4. Materials and Methods
Air- and moisture-sensitive reactions were performed under an atmosphere of argon in flame-dried apparatus. Tetrahydrofuran (THF), toluene, dichloromethane and diethyl ether were purified using a Pure-SolvTM 500 Solvent Purification System. Other dry solvents and starting materials were obtained from commercial sources and used as received unless stated otherwise. Petroleum ether (pet. ether) used for column chromatography was the 40–60 °C fraction. Triethylamine and 2,2,6,6-tetramethylpiperidine were distilled and stored under argon prior to use. n-Butyllithium solutions were titrated against diphenylacetic acid to obtain accurate molarity. 4 Å molecular sieves were oven dried prior to use.
Reactions were monitored by thin layer chromatography (TLC) using Merck silica gel 60 covered aluminium-backed plates F254. TLC plates were visualised under UV light and stained using potassium permanganate solution or acidic ethanolic anisaldehyde solution. Flash column chromatography was performed with silica gel (Geduran Si 60 35–70 µm) as solid support.
IR spectra were recorded using a Shimadzu FT IR-8400S ATR instrument (Shimadzu UK, Milton Keynes, UK). The IR spectrum of each compound (solid or liquid) was acquired directly on a thin layer at ambient temperature.
NMR spectra were recorded on Bruker Avance III 400 MHz and 500 MHz spectrometers (Bruker UK, Coventry, UK) at ambient temperature. 1H NMR chemical shifts are reported in ppm relative to CHCl3 (7.26) or CDCl2H (5.32) on the δ scale followed by integration, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br. = broad, app. = apparent or a combination of these) and coupling constant(s) J (Hz). 13C NMR spectra were recorded at 101 MHz and 126 MHz and chemical shifts are reported in ppm relative to CDCl3 (77.16) or CD2Cl2 (54.00) on the δ scale.
High resolution mass spectra (HRMS) were recorded by the University of Glasgow mass spectrometry service using a JEOL MStation JMS-700 instrument [positive electron impact ionisation (EI+)] (JEOL Ltd., Tokyo, Japan) or a Bruker micrOTOF-Q instrument [positive ion electrospray (ESI+)] (Bruker UK, Coventry, UK).
Optical rotations were recorded with an error of ≤ ±0.1 using an automatic polarimeter Autopol V (Rudolph Research Analytical, Hackettstown, USA) and melting points were recorded with an Electrothermal IA 9100 apparatus (Cole-Parmer, Stone, UK).
(1′S,3′R,9′S,11′R,15′Z)-4′-Methyl-2′,8′,12′-trioxaspiro(1,3-dioxolane-2,14′-tricyclo-[9.6.0.03,9]heptadecane)-4′,15′-dien-6′-one (7)
To a solution of the bis-enone 1 (870 mg, 3.13 mmol) in dichloromethane (63 mL) at −60 °C was added, 1,2-bis(trimethylsiloxy)ethane (0.81 mL, 3.3 mmol) and trimethylsilyl trifluoromethanesulfonate (0.06 mL, 0.3 mmol). The solution was allowed to warm to −25 °C and stirred for 16 h. The reaction mixture was diluted with diethyl ether (252 mL) and the mixture was washed with sat. aq. ammonium chloride (2 × 100 mL) and brine (100 mL). The organic phase was dried (MgSO4) and concentrated under reduced pressure to afford the acetal 7 (960 mg) as a brown oil. The unpurified acetal was used directly in the next reaction. For analytical purposes, a sample was purified by flash column chromatography (5–30% ethyl acetate in pet. ether) to afford 7 as a colourless solid. M.p. 145–146 °C; Rf = 0.43 (60% ethyl acetate in pet. ether); [α]D +22.6 (c = 0.50 in CHCl3, 29 °C); νmax 3028, 2953, 2932, 2884, 1655, 1443, 1348, 1115, 1086, 1074, 943, 893, 768 cm−1; 1H NMR (500 MHz, CDCl3) δ 5.90 (1H, dq, J = 1.2, 1.2 Hz), 5.82 (1H, ddd, J = 11.6, 9.5, 7.5 Hz), 5.63 (1H, d, J = 11.6 Hz), 4.23 (1H, d, J = 18.3 Hz), 4.13 (1H, d, J = 18.3 Hz), 3.94–3.83 (4H, m), 3.67 (1H, ddq, J = 9.0, 1.2, 1.2 Hz), 3.61 (1H, d, J = 12.7 Hz), 3.50 (1H, d, J = 12.7 Hz), 3.47 (1H, ddd, J = 11.7, 9.3, 4.4 Hz), 3.33 (1H, ddd, J = 9.3, 5.8, 4.0 Hz), 3.30 (1H, ddd, J = 11.7, 9.0, 4.7 Hz), 2.89 (1H, ddd, J = 13.5, 9.5, 4.0 Hz), 2.56 (1H, ddd, J = 13.5, 7.5, 5.8 Hz), 2.36 (1H, ddd, J = 12.1, 4.7, 4.4 Hz), 1.97 (3H, dd, J = 1.2, 1.2 Hz), 1.61 (1H, ddd, J = 12.1, 11.7, 11.7 Hz); 13C NMR (101 MHz, CDCl3) δ 201.4, 155.1, 133.9, 130.3, 127.1, 108.0, 82.8, 82.5, 78.1, 77.6, 77.4, 74.2, 64.8, 64.6, 38.3, 30.1, 22.9; HRMS (ESI+) [C17H22O6Na]+ found 345.1299, [M + Na]+ calcld. 345.1309.
(1′S,3′R,9′S,11′R,15′Z)-4′-Methyl-2′,8′,12′-trioxaspiro(1,3-dioxolane-2,14′-tricyclo-[9.6.0.03,9]heptadecane)-4′,6′,15′-trien-6′-yl prop-2-en-1-yl carbonate (8)
To a solution of the acetal 7 in THF (57 mL) was added, allyl chloroformate (1.58 mL, 14.9 mmol). The resultant solution was cooled to −78 °C and sodium bis(trimethylsilyl)amide (3.0 mL of a 2.0 M solution in THF, 6.0 mmol) was added dropwise. The reaction mixture was stirred for 4 h, diluted with 5% aq. potassium dihydrogen phosphate (240 mL) and the mixture was extracted with diethyl ether (3 × 60 mL). The combined organic extracts were washed with brine (3 × 60 mL), dried (MgSO4) and concentrated under reduced pressure. The residue was dissolved in toluene (100 mL) and then concentrated under reduced pressure and the procedure was repeated. Residual material was purified by flash column chromatography (5–40% diethyl ether in pet. ether) to give the allylic carbonate 8 (610 mg, 48% over 2 steps) as a colourless oil. Rf = 0.57 (60% ethyl acetate in pet. ether); [α]D +96.1 (c = 1.00 in C6H6, 29 °C); νmax 2951, 2891, 1759, 1663, 1451, 1364, 1246, 1225, 1198, 1126, 1090, 1055, 995, 947, 770 cm−1; 1H NMR (500 MHz, CD2Cl2) δ 6.62 (1H, d, J = 1.6 Hz), 5.96 (1H, ddt, J = 17.3, 10.5, 5.7 Hz), 5.86 (1H, ddd, J = 11.5, 9.6, 7.5 Hz), 5.67 (1H, d, J = 11.5 Hz), 5.54 (1H, dqd, J = 1.6, 1.6, 1.3 Hz), 5.37 (1H, ddt, J = 17.3, 1.4, 1.4 Hz), 5.29 (1H, dddd, J = 10.5, 1.4, 1.4, 1.4 Hz), 4.64 (2H, ddd, J = 5.7, 1.4, 1.4 Hz), 3.95–3.87 (4H, m), 3.74 (1H, ddq, J = 7.9, 1.3, 1.3 Hz), 3.65 (1H, ddd, J = 11.4, 7.9, 4.9 Hz), 3.63 (1H, d, J = 12.8 Hz), 3.54 (1H, d, J = 12.8 Hz), 3.52 (1H, ddd, J = 11.8, 9.4, 4.4 Hz), 3.32 (1H, ddd, J = 9.4, 5.7, 4.1 Hz), 2.92 (1H, ddd, J = 13.4, 9.6, 4.1 Hz), 2.56 (1H, ddd, J = 13.4, 7.5, 5.7 Hz), 2.49 (1H, ddd, J = 12.1, 4.9, 4.4 Hz), 1.89 (3H, dd, J = 1.6, 1.3 Hz), 1.67 (1H, ddd, J = 12.1, 11.8, 11.4 Hz); 13C NMR (126 MHz, CD2Cl2) δ 155.0, 141.2, 138.5, 135.0, 134.2, 132.0, 130.7, 119.3, 118.8, 108.4, 81.8, 80.1, 78.0, 77.9, 74.6, 69.5, 65.1, 65.0, 38.7, 30.4, 21.9; HRMS (ESI+) [C21H26O8Na]+ found 429.1507, [M + Na]+ calcld. 529.1520.
