Serum | Streptavidin-magnetic beads; biotinylated mAbs | 55 µg/mg; 16.5 µg/mg | 500 µL/20 µL | PBS + water | TFA 0.1% | [109] | ||||
| Ricin, SEB, BoTN A and B | Milk, orange and apple juices | M-280 tosyl- paramagnetic beads (250 µL); mAbs | - | 200 µL/8 µL | [110] | |||||
| Ricin, SEB, ETX | Milk, human urine, plasma | 1 mL/20 µL | PBS | Trypsin digestion | LC/HRMS (Q orbitrap) | [111] | ||||
| Shigatoxin (protein) + analogs | Cell culture | CH-Sepharose 4B (2 g); purified pAbs (4 mg) | Off-line SPE | /2 g | Tris HCl + NaCl | Glycine (pH 2.7), NaCl 0.5 M | SDS Page | [112] | ||
| Staphilococcal enterotoxins A and E (proteins) | Dialyzed cell culture supernatant | Affigel 10 (agarose) 1 mL; mAbs (5 mg) | 25 mL/1 mL | PB | AA, NaCl | UV | [113] | |||
| Target Analyte | Samples | Extraction Solvent; Dilution Factor, Solvent | MIP Synthesis: Monomer(s)/CL/Solvent; Polymerization Mode | Extraction Mode | Vsample/ MIP Amount | Washing | Elution | Analytical method | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| AFs (B1, B2, G1) | Maize | MeOH/water 7/3, NaCl; x3, PBS | MAA/EGDMA/EtOH SP on nanoporous carbon core | dSPE | -/ 80 mg | Water | ACN/water | LC/MS–MS | [116] |
| AFs (B1, B2, G1, G2) | Cereals | ACN/water 84/16; x12.5, PBS, Tween 20 | MAA/EGDMA/MeOH; PP on mesoporous silica surface | Off-line SPE | 50 mL/400 mg | MeOH | LC/Fluo | [117] | |
| AFs (B1, B2, G1, G2, M1) | Fish, mussel liver | ACN/PB 6/4; x2, PBS | MAA/DVB/ACN; PP | dSPE | 25 mL/40 mg | - | ACN/FA 2.5% | LC/MS–MS | [118] |
| Altenariol, altenariol monomethyl ether | Tomato juice, sesame oil | Water/ACN/salt; dry extract dil. in PB, 1% MeOH | 4-VP, MA/EGDMA/ACN; SP on silica microspheres | Off-line SPE | 25 mL/25 mg | ACN/water 5/95, ACN/ water 15/85 | MeOH/TFA 99/1 | LC/MS–MS | [119] |
| α-, β-amanitins | Human plasma | - | MAA,4-VP/EGDMA/DMSO; SP on vinylated silica microsphere | Off-line SPE/ dSPE | 1 mL/1.3 g (SPE), 4 mL/20 mg (dSPE) | NaCl, PBS | MeOH | LC/UV | [120] |
| BMAA | Cyanobacteria | TCA; SPE (mixed-mode); ACN, 1% FA | APTES/TEOS/EtOH-water-HCl; BP | Off-line SPE | 3 mL/25 mg | ACN/MeOH/ water 80/18/2 | LC/MS–MS | [121] | |
| Citrinine | Maize | MeOH/water 7/3 | DMAEDM/TRIM/Acetone-ACN; BP | 1 mL/300 mg | Water | MeOH/AA 98/2 | LC/Fluo | [122] | |
| Rice | MeOH/water 7/3; dil. HEPES | 4-VPU, MA/EGDMA/PVP-EtOH-water; SP on mNPs | dSPE | 5 mL/ 200 mg | ACN/water 5/95 | MeOH, TBA 50 mM | LC/UV | [123] | |
| Cereals, food supplement | MeOH; x2, water | AM/EGDMA/ACN; BP | SPE (on-line, 20 × 3 mm i.d.) | 50 µL/25 mg | Water/MeOH 75/25, AA 0.5% | Water/ACN 7/3, 0.5% AA (LC mobile phase) | LC/Fluo | [124] | |
| DA | Blue mussels | MeOH/water | 4-VP/EGDMA/toluene; SP on polystyrene beads (5 µm) | LC (150 × 4.6 mm i.d.) | 20 µL | - | ACN/water, 0.05%AA 7/3 | UV | [125] |
