Determination of Ochratoxin A and Ochratoxin B in Archived Tokaj Wines (Vintage 1959–2017) Using On-Line Solid Phase Extraction Coupled to Liquid Chromatography

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
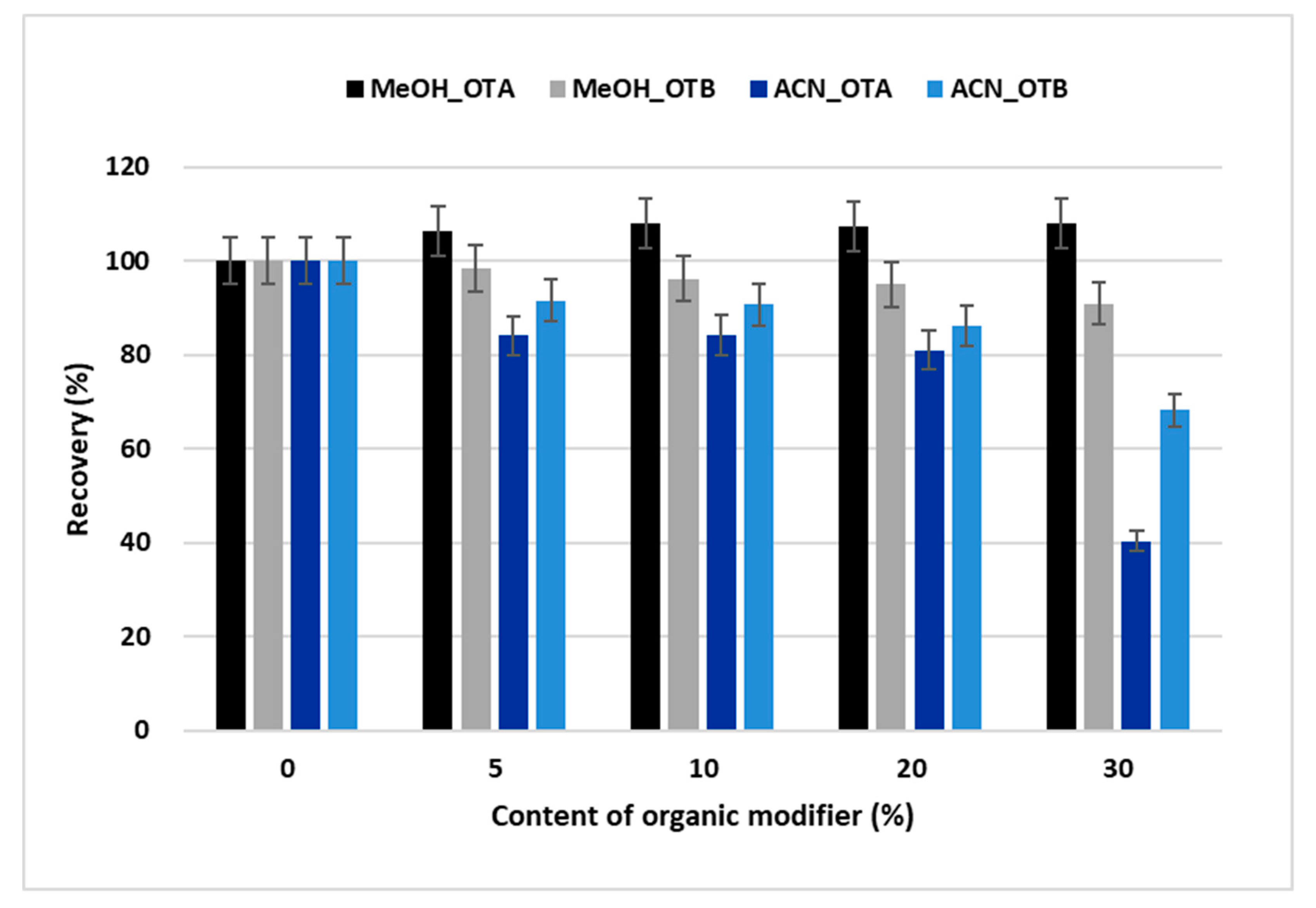
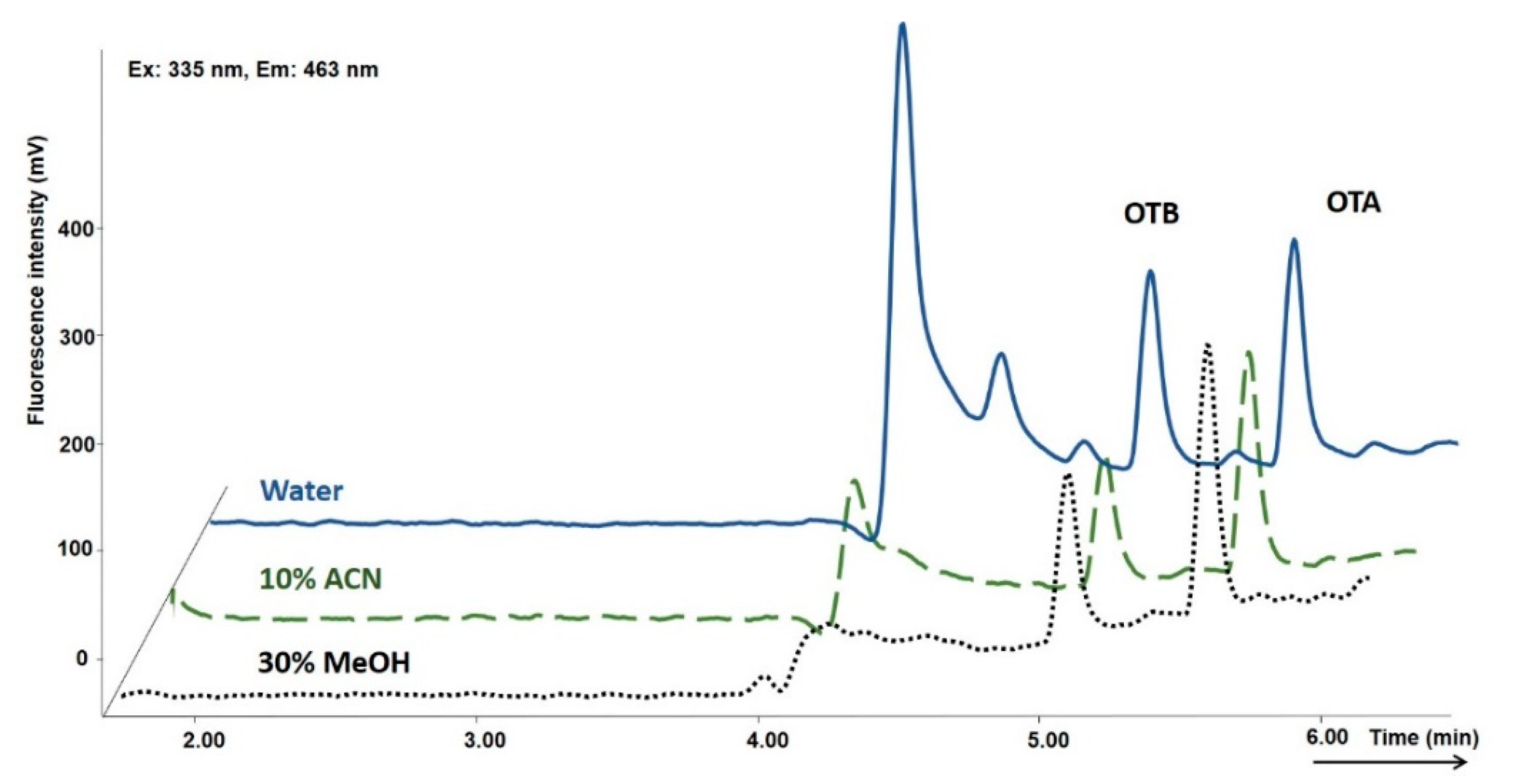
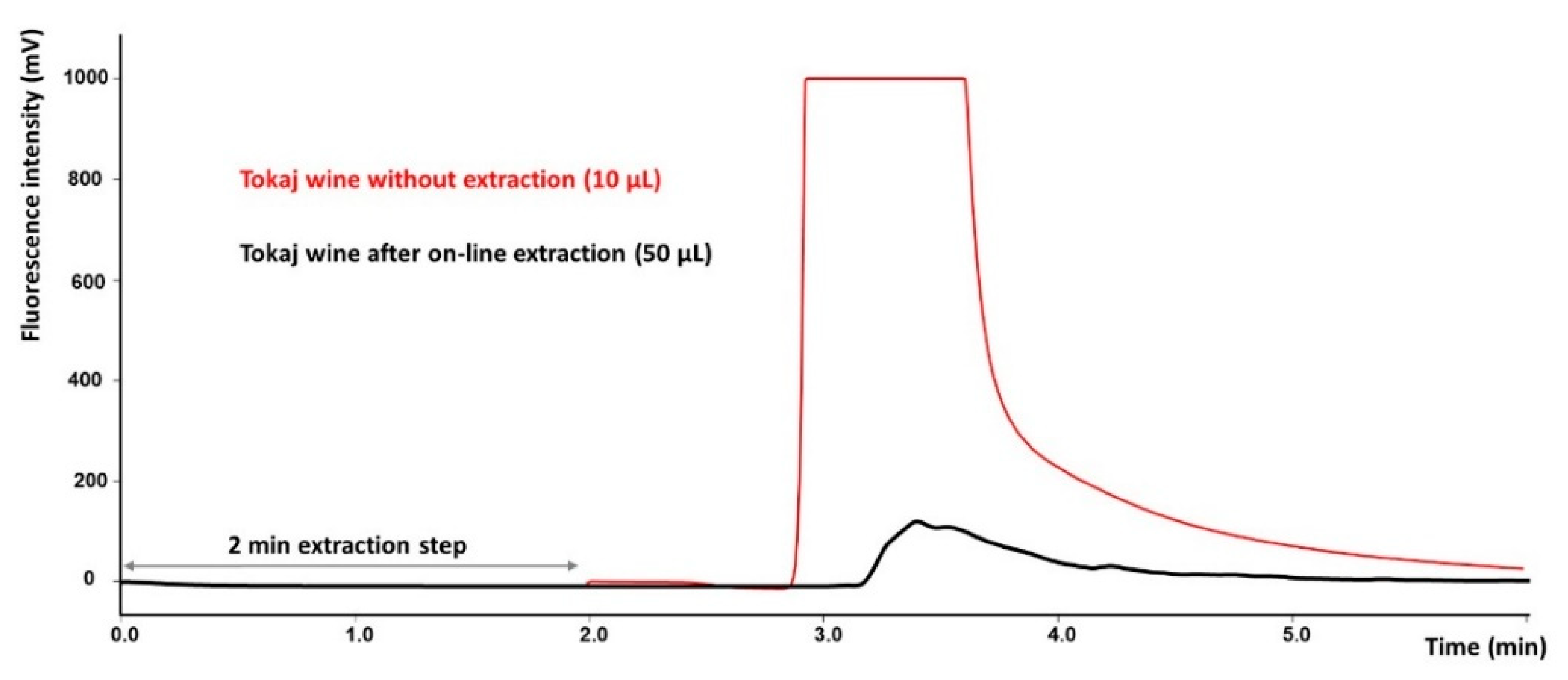
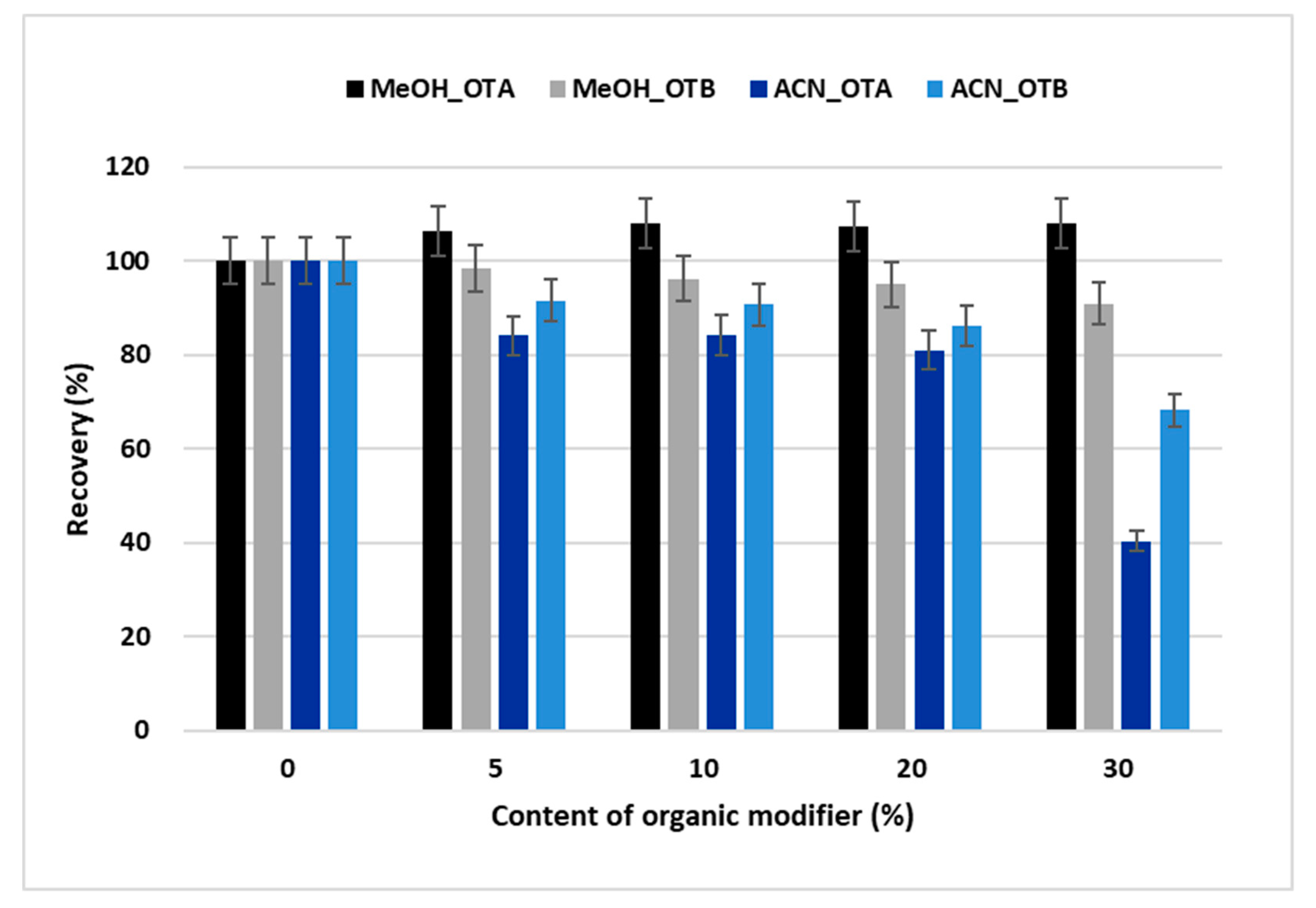
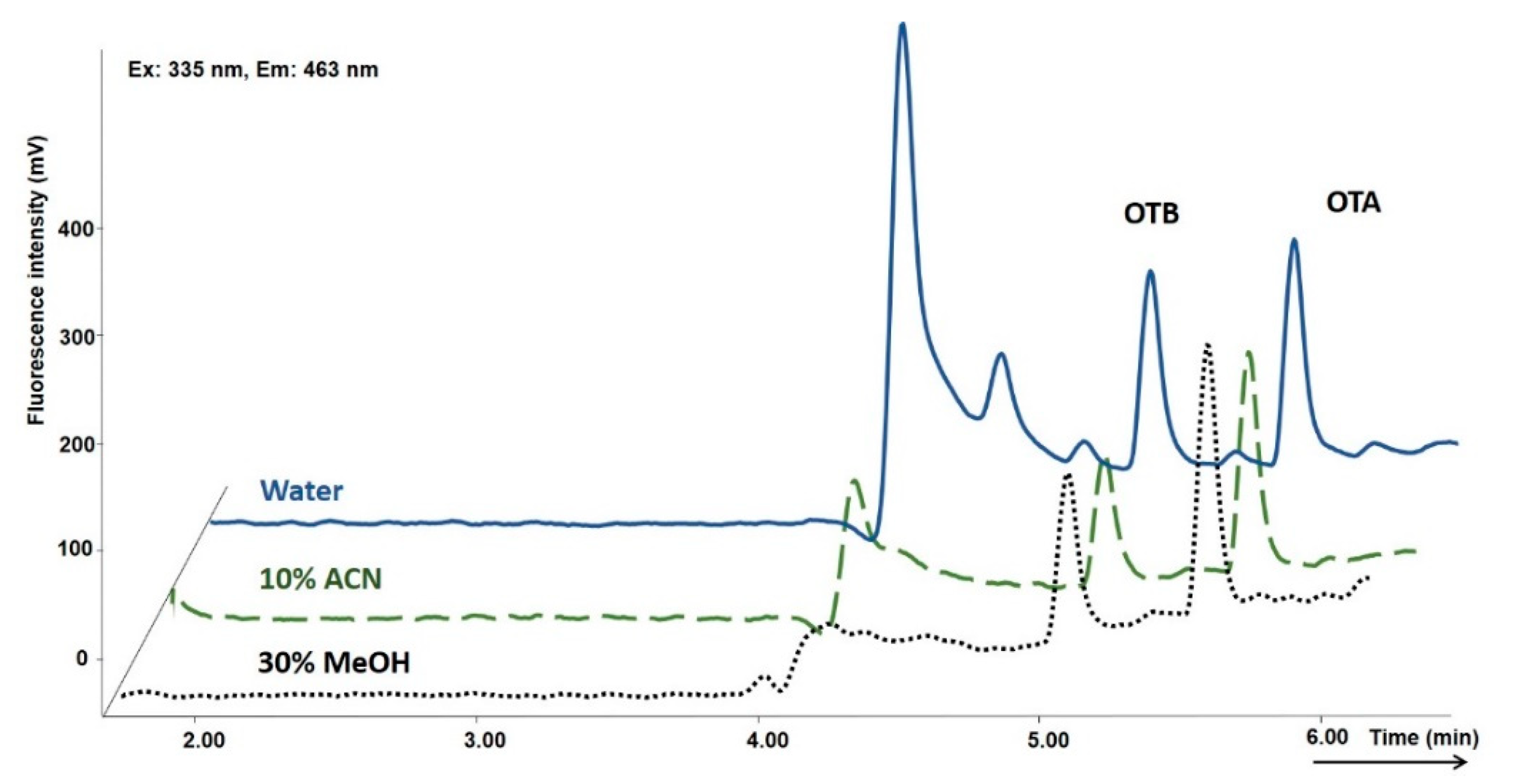
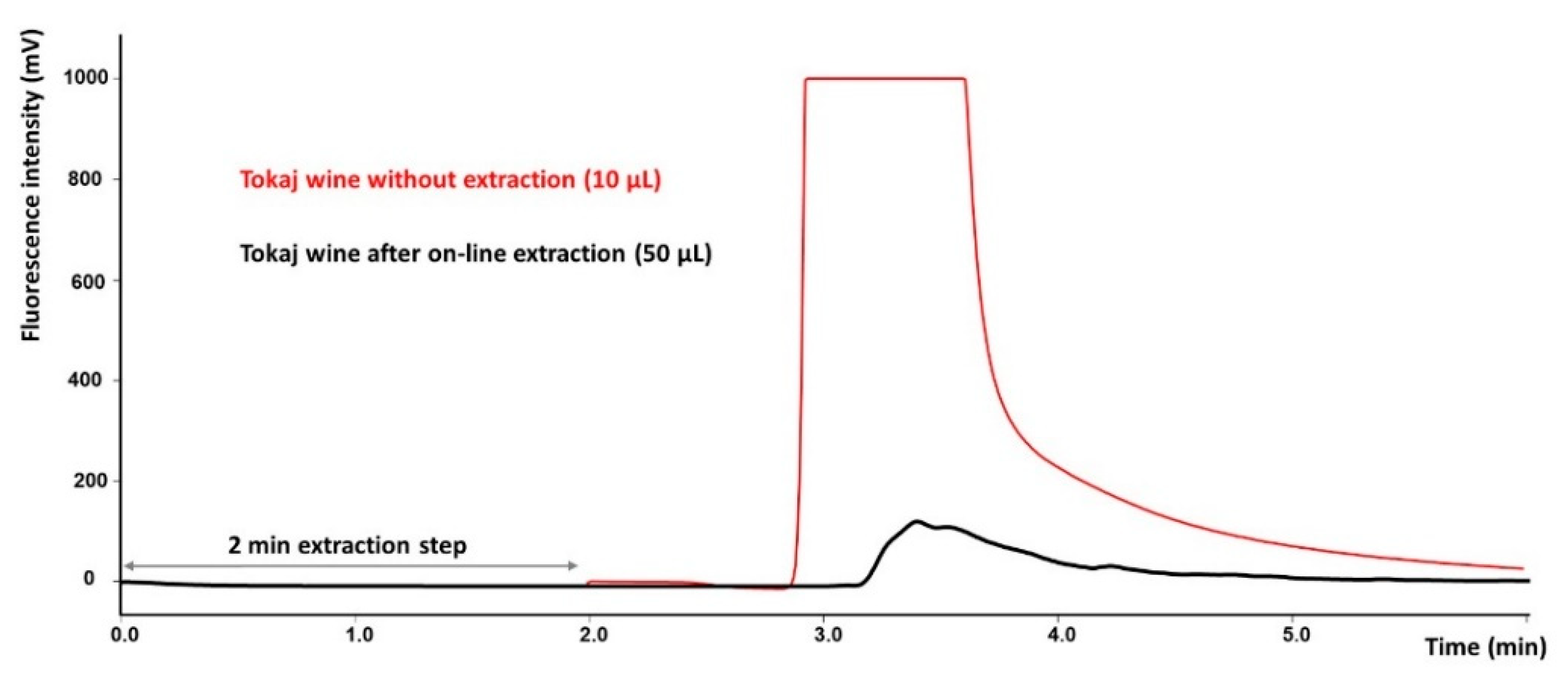
2.1. Optimization of the Online SPE-HPLC Procedure
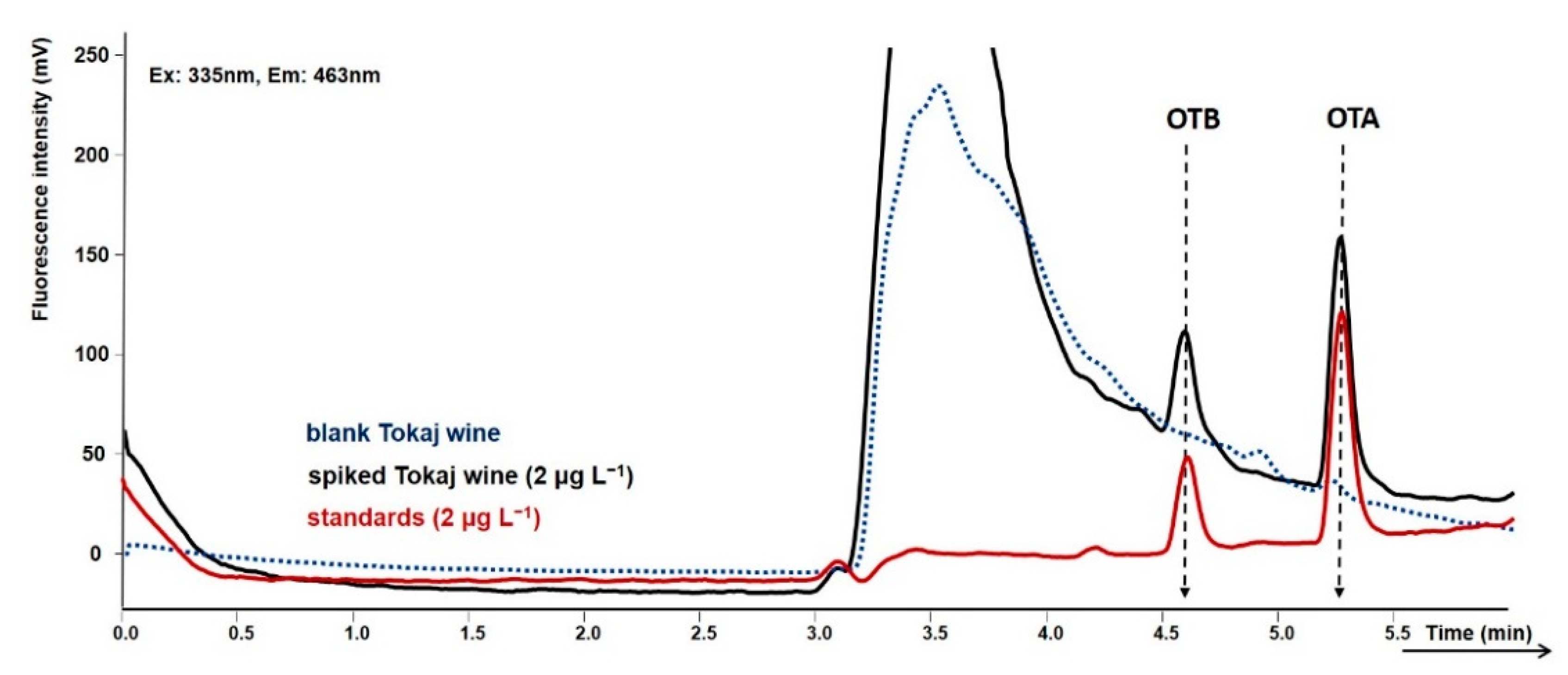
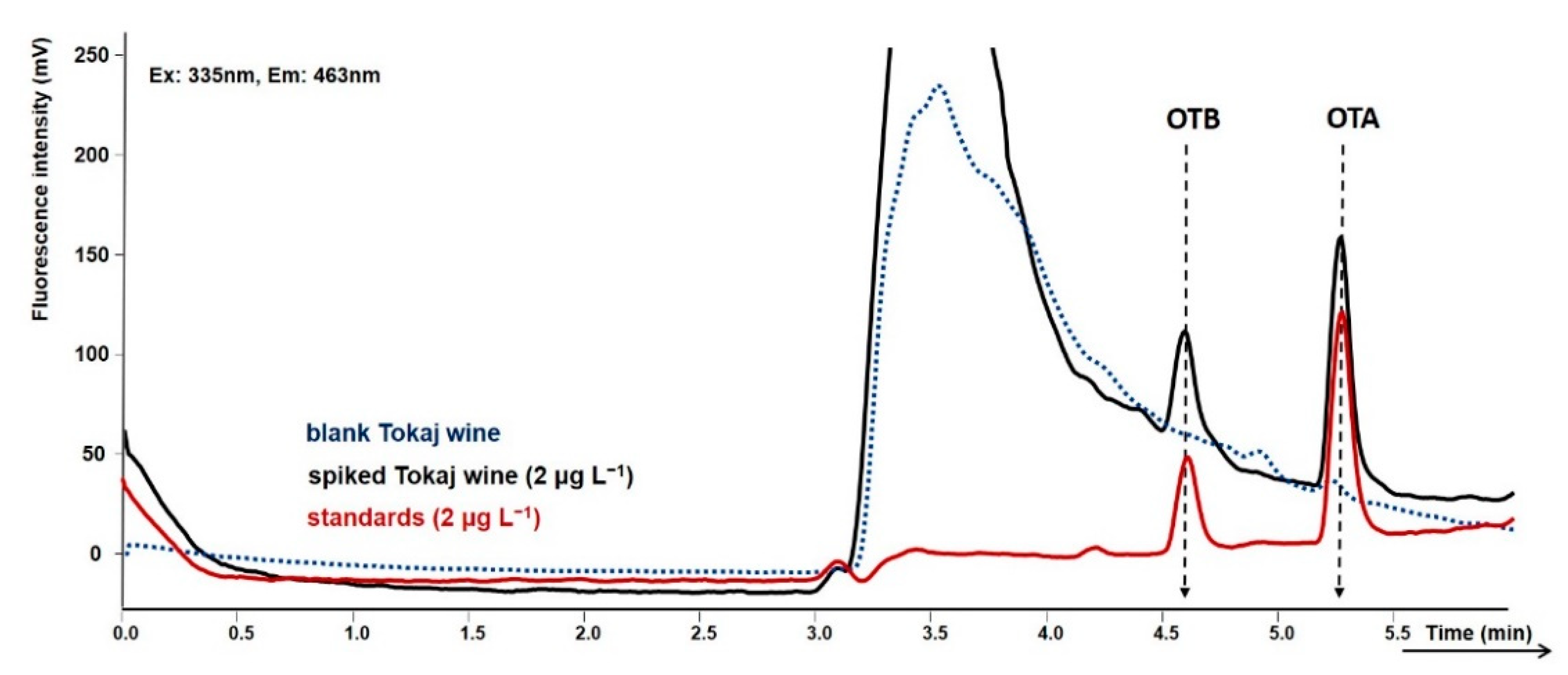
2.2. Validation
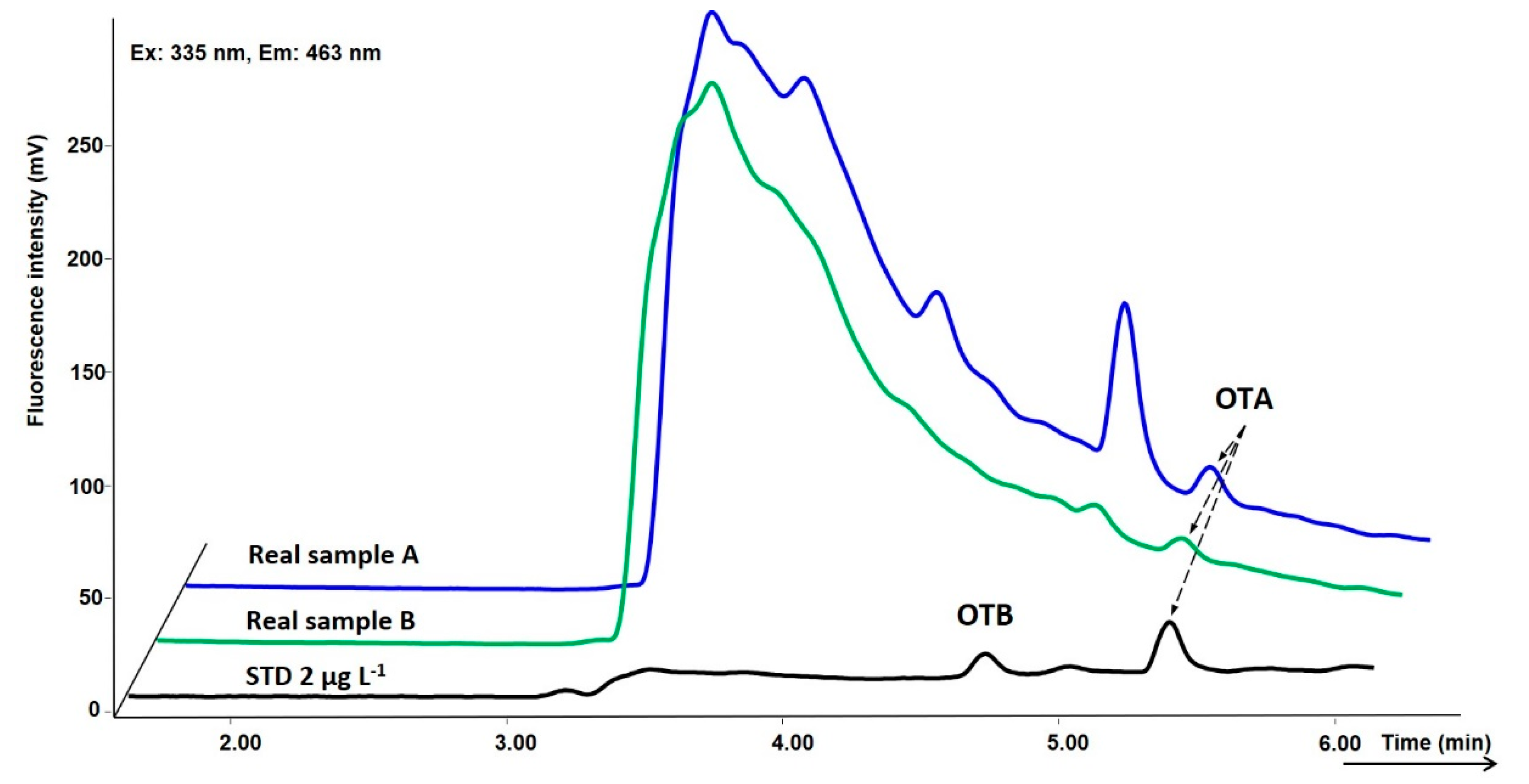
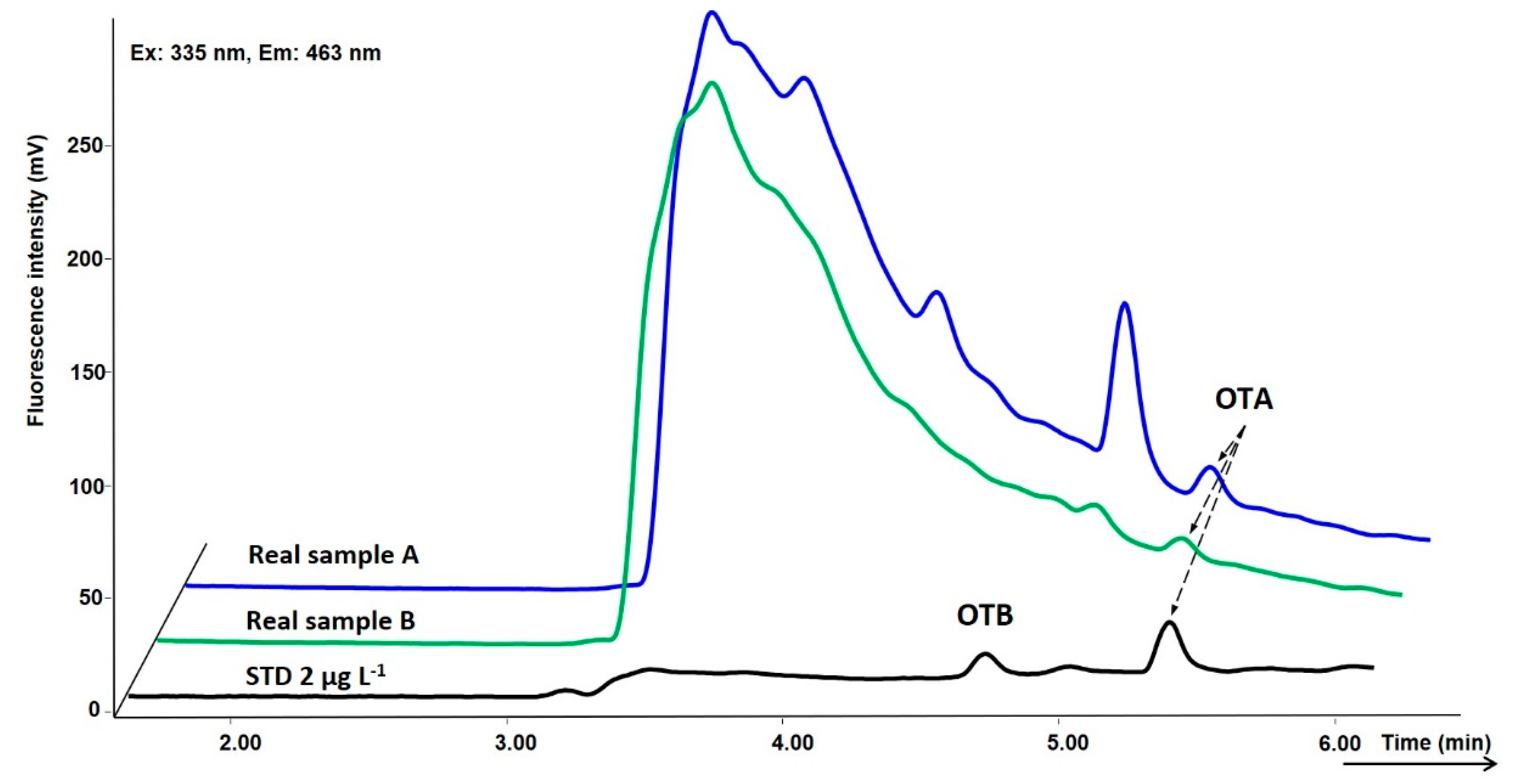
2.3. Tokaj Wine Analysis
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Reagents and Materials
5.2. Instrumentation and Software
5.3. Preparation of Standards Solutions
5.4. HPLC Column-Switching Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Travis, R.B.K.; Wu, F. Ochratoxin A and Human Health Risk: A Review of the Evidence. Crit. Rev. Food Sci. Nutr. 2015, 55, 1860–1869. [Google Scholar] [CrossRef] [Green Version]
- Mateo, R.; Medina, Á.; Mateo, E.M.; Mateo, F.; Jimenéz, M. An overview of ochratoxin a in beer and wine. Int. J. Food Microbiol. 2007, 119, 79–83. [Google Scholar] [CrossRef] [PubMed]