(1′S,3′R,7′R,9′S,11′R,15′Z)-4′-Methyl-7′-(prop-2-en-1-yl)-2′,8′,12′-trioxaspiro(1,3-dioxolane-2,14′-tricyclo[9.6.0.03,9]heptadecane)-4′,15′-dien-6′-one (10)
A mixture of tetrakis(triphenylphosphine)palladium(0) (67 mg, 0.058 mmol, 5 mol%) and (4R)-t-butyl-2-[2-(diphenylphosphino)phenyl]-4,5-dihydroxazole (9) (56 mg, 0.14 mmol, 13 mol%) was dissolved in THF (20 mL). The solution was stirred for 45 min and a solution of the allylic carbonate 8 (471 mg, 1.16 mmol) in THF (38 mL) was added. The mixture was stirred at rt for a further 3.5 h and then adsorbed onto Celite. The crude material was dry loaded on to a column of silica gel and purified by flash column chromatography (5–20% ethyl acetate in pet. ether) to afford the enone 10 (398 mg, 95%, >20:1 dr) as a pale yellow oil. Rf = 0.57 (60% ethyl acetate in pet. ether); [α]D +15.0 (c = 0.50 in CHCl3, 30 °C); νmax 2949, 2888, 2855, 1657, 1443, 1279, 1111, 1088, 999, 949, 918 cm−1; 1H NMR (500 MHz, CDCl3) δ 5.89 (1H, dq, J = 1.1, 1.1 Hz), 5.87 (1H, ddd, J = 11.5, 9.6, 7.5 Hz), 5.78 (1H, dddd, J = 17.0, 10.2, 6.8, 6.8 Hz), 5.70 (1H, d, J = 11.5 Hz), 5.12–5.00 (2H, m), 4.17 (1H, dd, J = 7.7, 4.4 Hz), 3.98–3.89 (4H, m), 3.68 (1H, ddq, J = 8.9, 1.1, 1.1 Hz), 3.67 (1H, d, J = 12.7 Hz), 3.55 (1H, d, J = 12.7 Hz), 3.50 (1H, ddd, J = 11.7, 9.3, 4.4 Hz), 3.40 (1H, ddd, J = 9.3, 5.5, 4.1 Hz), 3.33 (1H, ddd, J = 11.3, 8.9, 4.7 Hz), 2.97 (1H, ddd, J = 13.6, 9.6, 4.1 Hz), 2.59 (1H, ddd, J = 13.6, 7.5, 5.5 Hz), 2.49 (1H, ddd, J = 14.3, 6.8, 4.4 Hz), 2.41 (1H, ddd, J = 12.2, 4.7, 4.4 Hz), 2.36 (1H, ddd, J = 14.3, 7.7, 6.8 Hz), 2.01 (3H, dd, J = 1.1, 1.1 Hz), 1.70 (1H, ddd, J = 12.2, 11.7, 11.3 Hz); 13C NMR (126 MHz, CDCl3) δ 203.6, 153.7, 134.0, 133.5, 130.4, 126.7, 117.7, 107.9, 86.5, 82.5, 82.5, 77.6, 77.0, 74.2, 64.8, 64.6, 38.2, 38.1, 30.1, 22.7; HRMS (ESI+) [C20H26O6Na]+ found 385.1606, [M + Na]+ calcld. 385.1622.
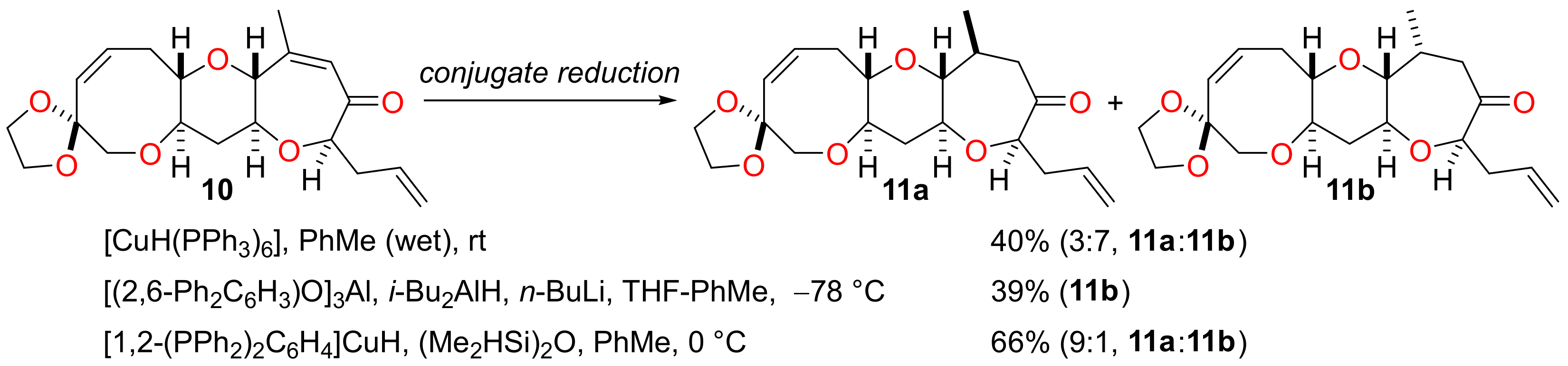
(1′S,3′R,4′S,7′R,9′S,11′R,15′Z)-4′-Methyl-7′-(prop-2-en-1-yl)-2′,8′,12′-trioxaspiro(1,3-dioxolane-2,14′-tricyclo[9.6.0.03,⁹]heptadecan)-15′-en-6′-one (11a) and (1′S,3′R,4′R,7′R,9′S,11′R,15′Z)-4′-Methyl-7′-(prop-2-en-1-yl)-2′,8′,12′-trioxaspiro(1,3-dioxolane-2,14′-tricyclo[9.6.0.03,9]heptadecan)-15′-en-6′-one (11b)
- (a)
Conjugate reduction of10with of ‘hot’ Stryker’s reagent
To 1,2-bis(diphenylphosphino)benzene (491 mg, 1.10 mmol) and copper(II) acetate hydrate (220 mg, 1.10 mmol) was added degassed toluene (19 mL). The resultant mixture was stirred vigorously and sparged continuously with argon for 15 min. The mixture was then sonicated for a further 30 min, at which point a pale blue suspension had formed. Tetramethyldisiloxane (0.80 mL, 4.4 mmol) was added to the mixture and it was stirred vigorously for a further 30 min, at which point the suspension developed a green colour. After refrigeration for 16 h, a red stock solution (0.055 M) had formed.
To the enone 10 (18 mg, 50 µmol) was added ‘hot’ Stryker’s reagent (2.0 mL of a 0.055 M solution in toluene, 0.11 mmol). The solution was stirred at 0 °C for 48 h and then concentrated under reduced pressure. The residue was dissolved in THF (5 mL) and sat. aq. potassium fluoride (2 mL) was added. The biphasic mixture was stirred vigorously for 16 h and then diluted with water (10 mL). The mixture was extracted with diethyl ether (3 × 5 mL) and the combined organic extracts were dried (MgSO4) and concentrated under reduced pressure. The residue was purified by flash column chromatography (10–20% ethyl acetate in pet. ether) to afford a diastereomeric mixture of the ketones 11a and 11b (12 mg, 66%, 9:1 a:b) as a colourless film.
