Phylogeny-Guided Selection of Priority Groups for Venom Bioprospecting: Harvesting Toxin Sequences in Tarantulas as a Case Study


Abstract

1. Introduction
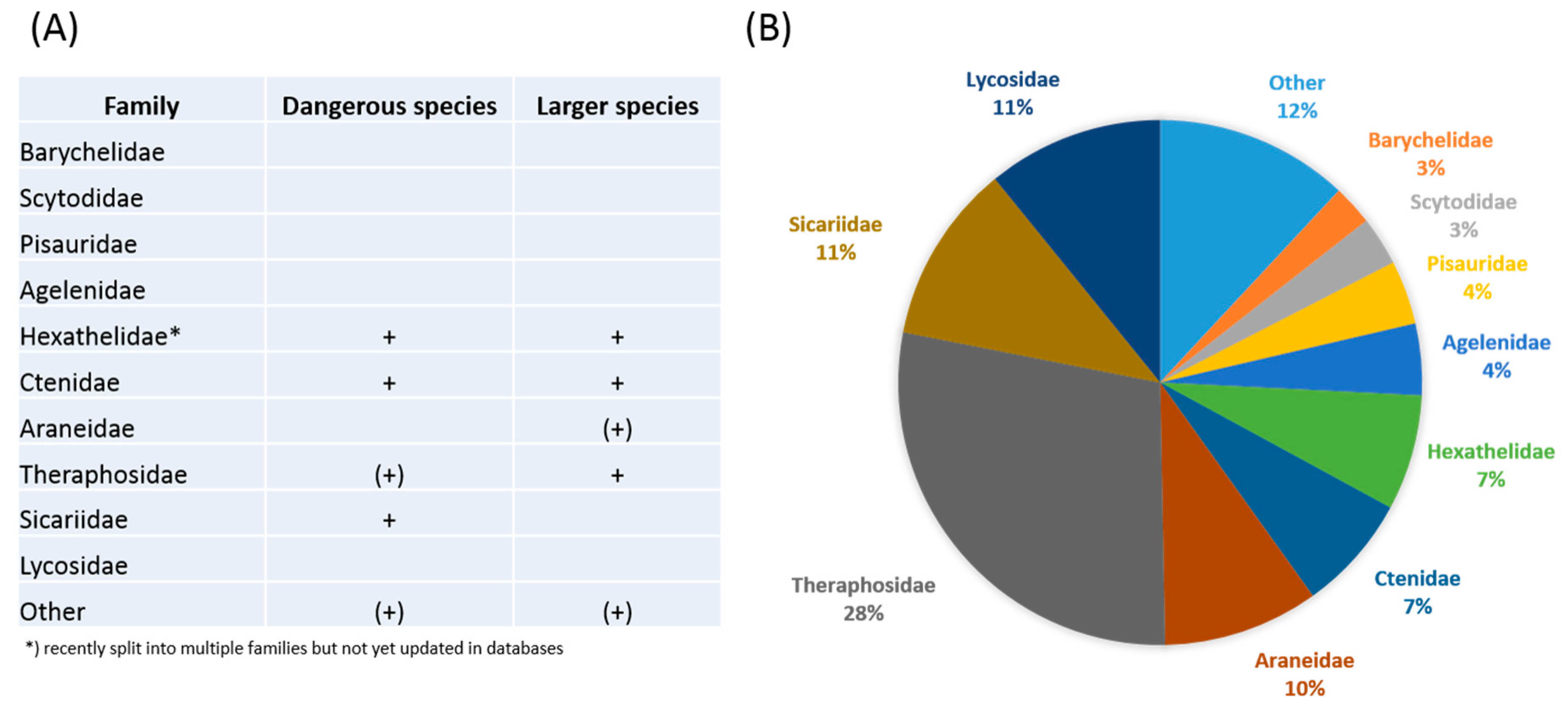
2. Novel Strategies in Venom Bioprospecting
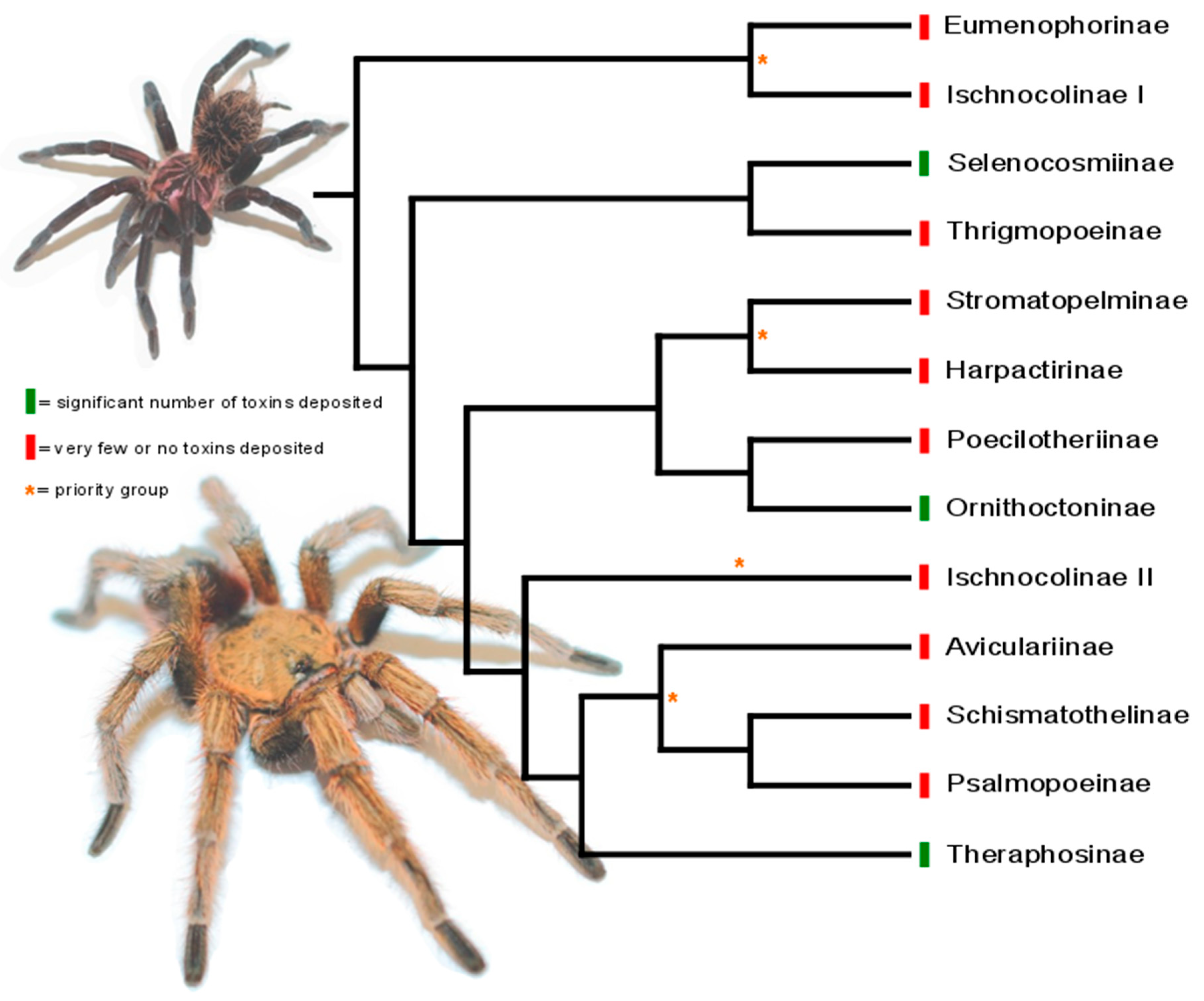
3. Phylogeny-Guided Selection of Priority Groups for Venom Bioprospecting: Tarantulas as a Case Study
4. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cragg, G.M.; Newman, D.J. Natural products: A continuing source of novel drug leads. Biochim. Biophys. Acta 2013, 1830, 3670–3695. [Google Scholar] [CrossRef] [PubMed]
- Fry, B.G.; Roelants, K.; Champagne, D.E.; Scheib, H.; Tyndall, J.D.; King, G.F.; Nevalainen, T.J.; Norman, J.A.; Lewis, R.J.; Norton, R.S.; et al. The toxicogenomic multiverse: Convergent recruitment of proteins into animal venoms. Annu. Rev. Genomics Hum. Genet. 2009, 10, 483–511. [Google Scholar] [CrossRef] [PubMed]
- Holford, M.; Daly, M.; King, G.F.; Norton, R.S. Venoms to the rescue. Science 2018, 6405, 842–844. [Google Scholar] [CrossRef] [PubMed]
- Von Reumont, B.M.; Campbell, L.I.; Jenner, R.A. Quo vadis venomics? A roadmap to neglected venomous invertebrates. Toxins 2014, 6, 3488–3551. [Google Scholar] [CrossRef] [PubMed]
- World Spider Catalog V.20.0. Natural History Museum Bern. 2019. Available online: www.wsc.nmbe.ch (accessed on 25 June 2019).
- Herzig, V.; King, G.F.; Undheim, E.A.B. Can we resolve the taxonomic bias in spider venom research? Toxicon X 2019, 1, 100005. [Google Scholar] [CrossRef]
- Hauke, T.J.; Herzig, V. Dangerous arachnids- Fake news or reality? Toxicon 2017, 138, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Pineda, S.S.; Chaumeil, P.A.; Kunert, A.; Kaas, Q.; Thang, M.W.C.; Le, L.; Nuhn, M.; Herzig, V.; Saez, N.J.; Cristofori-Armstrong, B.; et al. ArachnoServer 3.0: An online resource for automated discovery, analysis and annotation of spider toxins. Bioinformatics 2018, 34, 1074–1076. [Google Scholar] [CrossRef] [PubMed]