- IARC. Some naturally occurring substances: Food items and constituents, heterocyclic aromatic amines and mycotoxins. In Monographs on the Evaluation of Carcinogenic Risks to Humans; IARC: Lyon, France, 1993; Volume 56. [Google Scholar]
- Ringot, D.; Chango, A.; Schneider, Y.J.; Larondelle, Y. Toxicokinetics and toxicodynamics of ochratoxin A, an update. Chem. Biol. Interact. 2006, 159, 18–46. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Jia, X.; Gu, L.; Sung, C. Review on the qualitative and quantitative analysis of the mycotoxin citrinin. Food Control 2006, 17, 271–285. [Google Scholar] [CrossRef]
- Peraica, M.; Domijan, A.M.; Miletić-Medved, M.; Fuchs, R. The involvement of mycotoxins in the development of endemic nephropathy. Wien. Klin. Wochen. 2008, 120, 402–407. [Google Scholar] [CrossRef]
- Bayman, P.; Baker, J.L. Ochratoxins: A global perspective. Mycopatologia 2006, 162, 215–223. [Google Scholar] [CrossRef]
- Heussner, A.H.; Dietrich, D.R.; O’Brien, E. In vitro investigation of individual and combined cytotoxic effects of ochratoxin A and other selected mycotoxins on renal cells. Toxicol. Vitro 2006, 20, 332–341. [Google Scholar] [CrossRef] [Green Version]