- (b)
Conjugate reduction of10with lithium n-butyl(diisobutyl)aluminium hydride
To a solution of 2,6-diphenylphenol (380 mg, 1.50 mmol) in toluene (2 mL) at 0 °C was added dropwise trimethylaluminium (0.25 mL of a 2.0 M solution in toluene, 0.50 mmol) and the mixture was stirred for 10 min. After gas evolution had subsided, a solution of the enone 10 (145 mg, 0.400 mmol) in toluene (1 mL) was added and the resultant solution cooled to −78 °C and stirred for 10 min.
To a solution of diisobutylaluminium hydride (0.60 mL of a 1.0 M solution in heptane, 0.60 mmol) in THF (2 mL) at 0 °C was added dropwise n-butyllithium (0.26 mL of a 2.33 M solution in hexane, 0.60 mmol) and solution was stirred for 10 min. The solution was added to the aforementioned toluene solution of the enone 10 and aluminium tris(2,6-diphenylphenoxide) at −78 °C and the mixture was stirred for 2 h at this temperature. The mixture was then diluted with diethyl ether (50 mL) and sat. aq. Rochelle salt (50 mL) was added and the mixture was stirred vigorously for 16 h. The phases were separated and the organic phase was washed with water (25 mL) and brine (25 mL), then dried (MgSO4) and concentrated under reduced pressure. The residue was purified by flash column chromatography (2.5–20% ethyl acetate in pentane) to afford the ketone 11b (57 mg, 39%) as a colourless solid and the diketone (17 mg, 12%), resulting from deacetalation of the ketone 11b, as a colourless film. 11a Rf = 0.20 (20% ethyl acetate in pet. ether); [α]D +117.5 (c = 1.00 in CHCl3, 27 °C); νmax 2080, 3028, 2959, 2934, 2916, 2876, 1713, 1643, 1260, 1088, 1059, 949, 912, 891, 802, 769 cm−1; 1H NMR (500 MHz, CDCl3) δ 5.88 (1H, ddd, J = 11.6, 9.5, 7.5 Hz), 5.79 (1H, dddd, J = 17.1, 10.3, 6.9, 6.9 Hz), 5.67 (1H, d, J = 11.6 Hz), 5.11–5.04 (2H, m), 3.98–3.89 (4H, m), 3.80 (1H, dd, J = 7.9, 5.0 Hz), 3.66 (1H, d, J = 12.7 Hz), 3.54 (1H, d, J = 12.7 Hz), 3.46 (1H, ddd, J = 11.6, 9.3, 4.4 Hz), 3.28 (1H, ddd, J = 9.3, 5.8, 4.1 Hz), 2.96 (1H, ddd, J = 11.1, 9.3, 4.6 Hz), 2.89 (1H, ddd, J = 13.4, 9.5, 4.1 Hz), 2.88–2.78 (2H, m), 2.57 (1H, ddd, J = 13.4, 7.5, 5.8 Hz), 2.41–2.25 (3H, m), 2.12 (1H, dd, J = 11.5, 1.9 Hz), 1.69–1.57 (1H, m), 1.63 (1H, ddd, J = 12.4, 11.6, 11.1 Hz), 1.13 (3H, d, J = 6.6 Hz); 13C NMR (126 MHz, CDCl3) δ 215.2, 133.6, 133.1, 130.7, 118.0, 108.0, 86.5, 85.7, 81.6, 79.8, 77.9, 74.2, 64.8, 64.5, 45.1, 38.8, 37.2, 35.7, 30.1, 20.3; HRMS (ESI+) [C20H28O7Na]+ found 387.1766, [M + Na]+ calcld. 387.1778. 11b M.p. 108–110 °C; Rf = 0.25 (20% ethyl acetate in pet. ether); [α]D +87.1 (c = 2.00 in CHCl3, 29 °C); νmax 2957, 2936, 2884, 2855, 1711, 1641, 1288, 1090, 1061, 1042, 995, 949, 920, 770 cm−1; 1H NMR (500 MHz, CDCl3) δ 5.87 (1H, ddd, J = 11.5, 9.6, 7.4 Hz), 5.80 (1H, dddd, J = 17.1, 10.2, 6.9, 6.9 Hz), 5.70 (1H, d, J = 11.5 Hz), 5.11–5.02 (2H, m), 3.98–3.90 (4H, m), 3.71 (1H, dd, J = 8.3, 4.2 Hz), 3.67 (1H, d, J = 12.7 Hz), 3.54 (1H, d, J = 12.7 Hz), 3.41–3.34 (3H, m), 3.20 (1H, ddd, J = 11.1, 9.6, 5.0 Hz), 3.03–2.94 (1H, m), 3.00 (1H, dd, J = 11.7, 2.2 Hz), 2.50 (1H, ddd, J = 13.5, 7.4, 4.4 Hz), 2.42 (1H, ddddd, J = 13.4, 6.9, 4.2, 1.3, 1.3 Hz), 2.37 (1H, ddd, J = 12.1, 5.0, 4.2 Hz), 2.34–2.25 (3H, m), 1.65 (1H, ddd, J = 12.1, 11.3, 11.1 Hz), 0.86 (3H, d, J = 7.0 Hz); 13C NMR (126 MHz, CDCl3) δ 215.5, 133.9, 133.6, 130.9, 117.8, 107.8, 87.1, 83.1, 81.7, 77.7, 74.9, 74.4, 64.8, 64.6, 44.3, 38.5, 37.5, 31.9, 30.1, 12.3; HRMS (ESI+) [C20H28O6Na]+ found 387.1765, [M + Na]+ calcld. 387.1778.
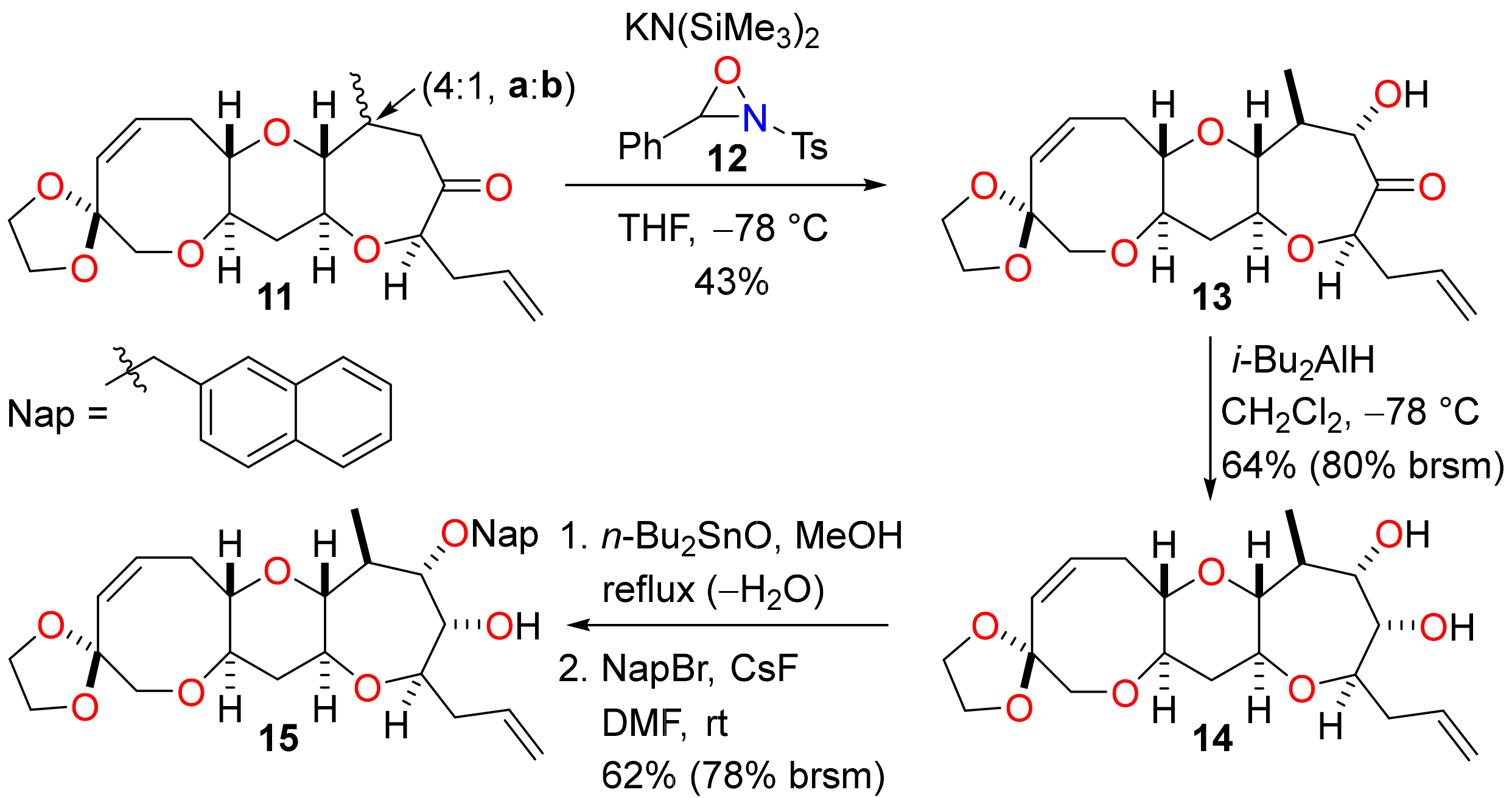
(1′S,3′R,4′S,5′S,7′R,9′S,11′R,15′Z)-5′-Hydroxy-4′-methyl-7′-(prop-2-en-1-yl)-2′,8′,12′-trioxaspiro(1,3-dioxolane-2,14′-tricyclo[9.6.0.03,9]heptadecan)-15′-en-6′-one (13)