- Herzig, V.; Wood, D.L.A.; Newell, F.; Chaumeil, P.; Kaas, Q.; Binford, G.J.; Nicholson, G.M.; Gorse, D.; King, G.F. Arachnoserver 2.0, an updated online resource for spider toxin sequences and structures. Nucl. Acids Res. 2011, 39, D653–D657. [Google Scholar] [CrossRef] [PubMed]
- Pineda, S.S.; Undheim, E.A.; Rupasinghe, B.E.; Ikonomopoulou, M.P.; King, G.F. Spider venomics: Implications for drug discovery. Future Med. Chem. 2014, 6, 1699–1714. [Google Scholar] [CrossRef]
- Fry, B.G.; Koludarov, I.; Jackson, T.N.W.; Holford, M.; Terrat, Y.; Casewell, N.R.; Undheim, E.A.B.; Vetter, I.; Ali, S.A.; Dolyce, H.W.; et al. Seeing the Woods for the Trees: Understanding Venom Evolution as a Guide for Biodiscovery. In Venoms to Drugs: Venom as a Source for the Development of Human Therapeutics; RSC Drug Discovery Series No.42; King, G.F., Ed.; The Royal Society of Chemistry: Cambridge, UK, 2015; pp. 1–36. [Google Scholar]
- Vidal, N.; Hedges, S.B. The phylogeny of squamate reptiles (lizards, snakes and amphisbaenians) inferred from nine nuclear protein-coding genes. Comptes Rendus Biol. 2005, 328, 1000–1008. [Google Scholar] [CrossRef]
- Bond, J.E.; Garrison, N.L.; Hamilton, C.A.; Godwin, R.L.; Hedin, M.; Agnarsson, I. Phylogenomics resolve a spider backbone phylogeny and rejects a prevailing paradigm for orb web evolution. Curr. Biol. 2014, 24, 1765–1771. [Google Scholar] [CrossRef] [PubMed]
- Vences, M.; Sanchez, E.; Hauswaldt, J.S.; Eikelmann, D.; Rodriguez, A.; Carranza, S.; Donaire, D.; Gehara, M.; Helfer, V.; Lötters, S.; et al. Nuclear and mitochondrial multilocus phylogeny and survey of alkaloid content in true salamanders of the genus Salamandra (Salamandridae). Mol. Phylogenet. Evol. 2014, 73, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Lüddecke, T.; Krehenwinkel, H.; Canning, G.; Glaw, F.; Longhorn, S.J.; Tänzler, R.; Wendt, I.; Vences, M. Discovering the silk road: Nuclear and mitochondrial data resolve the phylogenetic relationships among theraphosid spider subfamilies. Mol. Phylogenet. Evol. 2018, 119, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Pennignton, M.W.; Czerwinski, A.; Norton, R.S. Peptide therapeutics from venom: Current status and potential. Bioorg. Med. Chem. 2018, 26, 2738–2758. [Google Scholar] [CrossRef] [PubMed]