| Mussels | 4-VP/EGDMA/toluene; BP | Off-line SPE | 4 mL/150 mg | Water, ACN | MeOH, 5% AA | LC/UV, LC/HRMS | [126] | ||
| Clams | ACN+ ACN/water 1/1; x2, water, 0.2% AA | 4-VP/EGDMA/non-ionic surfactant; emulsion polymerization | 1 mL/50 mg (200 nm) | - | ACN/citric acid 4/1 | LC/UV | [127] | ||
| Sea water and shellfish | MeOH/water 1/1; x1.66, HCl 0.1 M | TFMA/EGDMA/ACN; PP | 500 mL (pH 1), 25 mL (shellfish extract)/100 mg | Water, FA 0.4% | MeOH, 1% FA | LC/UV | [128] | ||
| DON | Pasta | Water/EDTA | IA/EGDMA/DMF, BP | 1 mL/100 mg | PBS | MeOH | LC/UV | [129] | |
| DON, 3-ADON, 15-ADON, T-2, HT-2, FUS-X | Rice | MeOH/water, 7/3 | MAA/DVB/ACN; SP on mNPs | dSPE | 10 mL/30 mg (0,5 µm) | - | MeOH, 2% NaOH | LC/MS–MS | [130] |
| GTX 1,4 | Sea water | - | MAA/EGDMA/DMSO; BP | Off-line SPE | 1–50 mL/50 mg | AA 0.1 M | MeOH/water 95/5 | LC/Fluo | [131,132] |
| Microalgal culture | Water | MAA/EGDMA/CHCl3-PVA; suspension polymerization | 1 mL/100 mg | MeOH/water 95/5, water | AA 0.1 M | [133] | |||
| GTX 2,3 | AA; water | 1 mL/200 mg | MeOH/water 98/2 | [134] | |||||
| Sea water | - | LC/HRMS | [135] | ||||||
| MC-LR | Tap water, lake waters | x1.2, buffer | AMPSA, UAEE/EGDMA/DMSO; BP | Off-line SPE | 3, 20 or 100 mL/10 or 30 mg | - | MeOH | ELISA | [136] |
| - | Dopamine HCl/Tris; SP on magnetic GO | - | MeOH, AA | LC/UV | [137] | ||||
| - | MAA/EGDMA/toluene | MeOH/ water 1/9 | MeOH, 5% AA | Bioassay | [138] | ||||
| OTA | Red wine | C18 silica; MeOH extract | Acrylic monomers/EGDMA/CHCl3; BP | Off-line SPE | 3 mL/100 mg | MeOH | MeOH, 2% AA | LC/UV | [139] |
| Wine | Acidification (pH 1) | Pyrrole/EDMA/ACN; electropolymerization on stainless-steel frits | On-line SPE | 100 µL | Water | MeOH, 1% TEA (pulse elution) | LC/Fluo | [140,141] | |
| Wheat | MeOH/NaHCO3 3/7; x1.3, PBS, Tween 20 | MAA/EGDMA/CHCl3; BP | On-line SPE (50 × 6.6 mm i.d.) | 6 mL/45 mg | - | MeOH, 1%TBA | [142] | ||
| Coffee, grape juice, urine | Water, 1% NaHCO3 (Coffee); x2 water (urine), pH 1.5 | AFFINIMIP® SPE Ochratoxin (Polyintel, AffiniSep) | dSPE (PP envelope) | 10 mL/15 mg | Water | MeOH, 2%AA | [143] | ||
| Beer, red wine, grape juice | -; x2, acidified water (pH 1) | Off-line SPE | 20 mL | HCl 0.1 M/ACN 6/4 | LC/Fluo; LC–MS/MS | [144] | |||
| Wheat | ACN/water 6/4; x2, HCl 0.1 M | 4 mL/50–100 mg | LC/Fluo | [145] | |||||
| Wine | Precipitation with PEG 8000 | MAA/EGDMA/CHCl3; BP | 2 mL/250 mg | Water /ACN 4/1 | ACN, 2% AA | [146] | |||
| OTA, OTB, OTC | Rice, wine | ACN/water, 6/4 (Rice); dil. NaCl and NaHCO3 (wine) | Dopamine HCl; SP on mNPs | dSPE | 50 mL, pH 3/15 mg | ACN | [147] | ||
| Patulin | Apple juice | -; x2, water, 0.2% AA | AM/EGDMA/ACN; SP on silica beads | Off-line SPE | 2.5 mL/180 mg | Water, diethylether | Water, 1% AA | LC/UV | [148] |
| MAL/EGDMA/ACN; SP on a silica-gel | 1 mL/50 mg | NaHCO3, AA | ACN | [149] | |||||