To a solution of the ketones 11a and 11b (18 mg of a 4:1 mixture, 50 µmol) in THF (2.9 mL) at −78 °C, a solution of potassium bis(trimethylsilyl)amide (0.10 mL of a 0.50 M solution in toluene, 50 µmol) was added and the mixture was stirred for 1 h. A solution of the oxaziridine 12 (14 mg, 50 µmol) in THF (1 mL) was then added to the mixture dropwise and the resulting mixture was stirred for 1 h. The reaction mixture was diluted with sat. aq. sodium thiosulfate (20 mL) and the extracted with diethyl ether (3 × 10 mL). The combined organic extracts were washed with sat. aq. ammonium chloride (10 mL) and brine (2 × 10 mL), dried (MgSO4) and concentrated under reduced pressure. The residue was purified by flash column chromatography (10–30% diethyl ether in pet. ether) to give the α-hydroxyketone 13 (8 mg, 43%) and recovered starting material 11a (2 mg, 11%) as colourless oils. Rf = 0.73 (60% ethyl acetate in pet. ether); [α]D +75.8 (c = 0.60 in CHCl3, 26 °C); νmax 3474, 2926, 2874, 2855, 1713, 1643, 1283, 1096, 1047, 949, 922, 770 cm−1; 1H NMR (400 MHz, CDCl3) δ 5.88 (1H, ddd, J = 11.5, 9.5, 7.5 Hz), 5.78 (1H, ddd, J = 16.9, 9.8, 7.0 Hz), 5.67 (1H, d, J = 11.5 Hz), 5.13–5.06 (2H, m), 4.30 (1H, dd, J = 11.2, 6.4 Hz), 4.08 (1H, dd, J = 7.7, 5.0 Hz), 3.97–3.91 (4H, m), 3.66 (1H, d, J = 12.7 Hz), 3.54 (1H, d, J = 12.7 Hz), 3.46 (1H, ddd, J = 11.7, 9.3, 4.5 Hz), 3.33 (1H, d, J = 6.4 Hz), 3.28 (1H, ddd, J = 9.3, 5.8, 4.2 Hz), 3.01–2.94 (2H, m), 2.90 (1H, ddd, J = 13.5, 9.5, 4.2 Hz), 2.58 (1H, ddd, J = 13.5, 7.5, 5.8 Hz), 2.48–2.32 (3H, m), 1.63 (1H, app. q, J = 11.7 Hz), 1.54–1.44 (1H, m), 1.28 (3H, d, J = 6.3 Hz); 13C NMR (101 MHz, CDCl3) δ 215.5, 133.6, 132.4, 130.6, 118.7, 108.1, 85.5, 83.6, 81.7, 79.8, 77.7, 75.1, 74.2, 64.8, 64.5, 43.2, 38.6, 37.9, 30.1, 15.3; HRMS (ESI+) [C20H28O7Na]+ found 403.1715, [M + Na]+ calcld. 403.1727.
(1′S,3′R,4′S,5′S,6′S,7′R,9′S,11′R,15′Z)-4′-Methyl-7′-(prop-2-en-1-yl)-2′,8′,12′-trioxaspiro(1,3-dioxolane-2,14′-tricyclo[9.6.0.03,9]heptadecan)-15′-ene-5′,6′-diol (14)
To a solution of α-hydroxyketone 13 (31 mg, 81 µmol) in dichloromethane (2.9 mL) at −78°C, diisobutylaluminium hydride (0.32 mL of a 1.0 M solution in dichloromethane, 0.32 mmol) was added and the resultant solution stirred for 2 h. Further diisobutylaluminium hydride (0.32 mL of a 1.0 M solution in dichloromethane, 0.32 mmol) was added and the mixture was stirred for a further 2 h. The reaction mixture was diluted with sat. aq. Rochelle salt (20 mL) and ethyl acetate (20 mL) and stirred vigorously for 1 h. The phases were separated and the organic phase was washed with brine (2 × 10 mL), dried (MgSO4) and concentrated under reduced pressure. The residue was purified by flash column chromatography (20–100% diethyl ether in pet. ether) to afford the diol 14 (20 mg, 64%) and starting material 13 (6 mg, 19%) as colourless films. Rf = 0.30 (60% ethyl acetate in pet. ether); [α]D +18.4 (c = 0.50 in CHCl3, 27 °C); νmax 3447, 2926, 2874, 1647, 1456, 1285, 1090, 1042, 1013, 916, 770 cm−1; 1H NMR (400 MHz, CDCl3) δ 5.90 (1H, ddd, J = 11.4, 9.6, 7.6 Hz), 5.85 (1H, dddd, J = 17.1, 10.2, 6.9, 6.9 Hz), 5.67 (1H, d, J = 11.4 Hz), 5.17–5.04 (2H, m), 3.94 (4H, m), 3.86 (1H br s), 3.71 (1H, d, J = 9.4 Hz), 3.65 (1H, d, J = 12.7 Hz), 3.56 (1H, ddd, J = 7.6, 5.5, 5.5 Hz), 3.55 (1H, d, J = 12.7), 3.48 (1H, ddd, J = 11.6, 9.3, 4.4 Hz), 3.37 (1H, ddd, J = 11.4, 9.3, 4.6 Hz), 3.22 (1H, ddd, J = 9.3, 5.9, 4.1 Hz), 2.88 (1H, ddd, J = 13.6, 9.6, 4.1 Hz), 2.72 (1H, dd, J = 9.3, 9.0 Hz), 2.59 (1H, ddd, J = 13.6, 7.6, 5.9 Hz), 2.39–2.24 (3H, m), 2.21 (1H, br s), 2.12 (1H, ddq, 9.4, 9.0, 6.7 Hz), 1.94 (1H, br s), 1.48 (1H, ddd, J = 12.1, 11.6, 11.4 Hz), 1.15 (3H, d, J = 6.7 Hz); 13C NMR (101 MHz, CDCl3) δ 134.8, 133.4, 130.8, 117.5, 108.2, 84.8, 82.7, 81.7, 78.3, 78.2, 77.7, 74.1, 73.4, 64.8, 64.5, 39.4, 39.0, 38.0, 30.3, 15.4; HRMS (ESI+) [C20H30O7Na]+ found 405.1871, [M + Na]+ calcld. 405.1884.
