From Snake Venom’s Disintegrins and C-Type Lectins to Anti-Platelet Drugs



Abstract
1. Introduction
1.1. Integrins
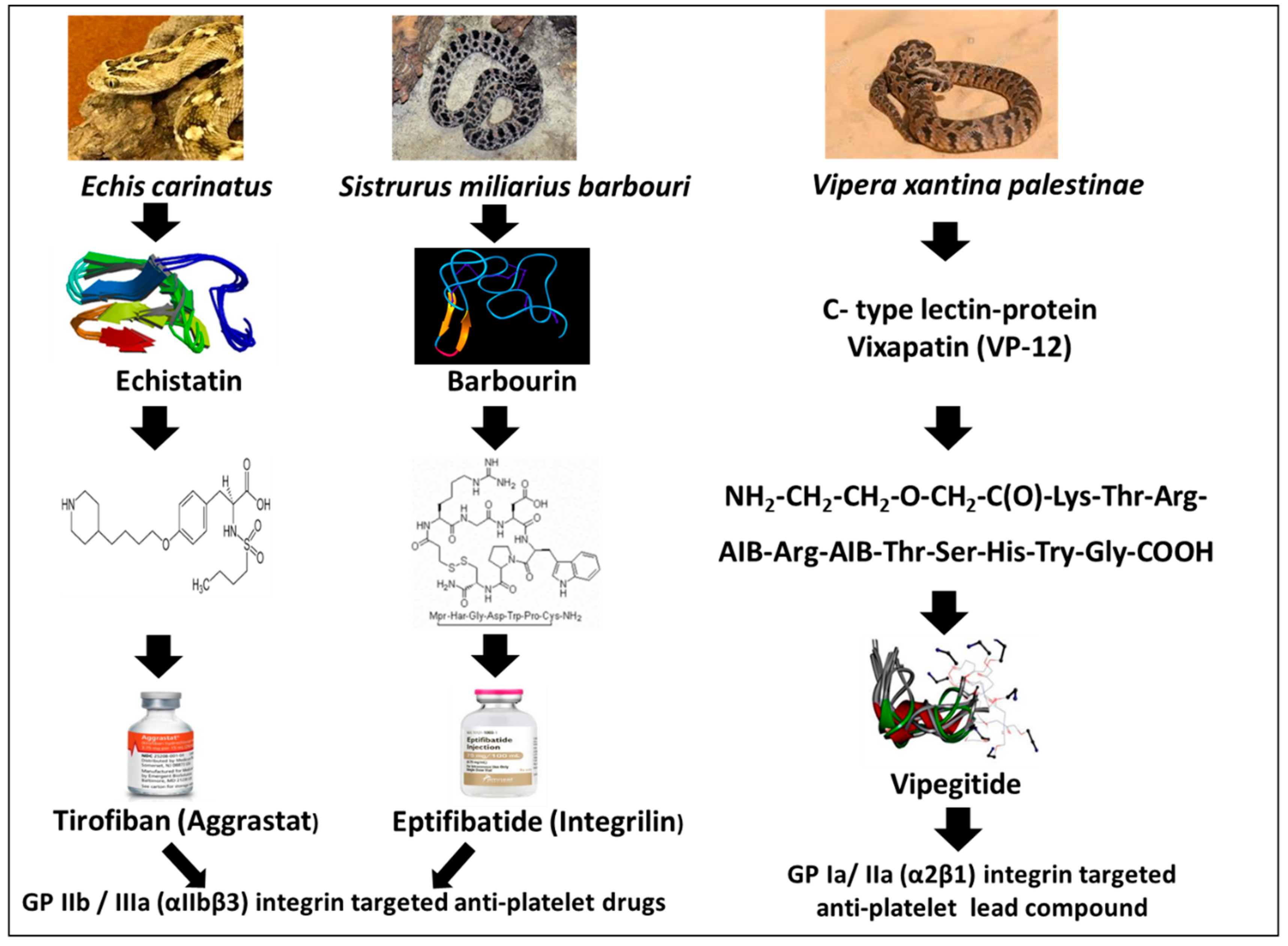
1.2. Disintegrins
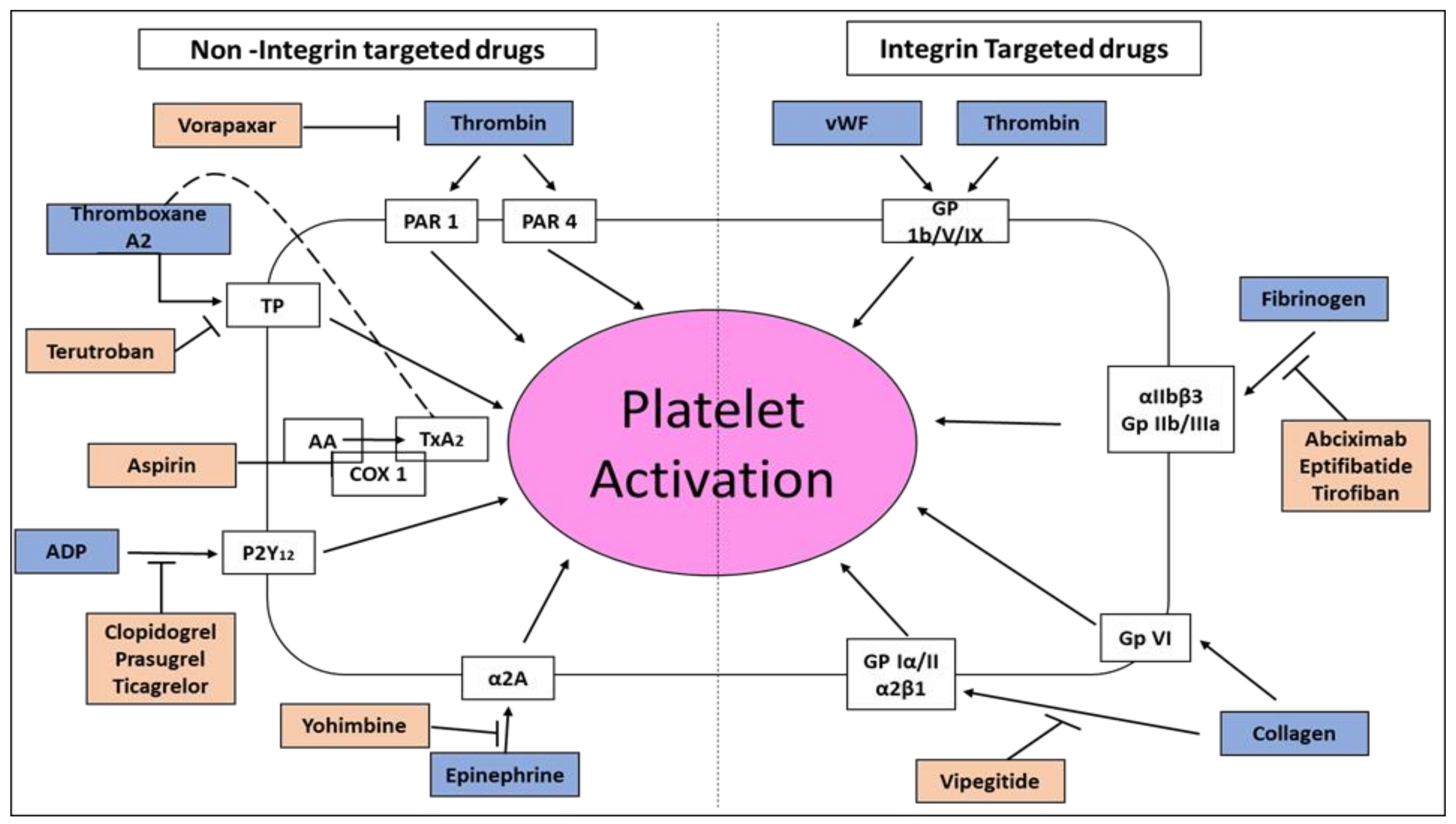
1.3. Platelets’ Integrins
1.4. Inhibition of Platelet Aggregation in Acute Coronary Syndromes and Percutaneous Coronary Intervention
1.5. Tirofiban (Aggrastat)
1.6. Eptifibatide (Integrilin)
1.7. Platelet’s GP Ia/IIa (α2β1) Integrin Receptor for Collagen
1.8. Vipegitide, a Partial Antagonist of α2β1 Integrin with Antiplatelet Activity
2. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CTL | C-type lectin-like protein |
| ECM | extracellular matrix |
| RGD | Arg-Gly-Asp |
| RTS | Arg-Thr-Ser |
| KTS | Lys-Thr-Ser |
| KGD | Lys-Gly-Asp |
| MVD | Met-Val-Asp |
| VGD | Val-Gly-Asp |
| WGD | Trp-Gly-Asp |
| MGD | Met-Gly-Asp |
| MLD | Met-Leu-Asp |
| ADP | adenosine diphosphate |
| ACS | acute coronary syndrome |
| PCI | percutaneous coronary intervention |
| STEMI | ST-segment elevation myocardial infarction |
| NSTEMI, | non STEMI |
| MI | myocardial infarct |
| vWF | von Willebrand factor |
| MALDI-TOF | matrix-assisted laser desorption/ionization- time-of-flight mass spectrometry |
| nM | nanomolar |
| kDa | kilodalton |
References
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef]
- Schwartz, M.A.; Ginsberg, M.H. Networks and crosstalk: Integrin signaling spreads. Nat. Cell Biol. 2002, 4, E65–E68. [Google Scholar] [CrossRef] [PubMed]
- Shattil, S.J.; Kim, C.; Ginsberg, M.H. The final steps of integrin activation: The end game. Nat. Rev. Mol. Cell Biol. 2010, 11, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Marcinkiewicz, C. Applications of snake venom components to modulate integrin activities in cell-matrix interactions. Int. J. Biochem. Cell Biol. 2013, 45, 1974–1986. [Google Scholar] [CrossRef]
- Arruda Macêdo, J.K.; Fox, J.W.; de Souza Castro, M. Disintegrins from snake venoms and their applications in cancer research and therapy. Curr. Protein Pept. Sci. 2015, 16, 532–548. [Google Scholar] [CrossRef]
- Kapp, T.G.; Rechenmacher, F.; Neubauer, S.; Maltsev, O.V.; Cavalcanti-Adam, E.A.; Zarka, R.; Reuning, U.; Notni, J.; Wester, H.J.; Mas-Moruno, C.; et al. A Comprehensive Evaluation of the Activity and Selectivity Profile of Ligands for RGD-binding Integrins. Sci. Rep. 2017, 7, 39805. [Google Scholar] [CrossRef] [PubMed]