| -; x2, water, 2% AA | Supel-MIP ® SPE Patulin, EASIMIP TM Patulin, AFFINIMIP® SPE Patulin | SPE (off-line, on-line) | 4 mL (off line); 50 µL (on-line)/70–80 mg | Off-line: NaHCO3, water, drying, diethylether, drying; on-line: NaHCO3 | Off-line: ethyl acetate; on-line: LC mobile phase (BF) | [150] | |||
| Juices | LLE; dried extract dil. in ACN/acetate buffer (pH 4) | APTES/TEOS/MeOH-water; SP on activated silica beads (125–180 µm) | On-line SPE (15 × 4 mm i.d.) | 50 mL/50 mg | - | LC mobile phase (BF) | [151] | ||
| Juices, fruits | ACN, MgSO4, NaCl; dry extract dil. in water | MAA/TRIM/MeOH; PP | Off-line SPE | 1 mL/30 mg | Water | MeOH | LC/MS–MS | [152] | |
| Pyrrolizidine alkaloids | Herbal plants | DCM/MeOH, NaOH; dry extract dil. in water, 0.1% FA | Allylsulfonate/EGDMA/ACN; SP on silica fiber | SPME fiber | 0.3 mL | MeOH, 0.1% FA | MeOH, NaOH | LC/MS | [153] |
| T-2 toxin | Maize, barley, oat | ACN/water 84/16 (+ LLE for oat); dry extract dil. in MeOH/water 2/8 | MA/EGDMA/CHCl3; BP | Off-line SPE | 1 mL/50 mg | MeOH/ water (6/4 -maize- or 2/8 – barley, oat-) | MeOH/AA 95/5 | [154] | |
| ZON | Beer | - | AFFINIMIP® SPE Zearalenone (AffiniSep) | On-line SPE | 50 µL | ACN/water 1/9 + AA 2% | LC mobile phase (ACN/water 35/65) | LC/Fluo | [155] |
| Cereals | ACN/water 8/2, dil. water 0.2% FA | 4-VP/EGDMA/dibutyl phtalate; SP on mNPs | dSPE | 10 mL, ACN 2%/100 mg | Water | MeOH x3, ACN x2 | LC/MS–MS | [156] | |
| Water extract | MAA/EGDMA/EtOH; PP with MOF | Off-line SPE | 10 mL/100 mg | ACN/ water 9/1 | LC/Fluo | [157] | |||
| Seed-strain | Extract with 60% ACN | 1-ALPP/TRIM/ACN; PP | 1 mL/100 mg | ACN/water 7/3 | MeOH/AA 95/5 | LC/MS–MS | [158] | ||
| ZON, alpha ZOL | Cereals, swine feed | Dried extracts dil. ACN | 1-ALPP/TRIM/ACN; BP | 5 mL/450 mg | Water | MeOH/phosphoric ac. 95/5 | LC/Fluo | [159] |
| Aptamer Modification/Spacer | Sorbent | Matrix | Extraction Solvent; Dilution Factor and Solvent | Extraction Mode | Vsample/Amount of OS | Washing | Elution | Analytical Method | Ref. | |
|---|---|---|---|---|---|---|---|---|---|---|
| AF B1 | biotin/- | Streptavidin-coated microtiter plate | Corn | Dried extract in BB | Partition | 500 µL | - | MeOH/water 8/2 | LC/Fluo | [170] |
| Magnetic streptavidin-NPs | Peanut oil | dSPE | - | - | - | Fluo | [171] | |||
| AF B2 | - | Magnetic NH2-NPs | [172] | |||||||
| NH2/C7 | CNBr-Sepharose | Peanut | MeOH/water 7/3, NaCl; ×10, BB | Off-line SPE | 5 mL/60 mg | BB | MeOH | LC/Fluo | [173] | |
| AFs B1, B2 | NHS-activated Sepharose | Peanut, maize, wheat, rice | MeOH/water 7/3, NaCl; ×9, BB | 30 mL/300 µl | [174] | |||||
| Biotin-Apt | Magnetic streptavidin-agarose beads (28 µm) | Maize | MeOH/water 7/3, NaCl; dry extract in BB | dSPE | 1 mL/200 µL | BB/MeOH 2/8 | [175] | |||
| AF M1 | - | Magnetic silanized NPs | Milk | LLE (hexane, MeOH, water); - | 15 mL/8 mg | MeOH | DCM, AA | [176] | ||