- Remiro, R.; Ibáñez-Vea, M.; González-Peñas, E.; Lizarraga, E. Validation of a liquid chromatography method for the simultaneous quantification of ochratoxin A and its analogues in red wines. J. Chromatogr. A 2010, 1217, 8249–8256. [Google Scholar] [CrossRef]
- Di Stefano, V.; Avellone, G.; Pitonzo, R.; Capocchiano, V.G.; Mazza, A.; Cicero, N.; Dugo, G. Natural co-occurrence of ochratoxin A, ochratoxin B and aflatoxins in Sicilian red wines. Food Addit. Contam. Part A 2015, 32, 1343–1351. [Google Scholar] [CrossRef]
- EC—European Commission. Commission Regulation (EC) No 1881/2006 of 19 December setting maximum levels for certain contaminants in foodstuffs. Off. J. Eur. Union L 2006, 364, 5–24. [Google Scholar]
- Bene, Z.; Mayar, I. Characterization of yeast and mould biota of botrytized grapes in Tokaj wine region in the years 2000 and 2001. Acta Aliment. 2004, 33, 259–267. [Google Scholar] [CrossRef]
- Felsociova, S.; Rybarik, L.; Tancinova, D.; Maskova, Z.; Kacaniova, M. Microfungi and mycotoxins of grapes from Tokaj wine region. J. Microbiol. Biotechnol. Food Sci. 2015, 4, 16–18. [Google Scholar] [CrossRef]
- Gil-Sena, J.; Vazques, C.; Gonzáles-Jaén, M.T.; Patino, B. Wine Contamination with Ochratoxins: A Review. Beverages 2018, 4, 6. [Google Scholar] [CrossRef] [Green Version]