- Waheed, H.; Moin, S.F.; Choudhary, M. Snake venom: From deadly toxins to life-saving therapeutics. Curr. Med. Chem. 2017, 24, 1874–1891. [Google Scholar] [CrossRef] [PubMed]
- Swenson, S.; Ramu, S.; Markland, F.S. Anti-angiogenesis and RGD-containing snake venom disintegrins. Curr. Pharm. Des. 2007, 13, 2860–2871. [Google Scholar] [CrossRef]
- Sanz, L.; Bazaa, A.; Marrakchi, N.; Pérez, A.; Chenik, M.; Bel Lasfer, Z.; El Ayeb, M.; Calvete, J.J. Molecular cloning of disintegrins from Cerastes vipera and Macrovipera lebetina transmediterranea venom gland cDNA libraries: Insight into the evolution of the snake venom integrin-inhibition system. Biochem. J. 2006, 395, 385–392. [Google Scholar] [CrossRef]
- Bazan-Socha, S.; Kisiel, D.G.; Young, B.; Theakston, R.D.; Calvete, J.J.; Sheppard, D.; Marcinkiewicz, C. Structural requirements of MLD-containing disintegrins for functional interaction with α4β1 and α9β1 integrins. Biochemistry 2004, 43, 1639–1647. [Google Scholar] [CrossRef]
- Brown, M.C.; Eble, J.A.; Calvete, J.J.; Marcinkiewicz, C. Structural requirements of KTS-disintegrins for inhibition of α1β1 integrin. Biochem. J. 2009, 417, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.S.; Berger, B.W.; Billings, P.C. The structure and function of platelet integrins. J. Thromb. Haemost. 2009, 7, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Bennett, J.S. Regulation of integrins in platelets. Biopolymers 2015, 104, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Li, X.; Shi, X.; Zhu, M.; Wang, J.; Huang, S.; Huang, X.; Wang, H.; Li, L.; Deng, H.; et al. Platelet integrin αIIbβ3: Signal transduction, regulation, and its therapeutic targeting. J. Hematol. Oncol. 2019, 12, 26. [Google Scholar] [CrossRef] [PubMed]
- Yeung, J.; Li, W.; Holinstat, M. Platelet signaling and disease: Targeted therapy for thrombosis and other related diseases. Pharmacol. Rev. 2018, 70, 526–548. [Google Scholar] [CrossRef]
- Andrews, R.K.; Gardiner, E.E.; Shen, Y.; Berndt, M.C. Structure-activity relationships of snake toxins targeting platelet receptors, glycoprotein Ib-IX-V and glycoprotein VI. Curr. Med. Chem. Cardiovasc. Hematol. Agents 2003, 1, 143–149. [Google Scholar] [CrossRef]