(1′S,3′R,4′S,5′S,6′R,7′R,9′S,11′R,15′Z)-4′-Methyl-5′-[(naphthalen-2-yl)methoxy]-7′-(prop-2-en-1-yl)-2′,8′,12′-trioxaspiro(1,3-dioxolane-2,14′-tricyclo[9.6.0.03,9]heptadecane)-15′-en-6′-ol (15)
To a solution of diol 14 (20 mg, 52 µmol) in methanol (5.2 mL), dibutyltin oxide (14 mg, 58 µmol) was added. The resultant suspension was stirred at reflux for 2 h to form a colourless solution. The mixture was concentrated under reduced pressure and dried by azeotropic distillation with toluene. The mixture was redissolved in DMF (5.2 mL) and 2-(bromomethyl)naphthalene (14 mg, 62 µmol) and caesium fluoride (9 mg, 0.06 mmol) were added. The reaction mixture was stirred for 16 h and then quenched with water carefully. The mixture was extracted with diethyl ether (3 × 10 mL) and the combined organic extracts were washed with brine (3 × 10 mL), dried (MgSO4) and concentrated under reduced pressure. The residue was purified by flash column chromatography (20–100% diethyl ether in pet. ether) to afford the naphthyl ether 15 (17 mg, 62%) and recovered diol 14 (4 mg, 20%) as colourless films. Rf = 0.39 (80% diethyl ether in pet. ether); [α]D +14.8 (c = 1.00 in CHCl3, 27 °C); νmax 3466, 2928, 2874, 2853, 1643, 1456, 1090, 949, 916, 1051, 818, 752 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.87–7.80 (3H, m), 7.76 (1H, s), 7.52–7.44 (3H, m), 5.91 (1H, ddd, J = 11.5, 9.6, 7.5 Hz), 5.83 (1H, dddd, J = 17.2, 10.3, 6.6, 6.6 Hz), 5.69 (1H, d, J = 11.5 Hz), 5.13–5.02 (2H, m), 4.74 (1H, d, J = 11.3 Hz), 4.65 (1H, d, J = 11.3 Hz), 3.98–3.92 (4H, m), 3.90 (1H, dd, J = 5.0, 2.2 Hz), 3.66 (1H, d, J = 12.7 Hz), 3.59 (1H, ddd, J = 7.9, 5.0, 5.0 Hz), 3.55 (1H, d, J = 12.7), 3.53 (1H, dd, J = 8.8, 2.2 Hz), 3.50 (1H, ddd, J = 11.6, 9.5, 4.3 Hz), 3.49 (1H, ddd, J = 11.6, 9.4, 4.5 Hz), 3.24 (1H, ddd, J = 9.4, 5.7, 4.1 Hz), 2.92 (1H, ddd, J = 13.7, 9.6, 4.1 Hz), 2.78 (1H, dd, J = 9.5, 7.7 Hz), 2.59 (1H, ddd, J = 13.7, 7.5, 5.7 Hz), 2.39–2.19 (4H, m), 1.46 (1H, ddd, J = 11.6, 11.6, 11.6 Hz), 1.18 (3H, d, J = 6.9 Hz); 13C NMR (101 MHz, CDCl3) δ 135.4, 134.9, 133.5, 133.3, 133.2, 130.9, 128.4, 128.0, 127.9, 127.1, 126.4, 126.3, 126.2, 117.2, 108.1, 85.4, 82.0, 81.8, 81.8, 78.2, 77.3, 74.3, 74.1, 72.6, 64.8, 64.5, 39.0, 39.0, 36.8, 30.3, 16.4; HRMS (ESI+) [C31H38O7Na]+ found 545.2497, [M + Na]+ calcld. 545.2510.
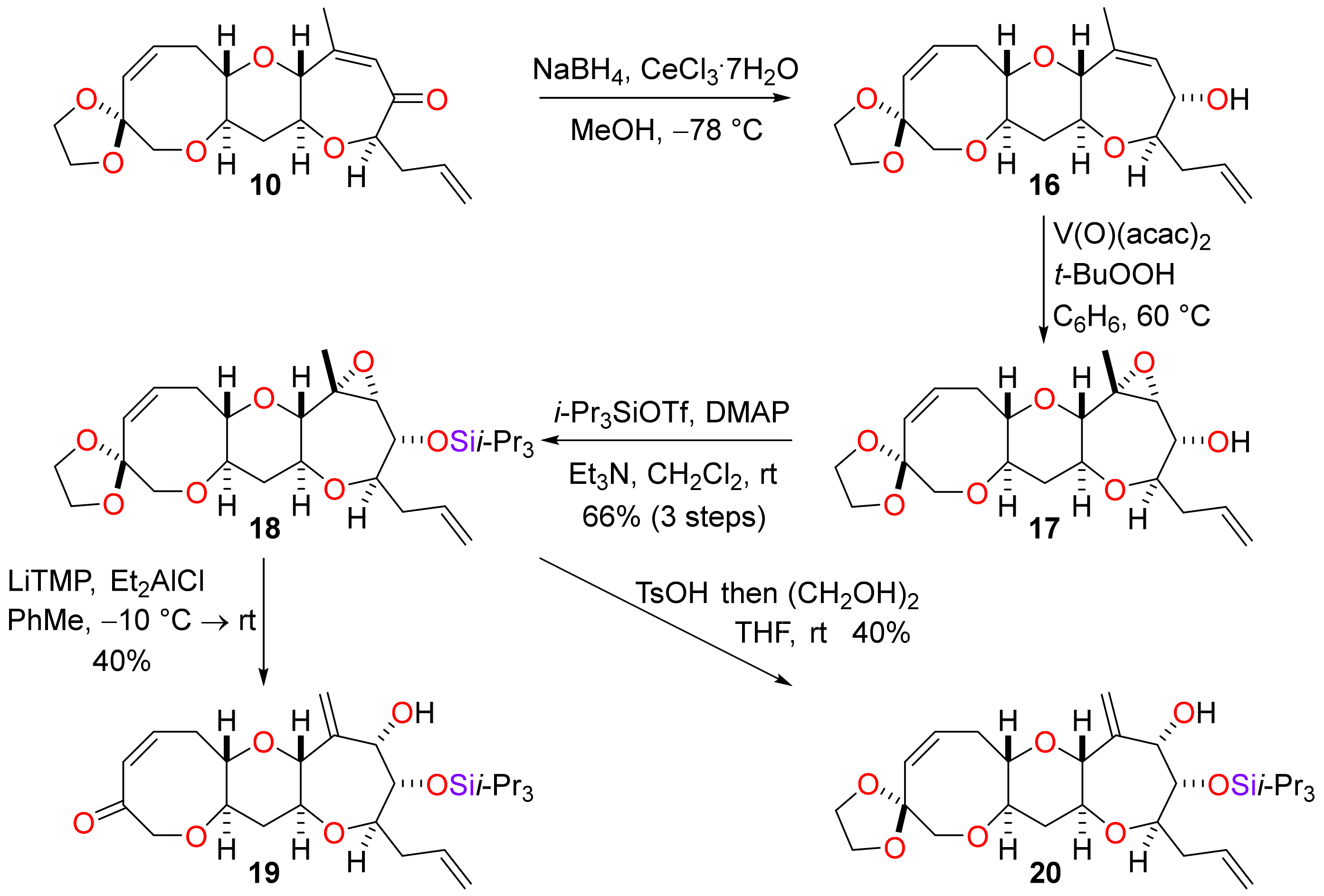
(1′S,3′R,6′S,7′R,9′S,11′R,15′Z)-4′-Methyl-7′-(prop-2-en-1-yl)-2′,8′,12′-trioxaspiro(1,3-dioxolane-2,14′-tricyclo[9.6.0.03,9]heptadecane)-4′,15′-dien-6′-ol (16)
To a solution of the enone 10 (292 mg, 0.806 mmol) in methanol (16 mL), cerium(III) chloride heptahydrate (361 mg, 0.969 mmol) was added. The solution was cooled to −78 °C and sodium borohydride (37 mg, 0.97 mmol) was added and the mixture was stirred for 2 h. The mixture was diluted with ethyl acetate (80 mL) and washed with sat. aq. ammonium chloride (3 × 40 mL). The organic phase was dried (MgSO4) and concentrated under reduced pressure to afford the crude allylic alcohol 16 (294 mg) as a colourless gum. The residue was used directly in the next reaction without purification. For analytical purposes, a sample of the product was purified by flash column chromatography (25–75% diethyl ether in pentane) to afford the allylic alcohol 16 as a colourless gum. Rf = 0.68 (60% ethyl acetate in pet. ether); [α]D +13.5 (c = 1.00 in CHCl3, 21 °C); νmax 2994, 2942, 2880, 1701, 1641, 1377, 1101, 1087, 984, 959, 854 cm−1; 1H NMR (400 MHz, CDCl3) δ 5.91 (1H, dddd, J = 17.1, 10.3, 6.9, 6.9 Hz), 5.88 (1H, ddd, J = 11.4, 9.6, 7.6 Hz), 5.69 (1H, d, J = 11.4 Hz), 5.47 (1H, ddq, J = 2.2, 1.6, 1.6 Hz), 5.17–5.00 (2H, m), 4.05 (1H, dd, J = 8.8, 2.2 Hz), 3.99–3.89 (4H, m), 3.66 (1H, ddq, J = 9.0, 1.6, 1.6 Hz), 3.66 (1H, d, J = 12.7 Hz), 3.55 (1H, d, J = 12.7 Hz), 3.45 (1H, ddd, J = 11.7, 9.2, 4.4 Hz), 3.35–3.23 (1H, m), 3.29 (1H, ddd, J = 11.2, 9.0, 4.7 Hz), 3.26 (1H, ddd, J = 9.2, 5.5, 4.1 Hz), 2.94 (1H, ddd, J = 13.6, 9.6, 4.1 Hz), 2.55 (1H, ddd, J = 13.6, 7.6, 5.5 Hz), 2.49 (1H, ddddd, J = 14.6, 6.9, 3.5, 1.3, 1.3 Hz), 2.30 (1H, ddd, J = 12.1, 4.7, 4.4 Hz), 2.20 (1H, ddddd, J = 14.6, 8.2, 6.9, 1.3, 1.3 Hz), 1.78 (3H, dd, J = 1.6, 1.6 Hz), 1.58 (1H, ddd, J = 12.1, 11.7, 11.2 Hz); 13C NMR (101 MHz, CDCl3) δ 137.7, 135.3, 133.6, 130.8, 129.0, 117.0, 108.0, 83.9, 82.2, 81.7, 78.2, 77.8, 74.2, 73.4, 64.7, 64.6, 38.9, 37.8, 30.1, 21.9; HRMS (ESI+) [C20H28O6Na]+ found 387.1771, [M + Na]+ calcld. 387.1778