| OTA | - | DADPA | Wheat extract | MeOH/water 6/4; x4, BB | Off-line SPE | 10–12 mL/300 µL | BB | Tris-HCl, EDTA, 20% MeOH | Fluo | [169] |
| MeOH/BB (2/8) | LC/Fluo | [177] | ||||||||
| NH2/C6 | CNBr-Sepharose | Red wine | pH adjusted | 1 mL/35 mg | BB/ACN 9/1 | Water/ACN (6/4) | LC/Fluo | [178] | ||
| NH2/C12 | Wheat extract | ACN/water 6/4; ×10, BB | Water/ACN (6/4) | LC/Fluo | [179] | |||||
| NH2/C6 | NHS-activated Sepharose | Ginger powder, TCM | ACN/water 6/4; ×10, BB | 2, 3 mL/200 µL | BB | MeOH | LC/Fluo LC/MS–MS | [180] [181] | ||
| SH/C6 | Au NPs/POSS-PEI monolith | Pure sample | - | 20 µL/100 × 0.1 mm i.d. capillary | Tris-HCl, EDTA | LC/Fluo | [182] | |||
| NH2/C6 | Magnetic carboxylated-NPs (100–250 nm) | Food | MeOH/water 5/5; ×2, Tris-HCl, Mg2+ | dSPE | 100 µL/100 µL | Tris-HCl | ACN/water (95/5) + 1% AA | LC/Fluo | [183] | |
| Biotin-Apt | Magnetic streptavidin-MOF | Corn, peanut | dry extract in HEPES, Mg2+ | -/10 mg | - | - | LC/MS–MS | [184] | ||
| SH/- | POSS-acrylate-based monolith (one-pot synthesis) | Beer | pH adjusted; ×2, BB | On-line SPE (loop) | 100 µL/100 × 0.1 mm i.d. capillary | BB | ACN/Tris-HCl, EDTA (3/7) | LC/Fluo | [185] | |
| SH/C6 | Poly(TMOS-co-MPTMS) monolith modified with AuNPs (25 nm) | Beer, wine | pH adjusted; ×9, BB | 20 µL/100 × 0.075 mm i.d. capillary | ACN/Tris-HCl, EDTA (7/3) | LC/Fluo | [186] | |||
| NH2/ C12 | Poly(APTES-co-TEOS) monolith | Beer | ×2, BB | On-line SPE | 250 nL/ 70 × 0.1 mm i.d. capillary | nano-LC mobile phase | NanoLC/LIF | [187] | ||
| SH/- | Poly(TMOS-co-MTMS) monolith | Beer and white wine | In-line SPE | 768 nL (beer), 5 µL (wine)/ 15 × 0.075 mm i. d. capillary | ACN/Tris, EDTA, 3/7 | CE/LIF | [188] | |||
| ZON | Biotin-Apt | Magnetic streptavidin beads | Beer | dSPE | - | - | 90 °C, BB | Fluo | [189] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delaunay, N.; Combès, A.; Pichon, V. Immunoaffinity Extraction and Alternative Approaches for the Analysis of Toxins in Environmental, Food or Biological Matrices. Toxins 2020, 12, 795. https://doi.org/10.3390/toxins12120795
Delaunay N, Combès A, Pichon V. Immunoaffinity Extraction and Alternative Approaches for the Analysis of Toxins in Environmental, Food or Biological Matrices. Toxins. 2020; 12(12):795. https://doi.org/10.3390/toxins12120795
Chicago/Turabian StyleDelaunay, Nathalie, Audrey Combès, and Valérie Pichon. 2020. "Immunoaffinity Extraction and Alternative Approaches for the Analysis of Toxins in Environmental, Food or Biological Matrices" Toxins 12, no. 12: 795. https://doi.org/10.3390/toxins12120795
APA StyleDelaunay, N., Combès, A., & Pichon, V. (2020). Immunoaffinity Extraction and Alternative Approaches for the Analysis of Toxins in Environmental, Food or Biological Matrices. Toxins, 12(12), 795. https://doi.org/10.3390/toxins12120795