- Ley, K.; Rivera-Nieves, J.; Sandborn, W.J.; Shattil, S. Integrin-based therapeutics: Biological basis, clinical use and new drugs. Nat. Rev. Drug Discov. 2016, 15, 173–183. [Google Scholar] [CrossRef]
- Huang, T.F.; Hsu, C.C.; Kuo, Y.J. Anti-thrombotic agents derived from snake venom proteins. Thromb. J. 2016, 14, 18. [Google Scholar] [CrossRef]
- Arora, R.R.; Rai, F. Antiplatelet intervention in acute coronary syndrome. Am. J. Ther. 2009, 16, 29–40. [Google Scholar] [CrossRef]
- Gan, Z.R.; Gould, R.J.; Jacobs, J.W.; Friedman, P.A.; Polokoff, M.A. A potent platelet aggregation inhibitor from the venom of the viper, Echis carinatus. J. Biol. Chem. 1988, 263, 19827–19832. [Google Scholar]
- Lynch, J.J.; Cook, J.J.; Sitko, G.R.; Holahan, M.A.; Ramjit, D.R.; Mellott, M.J.; Stranieri, M.T.; Stabilito, I.I.; Zhang, G.; Lynch, R.J. Nonpeptide glycoprotein IIb/IIIa inhibitors. Antithrombotic effects of MK-0383. J. Pharmacol. Exp. Ther. 1995, 272, 20–32. [Google Scholar]
- Barrett, J.S.; Murphy, G.; Peerlinck, K.; De Lepeleire, I.; Gould, R.J.; Panebianco, D.; Hand, E.; Deckmyn, H.; Vermylen, J.; Arnout, J. Pharmacokinetics and pharmacodynamics of MK-383, a selective nonpeptide platelet glyco-protein-IIb/IIIa receptor antagonist, in healthy men. Clin. Pharmacol. Ther. 1994, 56, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Vickers, S.; Theoharides, A.D.; Arison, B.; Balani, S.K.; Cui, D.; Duncan, C.A.; Ellis, J.D.; Gorham, L.M.; Polsky, S.L.; Prueksaritanont, T.; et al. In vitro and in vivo studies on the metabolism of tirofiban. Drug Metab. Dispos. 1999, 27, 1360–1366. [Google Scholar]
- EPIC investigators. Use of a monoclonal antibody directed against the platelet glycoprotein IIb/IIIa receptor in high-risk coronary angioplasty. N. Engl. J. Med. 1994, 330, 956–961. [Google Scholar] [CrossRef] [PubMed]
- EPILOG Investigators. Effect of the platelet glycoprotein IIb/IIIa receptor inhibitor Abciximab with lower heparin dosages on ischemic complications of percutaneous coronary revascularization. N. Engl. J. Med. 1997, 336, 1689–1696. [Google Scholar] [CrossRef] [PubMed]
- CAPTURE Investigators. Randomized placebo-controlled trial of Abciximab before and during coronary intervention in refractory unstable angina. Lancet 1997, 349, 1429–1435. [Google Scholar] [CrossRef]