(1′S,3′S,4′R,6′R,7′R,8′R,10′S,12′R,16′Z)-4′-Methyl-8′-(prop-2-en-1-yl)-2′,5′,9′,13′-tetraoxaspiro(1,3-dioxolane-2,15′-tetracyclo[10.6.0.03,10.04,6]octadecane)-16′-en-7′-ol (17)
To a stirred solution of allylic alcohol 16 (294 mg) in benzene (41 mL), vanadyl acetylacetonate (42 mg, 0.16 mmol, >20 mol%) was added. A teal solution formed after 10 min and then t-butyl hydroperoxide (0.32 mL of a 5.0 M in decane, 1.6 mmol) was added to form a red solution. The solution was stirred at 60 °C for 2 h during which time the solution turned yellow. The reaction mixture was diluted with diethyl ether (40 mL) and washed with sat. aq. sodium sulfite (2 × 40 mL), sat. aq. ammonium chloride / 10% aq. ammonium hydroxide (4:1, 2 × 40 mL) and brine (2 × 40 mL). The organic phase was dried (Na2SO4) and concentrated under reduced pressure to afford the epoxide 17 (290 mg) as a yellow oil. The residue was used directly in the next reaction without purification. For analytical purposes, a sample of the product was purified by flash column chromatography (25–75% diethyl ether in pentane) to afford the epoxide 17 as a colourless oil. Rf = 0.39 (60% ethyl acetate in pet. ether); [α]D +11.8 (c = 1.00 in CHCl3, 22 °C); νmax 3457, 2924, 2872, 2855, 1641, 1443, 1281, 1111, 1086, 1055, 1018, 882, 770 cm−1; 1H NMR (400 MHz, CD2Cl2) δ 5.93–5.81 (2H, m), 5.66 (1H, d, J = 11.5 Hz), 5.12–5.02 (2H, m), 3.95–3.86 (4H, m), 3.75 (1H, ddd, J = 8.9, 8.2, 1.0 Hz), 3.59 (1H, d, J = 12.7 Hz), 3.52 (1H, d, J = 12.7 Hz), 3.46 (1H, ddd, J = 11.6, 9.1, 4.5 Hz), 3.29 (1H, d, J = 9.1 Hz), 3.28–3.19 (2H, m), 3.15 (1H, ddd, J = 11.5, 9.1, 4.7 Hz), 3.08 (1H, d, J = 1.0 Hz), 2.86 (1H, ddd, J = 13.7, 9.6, 4.0 Hz), 2.60 (1H, ddd, J = 13.7, 6.9, 6.9 Hz), 2.53 (1H, ddd, J = 14.5, 6.9, 6.9 Hz), 2.26 (1H, ddd, J = 12.0, 4.7, 4.5 Hz), 2.12 (1H, ddd, J = 14.5, 7.7, 7.7 Hz), 2.02 (1H, d, J = 8.2 Hz), 1.42 (1H, ddd, J = 12.0, 11.6, 11.5 Hz), 1.40 (3H, s); 13C NMR (101 MHz, CD2Cl2) δ 135.8, 134.1, 130.7, 117.1, 108.5, 82.7, 82.2, 80.5, 78.0, 75.7, 74.4, 72.9, 67.2, 65.1, 64.9, 61.5, 38.9, 37.8, 30.5, 21.3; HRMS (EI+) [C20H28O7]+ found 380.1831, [M]+ calcld. 380.1835.
[(1′S,3′S,4′R,6′R,7′R,8′R,10′S,12′R,16′Z)-4′-Methyl-8′-(prop-2-en-1-yl)-2′,5′,9′,13′-tetraoxaspiro(1,3-dioxolane-2,15′-tetracyclo[10.6.0.03,10.04,6]octadecane)-16′-en-7′-yloxy]tris(propan-2-yl)silane (18)
The reaction was performed two batches. To a solution of the epoxy alcohol 17 (145 mg) in dichloromethane (20 mL), 4-dimethylaminopyridine (244 mg, 2.00 mmol), triethylamine (1.12 mL, 8.0 mmol) and triisopropylsilyl trifluoromethanesufonate (1.61 mL, 6.0 mmol) were added. The mixture was stirred for 16 h and diluted with diethyl ether (80 mL). The mixture was washed with sat. aq. ammonium chloride (3 × 50 mL), dried (Na2SO4) and concentrated under reduced pressure. The residue was purified by flash column chromatography (5–15% diethyl ether in pet. ether). The batches were combined to give the silyl ether 18 (284 mg, 66% over 3 steps) as a colourless oil. Rf = 0.51 (20% ethyl acetate in pet. ether); [α]D +39.2 (c = 0.50 in CHCl3, 27 °C); νmax 2943, 2893, 2866, 1641, 1464, 1109, 1089, 883, 912, 718 cm−1; 1H NMR (400 MHz, CD2Cl2) δ 5.89 (1H, ddd, J = 11.5, 9.6, 7.5 Hz), 5.86 (1H, dddd, J = 17.1, 10.3, 6.8, 6.8 Hz), 5.66 (1H, d, J = 11.5 Hz), 5.09–4.98 (2H, m), 3.97 (1H, dd, J = 9.2, 1.0 Hz), 3.94–3.86 (4H, m), 3.59 (1H, d, J = 12.7 Hz), 3.52 (1H, d, J = 12.7 Hz), 3.45 (1H, ddd, J = 11.6, 9.2, 4.4 Hz), 3.29–3.25 (1H, m), 3.27 (1H, d, J = 9.2 Hz), 3.21 (1H, ddd, J = 9.2, 6.0, 4.0 Hz), 3.14 (1H, ddd, J = 11.5, 9.2, 4.7 Hz), 3.01 (1H, d, J = 1.0 Hz), 2.87 (1H, ddd, J = 13.5, 9.6, 4.0 Hz), 2.65–2.53 (2H, m), 2.25 (1H, ddd, J = 11.9, 4.7, 4.4 Hz), 2.02 (1H, ddddd, J = 14.4, 10.1, 6.8, 1.3, 1.3 Hz), 1.40 (1H, ddd, J = 11.9, 11.6, 11.5 Hz), 1.38 (3H, s), 1.16–1.02 (21H, m); 13C NMR (101 MHz, CD2Cl2) δ 136.4, 134.1, 130.7, 116.7, 108.5, 82.8, 82.3, 81.2, 78.1, 75.6, 74.4, 74.4, 67.4, 65.1, 64.9, 61.2, 38.9, 37.8, 30.6, 21.5, 18.6, 18.5, 13.6; HRMS (ESI+) [C29H48O7SiNa]+ found 559.3047, [M + Na]+ calcld. 559.3062.