- Wongworapat, K.; Tu Ho, M.H.; Soontornjanagit, M.; Kawamura, O. Occurrence of ochratoxin A and ochratoxin B in commercial coffee in Vietnam and Thailand. JSM Mycotoxins 2016, 66, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Pena, A.; Cerejo, F.; Lino, C.; Silveira, I. Determination of ochratoxin A in Portuguese rice samples by high performance liquid chromatography with fluorescence detection. Anal. Bioanal. Chem. 2005, 382, 1288–1293. [Google Scholar] [CrossRef]
- Copetti, M.V.; Iamanaka, B.T.; Nester, M.A.; Efraim, P.; Taniwaki, M.H. Occurrence of ochratoxin A in cocoa by-products and determination of its reduction during chocolate manufacture. Food Chem. 2013, 136, 100–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maham, M.; Kiarostami, V.; Waqif-Husain, S.; Karami-Osboo, R.; Mirabolfathy, M. Analysis of Ochratoxin A in Malt Beverage Samples using Dispersive Liquid–Liquid Microextraction Coupled with Liquid Chromatography-Fluorescence Detection. Czech. J. Food Sci. 2013, 31, 520–525. [Google Scholar] [CrossRef] [Green Version]
- Aresta, A.; Palmisano, F.; Vatinno, R.; Zambonin, C.G. Ochratoxin A determination in beer by solid-phase microextraction coupled to liquid chromatography with fluorescence detection: A fast and sensitive method for assessment of noncompliance to legal limits. J. Agric. Food Chem. 2006, 54, 1594–1598. [Google Scholar] [CrossRef]
- Al-Hadithi, N.; Kössler, P.; Karlovsky, P. Determination of Ochratoxin A in Wheat and Maize by Solid Bar Microextraction with Liquid Chromatography and Fluorescence Detection. Toxins 2015, 7, 3000–3011. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Pei, F.; Liu, Q.; Fang, Y. Magnetic Solid-Phase Extraction for the Determination of Ochratoxin A in Wine and Beer by HPLC-FLD. Curr. Anal. Chem. 2018, 14, 129–134. [Google Scholar] [CrossRef]
- Iida, K.; Tabata, S.; Kimura, K.; Suzuki, J.; Ibe, A. Simultaneous Determination and Survey of Ochratoxin A, Ochratoxin B and Citrinin in Cereals by LC/MS/MS. ChemoBio Integr. Manag. 2009, 5, 24–31. [Google Scholar] [CrossRef]
- Zhao, Z.Y.; Liu, N.; Yang, L.C.; Wu, A.B.; Zhou, Z.L.; Deng, Y.F.; Song, S.Q.; Wang, J.H.; Hou, J.F. A new preparative method for simultaneous purification of ochratoxin A and ochratoxin B from wheat culture inoculated with Aspergillus ochraceous. World Mycotoxin J. 2016, 9, 31–40. [Google Scholar] [CrossRef]
- Prelle, A.; Sparado, D.; Garibaldi, A.; Gullino, M.L. Co-occurrence of aflatoxins and ochratoxin a in spices commercialized in Italy. Food Control 2014, 39, 192–197. [Google Scholar] [CrossRef]