- EPISTENT Investigators. Randomized, placebo-controlled and balloon angioplasty controlled trial to assess the safety of coronary stenting with the use of platelet glycoprotein IIb/IIIa blockade. Lancet 1998, 352, 87–92. [Google Scholar] [CrossRef]
- Alexander, J.H.; Harrington, R.A. Recent antiplatelet drug trials in the acute coronary syndromes. Clinical interpretation of PRISM, PRISM-PLUS, PARAGON A and PURSUIT. Drugs 1998, 56, 965–976. [Google Scholar] [CrossRef] [PubMed]
- PURSUIT investigators. Systematic adjudication of myocardial infarction end-points in an international clinical trial. Curr. Control. Trials Cardiovasc. Med. 2001, 2, 180–186. [Google Scholar] [CrossRef]
- IMPACT Investigators. Multicenter, randomized, double-blind, placebo-controlled trial of the platelet integrin glycoprotein IIb/IIIa blocker Integrilin in elective coronary intervention. Circulation 1995, 91, 2151–2157. [Google Scholar] [CrossRef]
- ESPRIT Investigators. Effect of glycoprotein IIb/IIIa receptor inhibition on angiographic complications during percutaneous coronary intervention in the ESPRIT trial. J. Am. Coll. Cardiol. 2001, 38, 653–658. [Google Scholar] [CrossRef]
- PRISM-PLUS Investigators. Inhibition of the platelet glycoprotein IIb/IIIa receptor with tirofiban in unstable angina and non-Q-wave myocardial infarction. N. Engl. J. Med. 1998, 338, 1488–1497. [Google Scholar] [CrossRef] [PubMed]
- The RESTORE Investigators. Effects of platelet glycoprotein IIb/IIIa blockade with tirofiban on adverse cardiac events in patients with unstable angina or acute myocardial infarction undergoing coronary angio-plasty. Circulation 1997, 96, 1445–1453. [Google Scholar] [CrossRef]
- TARGET Investigators. Comparison of two platelet glycoprotein IIb/IIIa inhibitors, tirofiban and abciximab, for the prevention of ischaemic events with percutaneous coronary revascularization. N. Engl. J. Med. 2001, 344, 1888–1894. [Google Scholar] [CrossRef] [PubMed]
- Scarborough, R.M.; Rose, J.W.; Hsu, M.A.; Phillips, D.R.; Fried, V.A.; Campbell, A.M.; Nannizzi, L.; Charo, I.F. Barbourin. A GPIIb-IIIa specific integrin antagonist from the venom of Sistrurus m. barbouri. J. Biol. Chem. 1991, 266, 9359–9362. [Google Scholar]