(1S,3R,5S,6S,7R,9S,11R,15Z)-5-Hydroxy-4-methylidene-7-(prop-2-en-1-yl)-6-{[tris(propan-2-yl)silyl]oxy}-2,8,12-trioxatricyclo[9.6.0.03,9]heptadec-15-en-14-one (19)
To a solution of 2,2,6,6-tetramethylpiperidine (0.05 mL, 0.3 mmol) in toluene (0.45 mL) at −10 °C, a solution of n-BuLi (0.5 mL of a 0.6 M solution in toluene and hexane, 3:2, 0.3 mmol) was added dropwise. The mixture was stirred for 30 min to produce an orange solution. Diethylaluminium chloride (0.30 mL of a 1.0 M solution in hexane, 0.30 mmol) was added dropwise and the mixture was stirred for a further 30 min resulting in the formation of a colourless solution. To the resultant solution of the complex was added a solution of the epoxide 18 (27 mg, 50 µmol) in toluene (0.70 mL). The reaction mixture was allowed to reach rt over 16 h and then diluted with diethyl ether (8 mL). Sat. aq. Rochelle salt (10 mL) was added and the mixture was stirred vigorously for 2 h. The phases were separated and the organic phase was washed with sat. aq. ammonium chloride (10 mL) and brine (10 mL), then dried (MgSO4) and concentrated under reduced pressure. The residue was purified by flash column chromatography (10–40% diethyl ether in pentane) to afford the allylic alcohol 19 (10 mg, 40%) as a yellow oil. Rf = 0.60 (60% ethyl acetate in pet. ether); [α]D +5.8 (c = 0.50 in CHCl3, 27 °C); νmax 3476, 2891, 2866, 1676, 1643, 1464, 1098, 1016, 997, 918, 883 cm−1; 1H NMR (400 MHz, CDCl3) δ 6.47 (1H, ddd, J = 12.4, 8.8, 7.6 Hz), 5.87 (1H, dddd, J = 17.1, 10.2, 6.9, 6.6 Hz), 5.86 (1H, dddd, J = 12.4, 1.6, 1.6, 1.1 Hz), 5.45 (1H, ddd, J = 1.3, 1.3, 1.3 Hz), 5.39 (1H, ddd, J = 1.3, 1.3, 1.3 Hz), 5.13–5.04 (2H, m), 4.51 (1H, dd, J = 17.8, 1.1 Hz), 4.47 (1H, ddd, J = 2.5, 1.3, 1.3 Hz), 4.20 (1H, d, J = 17.8 Hz), 3.80 (1H, dd, J = 6.7, 2.5 Hz), 3.68 (1H, ddd, J = 11.2, 9.6, 4.3 Hz), 3.64 (1H, ddd, J = 9.6, 1.3, 1.3 Hz), 3.55 (1H, ddd, J = 9.7, 6.7, 3.0 Hz), 3.41 (1H, ddd, J = 11.4, 9.1, 4.2 Hz), 3.32 (1H, ddd, J = 9.1, 9.1, 1.6 Hz), 2.70 (1H, dddd, J = 14.9, 7.6, 1.6, 1.6 Hz), 2.64 (1H, dddd, J = 14.9, 9.1, 8.8, 1.6 Hz), 2.46 (1H, ddddd, J = 14.4, 6.9, 3.0, 1.3, 1.3 Hz), 2.34 (1H, ddd, J = 11.6, 4.3, 4.2 Hz), 2.16 (1H, ddddd, J = 14.4, 9.7, 6.6, 1.3, 1.3 Hz), 1.65 (1H, ddd, J = 11.6, 11.4, 11.2 Hz), 1.15–1.03 (21H, m); 13C NMR (101 MHz, CDCl3) δ 203.7, 145.1, 138.1, 135.3, 129.0, 117.7, 117.0, 85.3, 83.1, 82.0, 78.7, 78.6, 77.4, 77.0, 76.3, 38.3, 38.2, 35.1, 18.3, 13.0; HRMS (ESI+) [C27H44O6SiNa]+ found 515.2775, [M + Na]+ calcld. 515.2799.
(1′S,3′R,5′S,6′S,7′R,9′S,11′R,15′Z)-4′-Methylidene-7′-(prop-2-en-1-yl)-6′-{[tris(propan-2-yl)silyl]oxy}-2′,8′,12′-trioxaspiro(1,3-dioxolane-2,14′-tricyclo[9.6.0.03,9]heptadecane)-15′-en-5′-ol (20)
To a solution of epoxide 18 (43 mg, 80 µmol) in THF (4 mL), p-toluenesulfonic acid (69 mg, 0.40 mmol) and 4 Å molecular sieves (3 g) were added. The resultant mixture was stirred for 16 h and dried (4 Å molecular sieves) ethylene glycol (1 mL) was added. The reaction mixture was stirred for a further 24 h and diluted with sat. aq. sodium bicarbonate (25 mL). The organic phase was extracted with diethyl ether (3 × 10 mL) and the combined organic extracts were washed with brine (3 × 10 mL), dried (MgSO4) and concentrated under reduced pressure. The residue was purified by flash column chromatography (10–30% diethyl ether in pet. ether) to afford allylic alcohol 20 (17 mg, 40%) and starting epoxide 18 (11 mg, 26%) as colourless films. Rf = 0.63 (60% ethyl acetate in pet. ether); [α]D +32.4 (c = 1.00 in CHCl3, 27 °C); νmax 3476, 2941, 2866, 1643, 1464, 1090, 1018, 901, 883 cm−1; 1H NMR (400 MHz, CDCl3) δ 5.92 (1H, ddd, J = 11.4, 9.5, 7.5 Hz), 5.87 (1H, dddd, J = 17.1, 10.3, 7.1, 6.5 Hz), 5.68 (1H, d, J = 11.4 Hz), 5.45 (1H, ddd, J = 1.4, 1.4, 1.4 Hz), 5.35 (1H, ddd, J = 1.4, 1.4, 1.4 Hz), 5.12–5.02 (2H, m), 4.44 (1H, dddd, J = 4.1, 2.7, 1.4, 1.4 Hz), 3.98–3.91 (4H, m), 3.79 (1H, dd, J = 6.9, 2.7 Hz), 3.66 (1H, d, J = 12.8 Hz), 3.61 (1H, ddd, J = 9.6, 1.4, 1.4 Hz), 3.56 (1H, d, J = 12.8 Hz), 3.54–3.46 (3H, m), 3.38 (1H, ddd, J = 9.3, 5.5, 4.1 Hz), 2.96 (1H, ddd, J = 13.4, 9.5, 4.1 Hz), 2.62 (1H, ddd, J = 13.4, 7.5, 5.5 Hz), 2.45 (1H, ddddd, J = 14.5, 7.1, 2.8, 1.3, 1.3 Hz), 2.31 (1H, d, J = 4.1 Hz), 2.30 (1H, ddd, J = 11.7, 4.4, 4.4 Hz), 2.15 (1H, ddddd, J = 14.5, 9.6, 6.5, 1.4, 1.4 Hz), 1.53 (1H, app. dt, J = 11.7, 11.4 Hz), 1.13–1.04 (21H, m); 13C NMR (101 MHz, CDCl3) δ 145.6, 135.4, 133.5, 130.9, 117.0, 116.9, 108.0, 83.2, 82.0, 82.0, 78.6, 78.2, 77.8, 76.1, 74.1, 64.8, 64.6, 38.7, 38.3, 30.3, 18.3, 13.1; HRMS (ESI+) [C29H48O7SiNa]+ found 559.3041, [M + Na]+ calcld. 559.3062.