- Karabín, M.; Lukočová, A.; Fiala, J.; Jelínek, L.; Hudcová, T.; Wang, D.; Dostálek, P. An immunochemical Method for Determination of Ochratoxin A in the Wine and its Applications. Kvasný Průmysl 2014, 60, 226–232. [Google Scholar] [CrossRef] [Green Version]
- Afrali, D.; Fathirad, F.; Ghaseminezhas, S. Determination of trace amounts of ochratoxin A in different food samples based on gold nanoparticles modified carbon paste electrode. J. Food Sci. Technol. 2016, 53, 909–914. [Google Scholar] [CrossRef] [Green Version]
- Mikulíková, R.; Běláková, S.; Benešová, K.; Svoboda, Z. Study of ochratoxin A content in South Moravian and foreign wines by the UPLC method with fluorescence detection. Food Chem. 2012, 133, 55–59. [Google Scholar] [CrossRef]
- Visconti, A.; Pascale, M.; Centonze, G. Determination of ochratoxin a in wine and beer by immunoaffinity column and liquid chromatographic analysis with fluorometric detection: Collaborative study. J. AOAC Int. 2001, 84, 1818–1827. [Google Scholar] [CrossRef] [Green Version]
- Lhotská, I.; Šatínský, D.; Havlíková, L.; Solich, P. A fully automated and fast method using direct sample injection combined with fused-core column online SPE-HPLC for determination of ochratoxin a and citrinin in lager beers. Anal. Bioanal. Chem. 2016, 408, 3319–3329. [Google Scholar] [CrossRef]
- Bacaloni, A.; Cavaliere, C.; Faberi, A.; Pastorini, E.; Samperi, R.; Laganá, A. Automated On-line Solid-Phase Extraction-Liquid Chromatography-Electrospray Tandem Mass Spectrometry Method for the Determination of Ochratoxin A in Wine and Beer. J. Agric. Food Chem. 2005, 53, 5518–5525. [Google Scholar] [CrossRef]
- Armutcu, C.; Uzun, L.; Denizli, A. Determination of Ochratoxin A traces in foodstuffs: Comparison of an automated on-line two-dimensional high-performance liquid chromatography and off-line immunoaffinity-high-performance liquid chromatography system. J. Chromatogr. A 2018, 1569, 139–148. [Google Scholar] [CrossRef]
- Campone, L.; Piccinelli, A.L.; Celano, R.; Pagano, I.; Russo, M.; Rastrelli, L. Rapid and automated on-line solid phase extraction HPLC–MS/MS with peak focusing for the determination of ochratoxin A in wine samples. Food Chem. 2018, 244, 128–135. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information. PubChem Database. Ochratoxin A, CID=442530. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Ochratoxin-A (accessed on 14 April 2020).