- Scarborough, R.M.; Rose, J.W.; Naughton, M.A.; Phillips, D.R.; Nannizzi, L.; Arfsten, A.; Campbell, A.M.; Charo, I.F. Characterization of the integrin specificities of disintegrins isolated from American pit viper venoms. J. Biol. Chem. 1993, 268, 1058–1065. [Google Scholar]
- Scarborough, R.M.; Naughton, M.A.; Teng, W.; Rose, J.W.; Phillips, D.R.; Nannizzi, L.; Arfsten, A.; Campbell, A.M.; Charo, I.F. Design of potent and specific integrin antagonists. Peptide antagonists with high specificity for glycoprotein IIb-IIIa. J. Biol. Chem. 1993, 268, 1066–1073. [Google Scholar] [PubMed]
- Phillips, D.R.; Scarborough, R.M. Clinical pharmacology of eptifibatide. Am. J. Cardiol. 1997, 80, 11–20. [Google Scholar] [CrossRef]
- Gilchrist, I.C. Platelet glycoprotein IIb/IIIa inhibitors in percutaneous coronary intervention: Focus on the pharmacokinetic-pharmacodynamic relationships of eptifibatide. Clin. Pharmacokinet. 2003, 42, 703–720. [Google Scholar] [CrossRef]
- Derex, L.; Paris, C.; Nighoghossian, N. Combining intravenous thrombolysis and antithrombotic agents in stroke: An update. J. Am. Heart Assoc. 2018, 7, e007454. [Google Scholar] [CrossRef]
- Rasican Surin, W.; Barthwal, M.K.; Dikshit, M. Platelet collagen receptors, signaling and antagonism: Emerging approaches for the prevention of intravascular thrombosis. Thrombo. Res. 2008, 122, 786–803. [Google Scholar] [CrossRef]
- Steinhubl, S.R.; Moliterno, D.J. The role of the platelet in the pathogenesis of atherothrombosis. Am. J. Cardiovasc. Drugs 2005, 5, 399–408. [Google Scholar] [CrossRef]
- Nieswandt, B.; Brakebusch, C.; Bergmeier, W.; Schulte, V.; Bouvard, D.; Mokhtari-Nejad, R.; Lindhout, T.; Heemskerk, J.W.; Zirngibl, H.; Fässler, R. Glycoprotein VI but not alpha2beta1 integrin is essential for platelet interaction with collagen. EMBO J. 2001, 20, 2120–2130. [Google Scholar] [CrossRef] [PubMed]
- Holtkotter, O.; Nieswandt, B.; Smyth, N.; Muller, W.; Hafner, M.; Schulte, V.; Krieg, T.; Eckes, B. Integrin alpha 2-deficient mice develop normally, are fertile, but display partially defective platelet interaction with collagen. J. Biol. Chem. 2002, 277, 10789–10794. [Google Scholar] [CrossRef]