- Von Reumont, B.M. Studying Smaller and Neglected Organisms in Modern Evolutionary Venomics Implementing RNASeq (Transcriptomics)—A Critical Guide. Toxins 2018, 10, 292. [Google Scholar] [CrossRef] [PubMed]
- Sunagar, K.; Morgenstern, D.; Reitzel, A.M.; Moran, Y. Ecological venomics: How genomics, transcriptomics and proteomics can shed new light on the ecology and evolution of venom. J. Proteom. 2016, 135, 62–72. [Google Scholar] [CrossRef]
- Jenner, R.A.; von Reumont, B.M.; Campbell, L.; Undheim, E.A.B. Parallel evolution of complex centipede venoms revealed by comparative proteotranscriptomic analyses. Mol. Biol. Evol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Von Reumont, B.M.; Blanke, A.; Richter, S.; Alvarez, F.; Bleidorn, C.; Jenner, R.A. The first venomous crustacean revealed by transcriptomics and functional morphology: Remipede venom glands express a unique toxin cocktail dominated by enzymes and a neurotoxin. Mol. Biol. Evol. 2014, 31, 48–58. [Google Scholar] [CrossRef]
- Von Reumont, B.M.; Undheim, E.A.B.; Jauss, R.T.; Jenner, R.A. Venomics of Remipede Crustaceans Reveals Novel Peptide Diversity and Illuminates the Venom’s Biological Role. Toxins 2017, 9, 234. [Google Scholar] [CrossRef]
- Santibáñez-López, C.E.; Ontano, A.Z.; Harvey, M.S.; Sharma, P.P. Transcriptomic Analysis of Pseudoscorpion Venom Reveals a Unique Cocktail Dominated by Enzymes and Protease Inhibitors. Toxins 2018, 10, 207. [Google Scholar] [CrossRef]
- Krämer, J.; Pohl, H.; Predel, R. Venom collection and analysis in the pseudoscorpion Chelifer cancroides (Pseudoscorpiones: Cheliferidae). Toxicon 2019, 162, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Paiva, A.L.B.; Mudadu, M.A.; Pereira, E.H.T.; Marri, C.A.; Guerra-Duarte, C.; Diniz, M.R.V. Transcriptome analysis of the spider Phoneutria reidyi venom gland reveals novel venom components for the genus Phoneutria. Toxicon 2019, 163, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Kuhn-Nentwig, L.; Langenegger, N.; Heller, M.; Koua, D.; Nentwig, W. The Dual Prey-Inactivation Strategy of Spiders-In-Depth Venomic Analysis of Cupiennius Salei. Toxins 2019, 11, 167. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Chen, B.; Xiao, Z.; Zhou, X.; Liu, Z. Transcriptomic Analysis of the Spider venom Gland Reveals Venom Diversity and Species Consanguinity. Toxins 2019, 11, 68. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, D.; Francke, O.F.; Bond, J.E. A tangle of forms and phylogeny: Extensive morphological homoplasy and molecular clock heterogeneity in Bonnetina and related tarantulas. Mol. Phylogenet. Evol. 2018, 127, 55–73. [Google Scholar] [CrossRef]
- Hüsser, M. A first phylogenetic analysis reveals a new arboreal tarantula genus from South America with description of a new species and two new species of Tapinauchenius Ausserer, 1871 (Araneae, Mygalomorphae, Theraphosidae). Zookeys 2018, 784, 59–93. [Google Scholar] [CrossRef]
- Foley, S.; Lüddecke, T.; Dong-Chiang, C.; Krehenwinkel, H.; Künzel, S.; Longhorn, S.J.; Wendt, I.; von Wirth, V.; Tänzler, R.; Vences, M.; et al. Tarantula phylogenomics: A robust phylogeny of multiple tarantula lineages inferred from transcriptome data sheds light on the prickly issue of urticating setae evolution. Mol. Phylogenet. Evol. 2019, 140. [Google Scholar] [CrossRef]
- Venomzone. SIB Swiss Institute of Bioinformatics. Available online: https://www.venomzone.expasy.org (accessed on 25 June 2019).
- Tarantupedia: An online Taxonomic Database for the World Largest Spiders. Available online: www.tarantupedia.com (accessed on 25 June 2019).
- Escoubas, P.; Rash, L. Tarantulas: Eight-legged pharmacists and combinatorial chemists. Toxicon 2004, 43, 555–574. [Google Scholar] [CrossRef]
- Oldrati, V.; Koua, D.; Allard, P.M.; Hulo, N.; Arrell, M.; Nentwig, W.; Lisacek, F.; Wolfender, J.L.; Kuhn-Nentwig, L.; Stöcklin, R. Peptidomic and transcriptomic profiling of four distinct spider venoms. PLoS ONE 2017, 12, e0172966. [Google Scholar] [CrossRef]
- Oldrati, V.; Arrell, M.; Violette, A.; Perret, F.; Sprüngli, X.; Wolfender, J.L.; Stöcklin, R. Advances in Venomics. Mol. Biosyst. 2016, 12, 3530–3543. [Google Scholar] [CrossRef]