- National Center for Biotechnology Information. PubChem Database. Ochratoxin B, CID=20966. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Ochratoxin-B (accessed on 14 April 2020).
- Freire, L.; Braga, P.A.C.; Furtado, M.M.; Delafiori, J.; Dias-Audibert, F.L.; Pereira, G.E.; Reyes, F.G.; Catharino, R.R.; Sant’Ana, A.S. From grape to wine: Fate of ochratoxin A during red, rose, and white winemaking process and the presence of ochratoxin derivatives in the final products. Food Control 2020, 113, 107167. [Google Scholar] [CrossRef]
- Yu, J.; Smith, I.N.; Mikiashivili, N. Reducing Ochratoxin A Content in Grape Pomace by Different Methods. Toxins 2020, 12, 424. [Google Scholar] [CrossRef]
- Bittner, A.; Cramer, B.; Humpf, H.U. Matrix Binding of Ochratoxin A during Roasting. J. Agric. Food Chem. 2013, 61, 12737–12743. [Google Scholar] [CrossRef] [PubMed]
- Remiro, R.; González-Peñas, E.; Lizarraga, E.; López de Cerain, A. Quantification of ochratoxin A and five analogs in Navarra red wines. Food Control 2012, 27, 139–145. [Google Scholar] [CrossRef]
- Amerine, M.A.; Joslyn, M.A. Table Wines: The Technology of Their Production, 2nd ed.; University of California Press: Berkeley, CA, USA; Los Angeles, CA, USA, 1970; pp. 37–38. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | Retention Time (min) | Ret. Time RSD (%) 1 | Peak Capacity 2 | Peak Symmetry 3 | Peak Areas Repeatability RSD (%) 4 | RS 5 |
|---|---|---|---|---|---|---|
| OTA | 5.27 | ≤0.1 | 11.44 | 1.203 | 0.84; 0.54; 0.92 | 4.14 |
| OTB | 4.59 | ≤0.1 | 11.23 | 1.211 | 1.21; 0.48; 1.08 |
| Validation Parameters | OTA | OTB |
|---|---|---|
| Standard linear calibration range (µg L−1) 1 | 0.1–75 | 0.2–75 |
| Slope | 74,443 ± 750 | 34,818 ± 904 |
| Intercept | −46,480 ± 22,627 | −4219 ± 28,743 |
| Regression coefficient (r2) | 0.9996 | 0.9977 |
| Matrix (Tokaj wine) linear calibration range (µg L−1) 1 | 0.1–75 | 0.2–75 |
| Slope | 73,837 ± 581 | 32,986 ± 786 |
| Intercept | −20,233 ± 17,547 | 22,092 ± 25,014 |
| Regression coefficient (r2) | 0.9998 | 0.9980 |
| LOD (µg L−1) | 0.03 | 0.06 |
| LOQ (µg L−1) | 0.10 | 0.20 |
| Precision (RSD, %) 2 | 1.60 | 0.63 |
| Interday precision (RSD, %) 3 | 6.81 | 1.79 |
| Accuracy-spike recovery (%) ± SD 4 | 91.69 ± 1.64 | 99.07 ± 0.58 |
| OTA (EU limit: 2 µg L−1) | Archívné víno 6-Put, Zlatý Strapec 1993 | 1.22 µg L−1 |
| Archívné víno Esencia, Ostrožovič 2000 | 0.85 µg L−1 | |
| Muškát žltý, Slámové víno, Ostrožovič 2010 | 0.72 µg L−1 | |
| Archívné víno 6-Put, Ostrožovič 1999 | 0.37 µg L−1 | |
| OTB | None found |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kholová, A.; Lhotská, I.; Uhrová, A.; Špánik, I.; Machyňáková, A.; Solich, P.; Švec, F.; Šatínský, D. Determination of Ochratoxin A and Ochratoxin B in Archived Tokaj Wines (Vintage 1959–2017) Using On-Line Solid Phase Extraction Coupled to Liquid Chromatography. Toxins 2020, 12, 739. https://doi.org/10.3390/toxins12120739
Kholová A, Lhotská I, Uhrová A, Špánik I, Machyňáková A, Solich P, Švec F, Šatínský D. Determination of Ochratoxin A and Ochratoxin B in Archived Tokaj Wines (Vintage 1959–2017) Using On-Line Solid Phase Extraction Coupled to Liquid Chromatography. Toxins. 2020; 12(12):739. https://doi.org/10.3390/toxins12120739
Chicago/Turabian StyleKholová, Aneta, Ivona Lhotská, Adéla Uhrová, Ivan Špánik, Andrea Machyňáková, Petr Solich, František Švec, and Dalibor Šatínský. 2020. "Determination of Ochratoxin A and Ochratoxin B in Archived Tokaj Wines (Vintage 1959–2017) Using On-Line Solid Phase Extraction Coupled to Liquid Chromatography" Toxins 12, no. 12: 739. https://doi.org/10.3390/toxins12120739
APA StyleKholová, A., Lhotská, I., Uhrová, A., Špánik, I., Machyňáková, A., Solich, P., Švec, F., & Šatínský, D. (2020). Determination of Ochratoxin A and Ochratoxin B in Archived Tokaj Wines (Vintage 1959–2017) Using On-Line Solid Phase Extraction Coupled to Liquid Chromatography. Toxins, 12(12), 739. https://doi.org/10.3390/toxins12120739