- Arlinghaus, F.T.; Eble, J.A. C-type lectin-like proteins from snake venoms. Toxicon 2012, 60, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Rath, D.; Schaeffeler, E.; Winter, S.; Levertov, S.; Müller, K.; Droppa, M.; Stimpfle, F.; Langer, H.F.; Gawaz, M.; Schwab, M.; et al. GPla Polymorphisms Are Associated with Outcomes in Patients at High Cardiovascular Risk. Front Cardiovasc. Med. 2017, 4, 52. [Google Scholar] [CrossRef]
- Handa, M.; Watanabe, K.; Kawai, Y.; Kamata, T.; Koyama, T.; Nagai, H.; Ikeda, Y. Platelet unresponsiveness to collagen: Involvement of glycoprotein Ia-IIa (alpha 2 beta 1 integrin) deficiency associated with a myeloproliferative disorder. Thromb. Haemost. 1995, 73, 521–528. [Google Scholar] [CrossRef]
- Goodman, S.L.; Picard, M. Integrins as therapeutic targets. Trends Pharmacol. Sci. 2012, 33, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Breuer, J.; Schneider-Hohendorf, T.; Ostkamp, P.; Herich, S.; Rakhade, S.; Antonijevic, I.; Klotz, L.; Wiendl, H.; Schwab, N. VLA-2 blockade in vivo by vatelizumab induces CD4+FoxP3+ regulatory T cells. Int. Immunol. 2019, 2019. [Google Scholar] [CrossRef]
- White, T.C.; Berny, M.A.; Robinson, D.K.; Yin, H.; DeGrado, W.F.; Hanson, S.R.; McCarty, O.J. The leech product saratin is a potent inhibitor of platelet integrin α2β1 and von Willebrand factor binding to collagen. FEBS J. 2007, 274, 1481–1491. [Google Scholar] [CrossRef]
- Miller, M.W.; Basra, S.; Kulp, D.W.; Billings, P.C.; Choi, S.; Beavers, M.P.; McCarty, O.J.; Zou, Z.; Kahn, M.L.; Bennett, J.S.; et al. Small molecule inhibitors of integrin alpha2beta1 that prevent pathological thrombus formation via an allosteric mechanism. Proc. Natl. Acad. Sci. USA 2009, 106, 719–724. [Google Scholar] [CrossRef] [PubMed]
- San Antonio, J.D.; Zoeller, J.J.; Habursky, K.; Turner, K.; Pimtong, W.; Burrows, M.; Choi, S.; Basra, S.; Bennett, J.S.; DeGrado, W.F.; et al. Key role for the integrin α2β1 in experimental and developmental angiogenesis. Am. J. Pathol. 2009, 175, 1338–1347. [Google Scholar] [CrossRef]
- Choi, S.; Vilaire, G.; Marcinkiewicz, C.; Winkler, J.D.; Bennett, J.S.; DeGrado, W.F. Small-molecule inhibitors of integrin alpha2beta1. J. Med. Chem. 2007, 50, 5457–5462. [Google Scholar] [CrossRef]