- Drukewitz, S.H.; von Reumont, B.M. The significance of comparative genomics in modern evolutionary venomics. Front. Ecol. Evol. 2019. [Google Scholar] [CrossRef]
- Piacentini, L.N.; Ramirez, M.J. Hunting the wolf: A molecular phylogeny of the wolf spiders (Araneae, Lycosidae). Mol. Phylogenet. Evol. 2019, 136, 227–240. [Google Scholar] [CrossRef] [PubMed]
- Huber, B.A.; Eberle, J.; Dimitrov, D. The phylogeny of pholcid spiders: A critical evaluation of relationships suggested by molecular data (Araneae, Pholcidae). Zookeys 2018, 789, 51–101. [Google Scholar] [CrossRef] [PubMed]
- Godwin, R.L.; Opatova, V.; Garrison, N.L.; Hamilton, C.A.; Bond, J.E. Phylogeny of a cosmopolitan family of morphologically conserved trapdoor spiders (Mygalomorphae, Ctenizidae) using Anchored Hybrid Enrichment, with a description of the family, Halonoproctidae Pocock 1901. Mol. Phylogenet. Evol. 2018, 126, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Kallal, R.J.; Fernandez, R.; Giribet, G.; Hormiga, G. A phylotranscriptomic backbone of the orb-weaving spider family Araneidae (Arachnida, Araneae) supported by multiple methodological approaches. Mol. Phylogenet. Evol. 2018, 126, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.Q.; Piel, W.H. The origins of Psechridae: Web-building lycosoid spiders. Mol. Phylogenet. Evol. 2018, 125, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Barnosky, A.D.; Matzke, N.; Tomiya, S.; Wogan, G.O.; Swartz, B.; Quental, T.B.; Marshall, C.; McGuire, J.L.; Lindsey, E.L.; Maguire, K.C.; et al. Has the Earth’s sixth mass extinction already arrived? Nature 2011, 471, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Dirzo, R.; Young, H.S.; Galetti, M.; Ceballos, G.; Isaac, N.J.; Collen, B. Defaunation in the Anthropocene. Science 2014, 345, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Régnier, C.; Achaz, G.; Lambert, A.; Cowie, R.H.; Bouchet, P.; Fontaine, B. Mass extinction in poorly known taxa. Proc. Natl. Acad. Sci. USA 2015, 112, 7761–7766. [Google Scholar]
- Wake, D.B.; Koo, M.S. Amphibians. Curr. Biol. 2018, 28, R1237–R1241. [Google Scholar] [CrossRef]