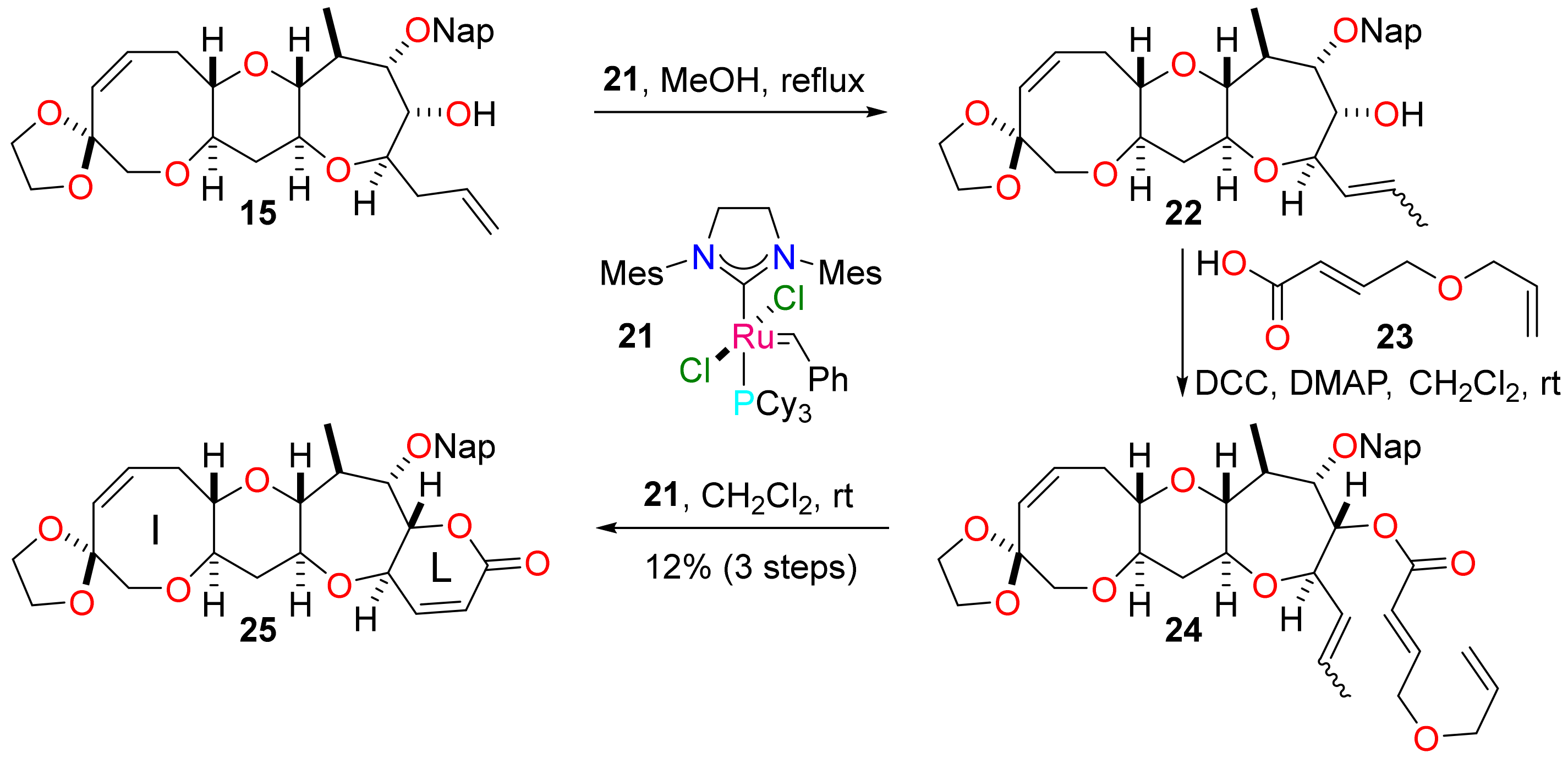
(1′R,3′S,5′Z,10′R,12′S,14′R,19′S,20′S,21′S)-21′-Methyl-20′-[(naphthalen-2-yl)methoxy]-2′,9′,13′,18′-tetraoxaspiro[1,3-dioxolane-2,7′-tetracyclo(10.9.0.03,10.014,19]henicosane)-5′,15′-dien-17′-one (25)
To a solution of alcohol 15 (8 mg, 0.02 mmol) in methanol (0.64 mL), the ruthenium complex 21 (3 mg, 0.03 mmol, 20 mol%) was added. The solution was stirred at reflux for 1 h and concentrated under reduced pressure. The residue was purified by flash column chromatography to afford an E / Z mixture of alcohol 22 (4 mg) as a colourless film. The residue was used directly in the next reaction without purification.
To a solution of (2E)-4-(prop-2-en-1-yloxy)but-2-enoic acid 23 (2 mg, 0.02 mmol) in dichloromethane (0.8 mL), N,N′-dicyclohexylcarbodiimide (3 mg, 0.02 mmol) and 4-dimethylaminopyridine (0.2 mg, 0.02 mmol) were added. The resultant suspension was added to 22 (4 mg) and the mixture stirred for 16 h. The mixture was diluted with diethyl ether (4.2 mL) and filtered. The filtrate was concentrated under reduced pressure and the residue was purified by flash column chromatography (5–60% diethyl ether in pet. ether) to give an isomeric mixture of esters 24 (2 mg) as a colourless film. The residue was used directly in the next reaction without purification.
To a solution of ester 24 (2 mg) in dichloromethane (16 mL), the ruthenium complex 21 (1 mg, 1 µmol) was added. The solution was stirred for 6 h at reflux and concentrated under reduced pressure. The residue was purified by flash column chromatography (0–25% diethyl ether in dichloromethane) to deliver the tetracyclic lactone 25 (1 mg, 12% over 3 steps) as a colourless film. Rf = 0.23 (80% diethyl ether in pet. ether); [α]D +48 (c = 0.08 in CHCl3, 36 °C); νmax 2922, 2851, 1734, 1636, 1559, 1119, 1042, 818 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.88–7.81 (3H, m), 7.80 (1H, s), 7.54–7.42 (3H, m), 6.82 (1H, dd, J = 9.9, 1.6 Hz), 5.93 (1H, dd, J = 9.9, 2.5 Hz), 5.91 (1H, ddd, J = 11.4, 9.7, 7.4 Hz), 5.73 (1H, d, J = 11.4 Hz), 4.87 (1H, d, J = 11.5 Hz), 4.86 (1H, ddd, J = 10.5, 2.5, 1.6 Hz), 4.81 (1H, d, J = 11.5 Hz), 4.26 (1H, dd, J = 10.5, 0.7 Hz), 3.96 (4H, m), 3.90 (1H, ddd, J = 11.0, 9.3, 5.5 Hz), 3.83 (1H, dd, J = 2.6, 0.7 Hz), 3.67 (1H, d, J = 12.7 Hz), 3.54 (1H, d, J = 12.7 Hz), 3.43 (1H, ddd, J = 12.4, 9.1, 4.3 Hz), 3.23 (1H, ddd, J = 9.1, 7.2, 5.5 Hz), 2.97 (1H, ddd, J = 13.5, 9.7, 7.2 Hz), 2.93 (1H, dd, J = 9.3, 5.7 Hz), 2.54 (1H, ddd, J = 13.5, 7.4, 5.5 Hz), 2.33 (1H, ddd, J = 11.8, 5.5, 4.3 Hz), 2.27 (1H, qdd, J = 7.4, 5.7, 2.6 Hz), 1.47 (1H, ddd, J = 12.4, 11.8, 11.0 Hz), 1.18 (3H, d, J = 7.4 Hz); 13C NMR (126 MHz, CDCl3) δ 163.4, 149.9, 135.6, 133.8, 133.3, 133.1, 130.8, 128.3, 128.1, 127.9, 126.8, 126.3, 126.1, 126.1, 119.4, 107.9, 86.9, 82.9, 81.8, 81.0, 77.7, 75.4, 74.4, 73.0, 69.8, 64.8, 64.7, 39.9, 39.0, 32.1, 19.9; HRMS (ESI+) [C31H34O8Na]+ found 557.2124, [M + Na]+ calcld. 557.2146.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