- Eble, J.A.; Tuckwell, D.S. The alpha2beta1 integrin inhibitor rhodocetin binds to the A-domain of the integrin alpha2 subunit proximal to the collagen-binding site. Biochem. J. 2003, 376, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Marcinkiewicz, C.; Lobb, R.R.; Marcinkiewicz, M.M.; Daniel, J.L.; Smith, J.B.; Dangelmaier, C.; Weinreb, P.H.; Beacham, D.A.; Niewiarowski, S. Isolation and characterization of EMS16, a C-lectin type protein from Echis multisquamatus venom, a potent and selective inhibitor of the alpha2beta1 integrin. Biochemistry 2000, 39, 9859–9867. [Google Scholar] [CrossRef]
- Momic, T.; Cohen, G.; Reich, R.; Arlinghaus, F.T.; Eble, J.A.; Marcinkiewicz, C.; Lazarovici, P. Vixapatin (VP12), a c-type lectin-protein from Vipera xantina palestinae venom: Characterization as a novel anti-angiogenic compound. Toxins 2012, 4, 862–877. [Google Scholar] [CrossRef]
- Arlinghaus, F.T.; Momic, T.; Ammar, N.A.; Shai, E.; Spectre, G.; Varon, D.; Marcinkiewicz, C.; Heide, H.; Lazarovici, P.; Eble, J.A. Identification of α2β1 integrin inhibitor VP-i with anti-platelet properties in the venom of Vipera palaestinae. Toxicon 2013, 64, 96–105. [Google Scholar] [CrossRef]
- Momic, T.; Arlinghaus, F.T.; Arien-Zakay, H.; Katzhendler, J.; Eble, J.A.; Marcinkiewicz, C.; Lazarovici, P. Pharmacological aspects of Vipera xantina palestinae venom. Toxins 2011, 3, 1420–1432. [Google Scholar] [CrossRef] [PubMed]
- Momic, T.; Katzhendler, J.; Shai, E.; Noy, E.; Senderowitz, H.; Eble, J.A.; Marcinkiewicz, C.; Varon, D.; Lazarovici, P. Vipegitide: A folded peptidomimetic partial antagonist of α2β1 integrin with antiplatelet aggregation activity. Drug Des. Devel. Ther. 2015, 9, 291–304. [Google Scholar]



{kind=link}
{kind=link}
| Drug | Product Availability | FDA-Approved Indications 4 | Clinical Trial |
|---|---|---|---|
| Abciximab 1 (ReoPro) | 5 mL vial (2 mg/mL) | PCI only | EPIC |
| Eptifibatide 2 (Integrilin) | a. 100 mL (750 µg/mL) | Unstable Angina/NSTEMI/PCI | PURSUIT |
| b. 10 mL/vial (2000 µg/mL) | ESPRIT | ||
| c. 100 mL/vial (2000 µg/mL) | IMPACT-II | ||
| Tirofiban 3 (Aggrastat) | a. 250 mL (50 µg/mL) | Unstable Angina / NSTEMI only | PRISM |
| b. 500 mL (50 µg/mL) | PRISM-PLUS | ||
| c. 25 mL/vial (250 µg/mL) | RESTORE | ||
| d. 50 mL /vial (250 µg/mL) | TARGET |
| Drug | Description | Indication | Major Clinical Trial Findings * | Major Side Effects | References |
|---|---|---|---|---|---|