- Collins, J.P. Amphibian decline and extinction: What we know and what we need to learn. Dis. Aquat. Organ. 2010, 92, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Wake, D.B.; Vredenburg, V.T. Are we in the midst of the sixth mass extinction? A view from the world of amphibians. Proc. Natl. Acad. Sci. USA 2008, 105, 11466–11473. [Google Scholar] [CrossRef] [PubMed]
- Overmann, J.; Hartmann Scholz, A. Microbiological Research Under the Nagoya Protocol: Facts and Fiction. TIMI 2016, 1385. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subfamily | Number of Species WSC | Number of Toxins AS | Number of Toxins VZ |
|---|---|---|---|
| Acanthopelminae 1 | 2 | 0 | 0 |
| Aviculariinae | 31 | 0 | 1 |
| Eumenophorinae | 62 | 10 | 1 |
| Harpactirinae | 62 | 8 | 7 |
| Ischnocolinae 2 | 85 | 3 | 0 |
| Ornithoctoninae | 27 | 247 | 339 |
| Poecilotheriinae 3 | 14 | 0 | 0 |
| Psalmopoeinae | 27 | 6 | 7 |
| Schismatothelinae 2 | 21 | 0 | 0 |
| Selenocosmiinae | 114 | 76 | 120 |
| Selenogyrinae 1 | 10 | 0 | 0 |
| Stromatopelminae | 10 | 5 | 6 |
| Theraphosinae | 526 | 95 | 51 |
| Thrigmopoeinae | 9 | 0 | 0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lüddecke, T.; Vilcinskas, A.; Lemke, S. Phylogeny-Guided Selection of Priority Groups for Venom Bioprospecting: Harvesting Toxin Sequences in Tarantulas as a Case Study. Toxins 2019, 11, 488. https://doi.org/10.3390/toxins11090488
Lüddecke T, Vilcinskas A, Lemke S. Phylogeny-Guided Selection of Priority Groups for Venom Bioprospecting: Harvesting Toxin Sequences in Tarantulas as a Case Study. Toxins. 2019; 11(9):488. https://doi.org/10.3390/toxins11090488
Chicago/Turabian StyleLüddecke, Tim, Andreas Vilcinskas, and Sarah Lemke. 2019. "Phylogeny-Guided Selection of Priority Groups for Venom Bioprospecting: Harvesting Toxin Sequences in Tarantulas as a Case Study" Toxins 11, no. 9: 488. https://doi.org/10.3390/toxins11090488
APA StyleLüddecke, T., Vilcinskas, A., & Lemke, S. (2019). Phylogeny-Guided Selection of Priority Groups for Venom Bioprospecting: Harvesting Toxin Sequences in Tarantulas as a Case Study. Toxins, 11(9), 488. https://doi.org/10.3390/toxins11090488