| Abciximab | Mab targeting αIIbβ3 and inhibiting fibrinogen binding | Prevention of cardiac ischemic effects in patients with ACS undergoing PCI | EPIC (2099 patients): 35% fewer events ** | Major bleeding Thrombocytopenia | [24] |
| EPILOG (2972 patients): 65% fewer events, benefit 1 year | [25] | ||||
| CAPTURE (1265 patients): 29% fewer events | [26] | ||||
| EPISTENT (2399 patients): 51% fewer events | [27] | ||||
| Eptifibatide | A cyclic peptide that blocks the binding of fibrinogen to αIIbβ3 integrin | Patients with NSTEMI who are undergoing PCI | PURSUIT (10,948 patients): 10% fewer events, benefit 6 months | Major bleeding unchanged, minor bleeding slightly increased with Eptifibatide treatment in IMPACT‑II; | [28,29] |
| IMPACT II (4010 patients): 19% fewer events | [30] | ||||
| ESPRIT (2064 patients): significantly fewer events, benefit 6 months and 1 year | [31] | ||||
| Tirofiban | A small molecule that that blocks the binding of fibrinogen to αIIbβ3 integrin | Patients with unstable angina or NSTEMI who are undergoing PCI | PRISM (3232 patients): 32% fewer events | Bleeding similar in both groups in PRISM, PRISM-PLUS and RESTORE; Reversible thrombocytopenia was three times more common in Tirofiban-treated patients than placebo-treated patients in PRISM, no difference was observed between these groups in RESTORE | [28] |
| PRISM-PLUS (1915 patients): 28% fewer events compared to heparin and aspirin) | [28,32] | ||||
| RESTORE (2139 patients): reduction of events at 2- and 7-days post-treatment | [33] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lazarovici, P.; Marcinkiewicz, C.; Lelkes, P.I. From Snake Venom’s Disintegrins and C-Type Lectins to Anti-Platelet Drugs. Toxins 2019, 11, 303. https://doi.org/10.3390/toxins11050303
Lazarovici P, Marcinkiewicz C, Lelkes PI. From Snake Venom’s Disintegrins and C-Type Lectins to Anti-Platelet Drugs. Toxins. 2019; 11(5):303. https://doi.org/10.3390/toxins11050303
Chicago/Turabian StyleLazarovici, Philip, Cezary Marcinkiewicz, and Peter I. Lelkes. 2019. "From Snake Venom’s Disintegrins and C-Type Lectins to Anti-Platelet Drugs" Toxins 11, no. 5: 303. https://doi.org/10.3390/toxins11050303
APA StyleLazarovici, P., Marcinkiewicz, C., & Lelkes, P. I. (2019). From Snake Venom’s Disintegrins and C-Type Lectins to Anti-Platelet Drugs. Toxins, 11(5), 303. https://doi.org/10.3390/toxins11050303